
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Ultra-fast Suzuki and Heck reactions for the synthesis of styrenes and stilbenes using arenediazonium salts as super-electrophiles†
Marina E.
Trusova
a,
Mireia
Rodriguez-Zubiri
b,
Ksenia V.
Kutonova
ac,
Nicole
Jung
cd,
Stefan
Bräse
cd,
François-Xavier
Felpin
 *be and
Pavel S.
Postnikov
*f
*be and
Pavel S.
Postnikov
*f
aDepartment of Biotechnology and Organic Chemistry, National Research Tomsk Polytechnic University, 43 Lenin ave., 634050 Tomsk, Russia. Tel: +7(3822)563-861
bUniversité de Nantes, UFR des Sciences et des Techniques, CNRS UMR 6230, CEISAM, 2 rue de la Houssinière, 44322 Nantes Cedex 3, France. E-mail: fx.felpin@univ-nantes.fr
cInstitute of Organic Chemistry, Karlsruhe Institute of Technology (KIT), Fritz-Haber-Weg 6, 76131 Karlsruhe, Germany
dInstitute of Toxicology and Genetics, Karlsruhe Institute of Technology (KIT), Hermann-von-Helmholtz-Platz 1, D-76344 Eggenstein-Leopoldshafen, Germany
eInstitut Universitaire de France, 1 rue Descartes, 75231 Paris Cedex 05, France
fDepartment of Technology of Organic Substances and Polymer Materials, Tomsk Polytechnic University, 43 Lenin ave., 634050 Tomsk, Russia. E-mail: postnikov@tpu.ru
First published on 14th September 2017
Abstract
The super-electrophilic properties of arenediazonium salts have been exploited for achieving ultra-fast palladium-catalyzed coupling reactions with turnover frequencies up to 16![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 200 h−1. These ultra-fast coupling reactions have been exemplified with the synthesis of prone-to-polymerization styrenes within seconds through Suzuki cross-couplings with potassium vinyltrifluoroborate. Heterocycles and functional groups such as halides were well tolerated. The ambivalent properties of potassium vinyltrifluoroborate also allowed the development of ultra-fast sequential Suzuki–Heck reactions for the preparation of symmetrical and unsymmetrical stilbenes within minutes.
200 h−1. These ultra-fast coupling reactions have been exemplified with the synthesis of prone-to-polymerization styrenes within seconds through Suzuki cross-couplings with potassium vinyltrifluoroborate. Heterocycles and functional groups such as halides were well tolerated. The ambivalent properties of potassium vinyltrifluoroborate also allowed the development of ultra-fast sequential Suzuki–Heck reactions for the preparation of symmetrical and unsymmetrical stilbenes within minutes.
Introduction
Arenediazonium salts are a class of powerful electrophiles allowing for a plethora of organic transformations.1 For instance, the use of arenediazonium salts as aryl halide surrogates in palladium-catalyzed reactions allowing C–C bond formation is certainly one of the most intriguing applications.2 Indeed, the high polarization of the C–N2+ bond allows facile oxidative additions of Pd(0) complexes in mild conditions at moderate temperature, usually ranging from 25 to 60 °C, and most often under base-, ligand- and additive-free conditions.3 The peculiar properties of these super-electrophiles are salient advantages for the development of palladium-catalyzed sustainable processes since they allow working under experimentally simple conditions, reducing both energy expenses and waste generation.4By contrast, exploiting the high reactivity of arenediazonium salts with Pd(0) complexes has been mostly overlooked for the development of ultra-fast reactions speeding up the productivity.5 With no doubt, the Heck and Suzuki coupling reactions are the most popular palladium-catalyzed transformations in the toolbox of synthetic chemists. Under traditional conditions, when using aryl halides as electrophiles, these reactions usually require extensive reaction times (≫1 hour), mostly under heating, especially for the more demanding Heck reaction although few catalytic systems allowing high turnover frequencies (TOFs) have been reported.6 Developing ultra-fast palladium-catalyzed coupling reactions, completed within minutes or even seconds, would be of great interest to speed up processes, either in multi-step synthesis where reaction time contributes to a large extent to the global time required for the synthesis of complex architectures, or with unstable products not supporting extended stirring and heating. In this contribution, we report our findings on catalyst systems allowing ultra-fast palladium-catalyzed coupling reactions by taking advantage of the high reactivity of arenediazonium salts. Our efforts have culminated with the synthesis of highly sensitive styrenes within seconds through the coupling of arenediazonium salts with vinyl potassium trifluoroborate. Further studies allowed the preparation of symmetrical and unsymmetrical stilbenes within minutes through sequential Suzuki–Heck reactions under microwave heating.
Results and discussion
The development of ultra-fast reactions is particularly relevant for the preparation of highly sensitive architectures which are prone to degradation upon exposure to external stimuli such as light, oxygen or high temperature. In this regard, we reasoned that the synthesis of styrenes, which are intermediates of utmost importance in medicinal and polymer chemistry, could serve as illustrative examples for showing the potential of ultra-fast reactions. Our approach for the synthesis of styrene consists in the coupling of arenediazonium salts with potassium vinyltrifluoroborate 1. While this process could be hastily considered as trivial, it required a fine tuning of experimental conditions since the ambivalent properties of vinyltrifluoroborate could lead to divergent reaction outcomes with either a Suzuki (pathway A) or Heck coupling (pathway B), eventually followed by a second coupling (pathway C and D) as depicted in Scheme 1. However, we anticipated that pathway A would be favored since Suzuki couplings are usually much less demanding processes than Heck reactions.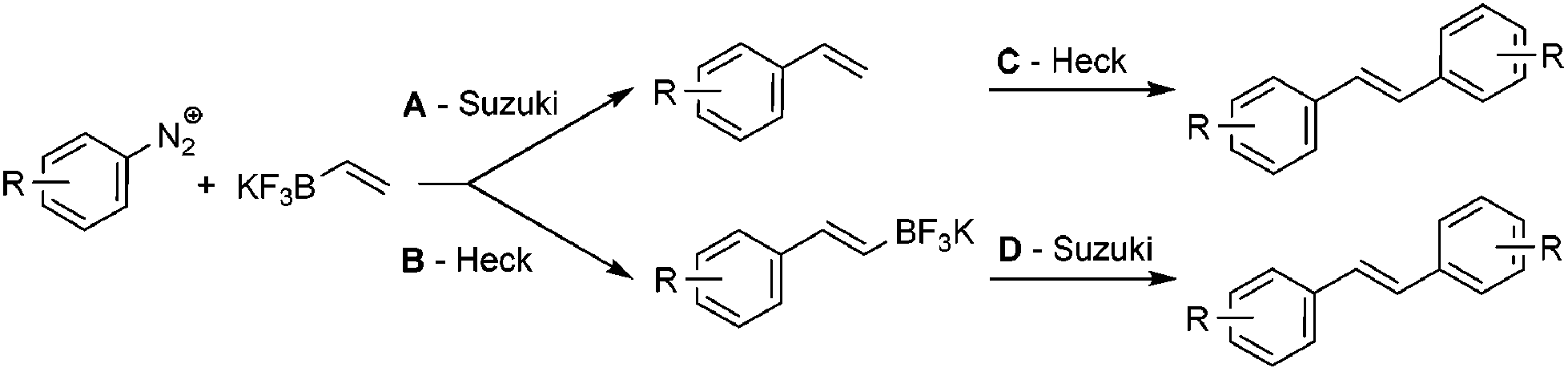 | ||
| Scheme 1 Divergent pathways for the coupling of arenediazonium salts with vinyltrifluoroborate. | ||
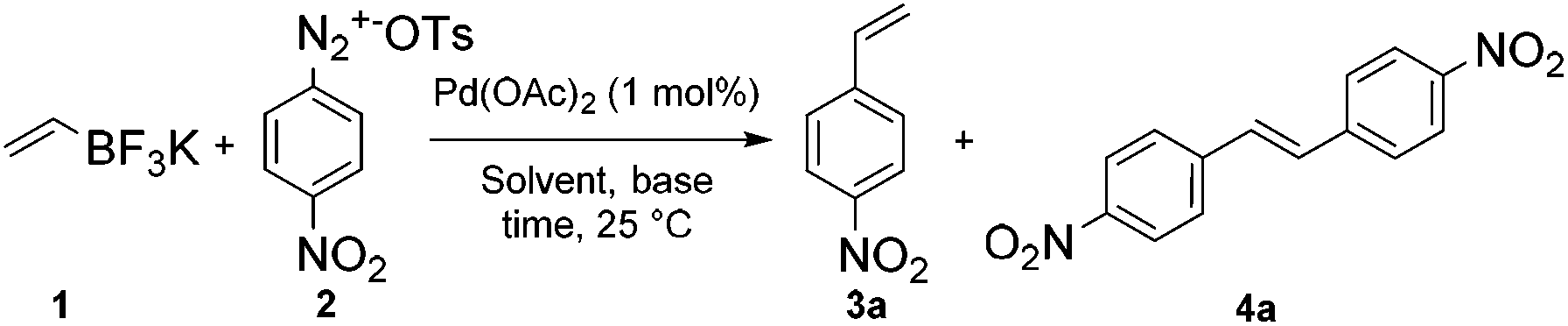
We studied these different reaction outcomes on a benchmark reaction involving the coupling of bench-stable 4-nitrobenzenediazonium tosylate 2 with a slight excess of potassium vinyltrifluoroborate 1 (1.5 eq.) using 1 mol% of Pd(OAc)2 as a catalyst. While most Pd-catalyzed coupling reactions have been described with diazonium salts having a tetrafluoroborate counterion, we recently highlighted the benefits of diazonium tosylates since they usually display a higher reactivity and stability and do not produce highly harmful HF as by-product.4f We initially carried out the reaction in water and at room temperature without any additive such as base or ligand (entry 1). Under these conditions styrene 3a was isolated as the major product, although significant amounts of stilbene 4a, resulting from a sequential Suzuki–Heck process (pathways A → C), were also isolated. We reasoned that the increase of hydroxyl ion concentration with the use of NaOAc as a base, could favor the Suzuki coupling through the formation of palladium hydroxo intermediates,7 but unfortunately, our hypothesis was invalidated since the distribution of products 3a/4 was unchanged under these conditions (entry 2). Suspecting that the low selectivity for 3a could be due to solubility issues, we switched the reaction solvent from H2O to MeOH and CH3CN. While MeOH is usually an excellent solvent for Pd-catalyzed reactions involving arenediazonium salts,3,8 it proved to be unsuitable for our methodology since 3a was isolated in only 23% (entry 3).
Actually, nitrobenzene, resulting from the reduction of 2, was the main side-product formed with 63% yield. By contrast, when the reaction was carried out in CH3CN the selectivity and the reaction rate were notably improved since the expected styrene 3a was formed as the only isolable product with 95% yield after only 80 seconds (entry 5).
While these results were already impressive on their own, we discovered that the reaction time could be reduced to 25 seconds, allowing a remarkable turn over frequency (TOF) of 14000 h−1, in a CH3CN/H2O (1/1) co-solvent media and still with a complete selectivity for 3a (entry 6). The process efficiency was highly dependent on the source of palladium since the switch of Pd(OAc)2 for PdCl2(Ph3P)2 considerably slowed down the reaction rate (entry 7). The very high reaction rates observed on this benchmark reaction significantly differ from literature precedents.5b,c We attribute this impressive feature to the higher solubility of arenediazonium tosylates with respect to traditional arenediazonium tetrafluoroborates and the increased reactivity of potassium vinyltrifluoroborate 1 with respect to potassium aryltrifluoroborates.9 As background reaction, we carried out the coupling between potassium vinyltrifluoroborate 1 and 4-nitrobenzenediazonium tetrafluoroborate under optimized conditions. The result reveals that the cross-coupling requires 120 seconds to reach completion, corresponding to a disappointing TOF of 2550 h−1 (entry 8). Moreover, the process is notably plagued by the formation of the undesired stilbene 4a with 6% yield.
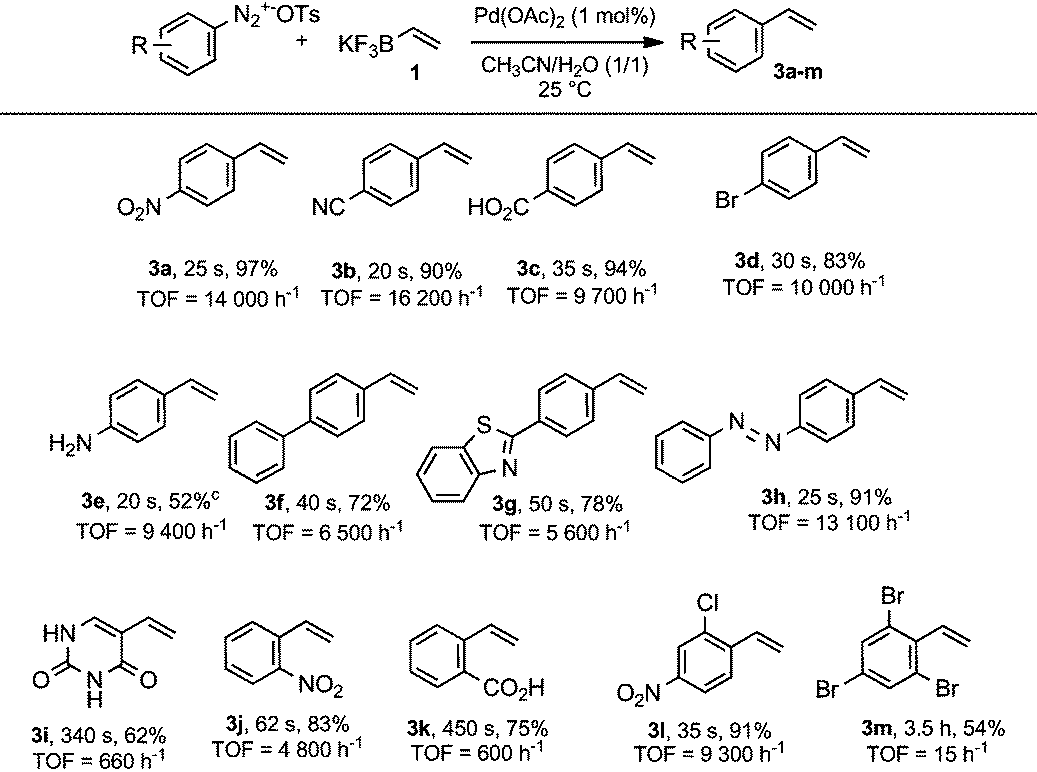
With an optimized procedure in hands, we demonstrated the robustness of our methodology through the screening of variously decorated arenediazonium tosylates (Table 2). We were pleased to find that both para- and ortho-substituted arenediazonium salts were coupled with vinyltrifluoroborate 1 with very high rates corresponding to reaction times ranging from ca. 20 to 60 seconds. These ultra-fast reaction times correspond to remarkably high TOF from 4800 h−1 up to 16200 h−1. The only exception to our survey was observed for styrene 3k, bearing a carboxylate group in ortho position, which required 7.5 minutes of reaction time (TOF = 600 h−1). This behavior was likely associated to an internal chelation between the carboxylate group and the cationic palladium atom after the oxidative addition which slowed down the reactivity of palladium intermediates. The reaction also proved to be sensitive to steric hindrance when ortho–ortho′ positions were substituted by bromine atoms (compound 3m). However, while the reaction time increased to 3.5 hours, the steric hindrance was not prohibitive for producing 3m in a synthetically useful yield. It should be noted, that this described methodology is fully compatible with heterocycle- and azo-containing diazonium salts (compounds 3g–i). For instance, we were able to prepare 4-vinyluracyl 3i with a good yield (62%) in only 5.5 minutes (TOF = 660 h−1); 3i has unique electronic properties and can be used for the preparation of labeled nucleosides.7,8b
|
|
||||
|---|---|---|---|---|
| Entrya | Solvent | Base (eq.) | Reaction time (seconds) | Yield 3a/4ab (%) |
| a Reaction conditions: 4-Nitrobenzenediazonium tosylate 2 (0.2 mmol), potassium vinyltrifluoroborate 1 (0.3 mmol) and Pd(OAc)2 (1 mol%) were stirred in the solvent (5 ml) at room temperature. The progress of the reaction was monitored by TLC analysis. b Isolated yield. c Nitrobenzene was isolated with 63% yield. d A 1/1 ratio of starting materials was used. e Pd(PPh3)2Cl2 (1 mol%) was used as catalyst. f 4-Nitrobenzenediazonium tetrafluoroborate was used instead of 4-nitrobenzenediazonium tosylate. | ||||
| 1 | H2O | — | 900 | 54/20 |
| 2 | H2O | NaOAc (1) | 1920 | 52/18 |
| 3 | MeOH | — | 540 | 23/5c |
| 4 | CH3CN | — | 80 | 95/0 |
| 5 | CH3CN | NaOAc (1) | 80 | 93/1 |
| 6 | CH3CN/H2Od | — | 25 | 97/0 |
| 7e | CH3CN/H2Od | — | 2220 | 95/0 |
| 8f | CH3CN/H2O | — | 120 | 85/6 |
| a Reaction conditions: Diazonium salt (0.2 mmol), potassium vinyltrifluoroborate 1 (0.3 mmol) and Pd(OAc)2 (1 mol%) were mixed in CH3CN/H2O (1/1, v/v, 5 ml) and stirred at room temperature. The reaction was stopped after full conversion of arenediazonium tosylate as determined by β-naphtol test. b Isolated yield. c NaOAc (0.2 mmol) was added to the reaction mixture. |
|---|
|
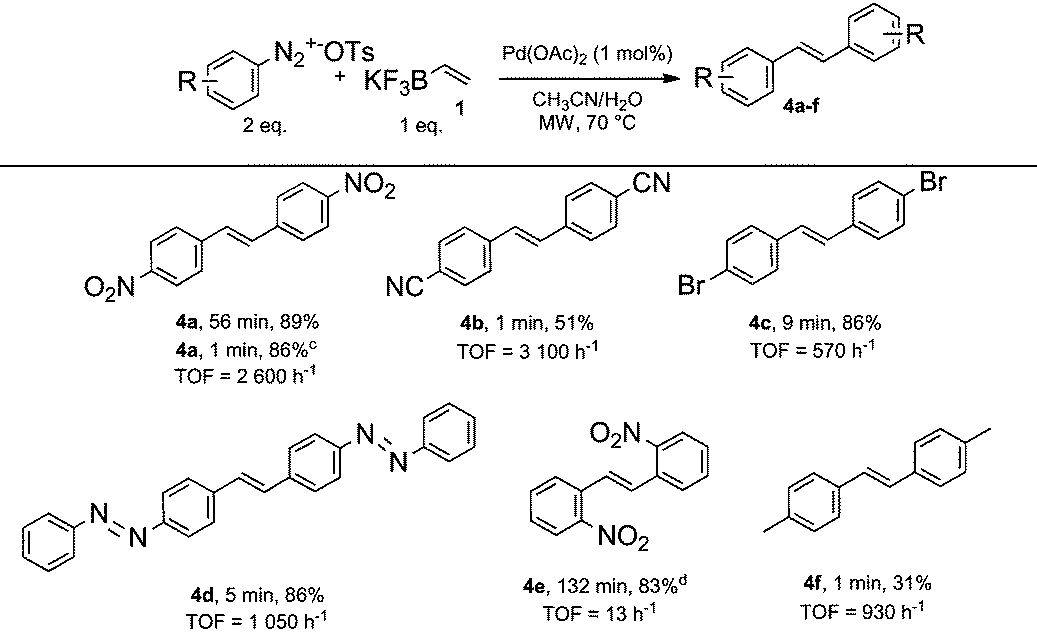
As discussed above, the formation of symmetrical stilbene 4a as side-product was observed in some circumstances during our optimization studies due to the ambivalent properties of vinyltrifluoroborate 1 (Table 1, entries 1–3 and 5). Therefore, we wondered if the use of an excess of diazonium salt 2 with respect to vinyltrifluoroborate 1 could allow an ultra-fast preparation of symmetrical stilbenes through a sequential Suzuki/Heck process. While similarities in reaction conditions for conducting Suzuki and Heck couplings with diazonium salts would help us for succeeding in developing an ultra-fast sequential process, we reasoned that the demanding nature of the Heck reaction might require to fine tune experimental conditions. With this objective in mind, we explored the coupling of two equivalents of 4-nitrobenzenediazonium salt 2 with one equivalent of vinyltrifluoroborate 1 in aqueous acetonitrile in the presence of 1 mol% of Pd(OAc)2 as a catalyst. The double coupling reached completion after being stirred for 56 minutes, giving the expected symmetrical stilbene 4a with 89% yield. Since the Suzuki cross-coupling occurred in a matter of seconds, we clearly identified the Heck coupling as the rate limiting reaction. In order to speed-up the sequential process and especially the Heck reaction, we stirred the reaction mixture under microwave heating at 70 °C. Under these conditions it was gratifying to record a striking decrease of the reaction time to only 1 minute for achieving 4a with an excellent yield (86%, TOF = 2600 h−1). With these conditions in hand we prepared a collection of representative structures. The results depicted in Table 3 deserve few comments.
| a Reaction conditions: Diazonium salt (0.4 mmol), potassium vinyltrifluoroborate 1 (0.2 mmol) and Pd(OAc)2 (1 mol%) were mixed in CH3CN/H2O (1/1, v/v, 5 ml) and immediately placed in microwave reactor (70 °C, 30 bars). The reaction was stopped after full conversion of arenediazonium tosylate as determined by β-naphtol test. b Isolated yield. c 2 mol% of Pd(OAc)2 were used. d 3 mol% of Pd(OAc)2 were used. |
|---|
|
First, all symmetrical stilbenes prepared in this study required less than 9 minutes of reaction time corresponding to remarkable TOF ranging from 570 to 5500 h−1 for both sequential chemical reactions, in order to achieve completion with the exception of stilbene 4e which required 132 minutes. While steric considerations might explain, at least in part, this behavior, the preparation of 4f in only 1 minute (TOF = 930 h−1), suggests that electronic factors might also be involved.
Secondly, bromine atoms of stilbene 4c remarkably survived to the microwave heating and no evidence for a competing coupling at the bromine atom was detected. The high chemoselectivity for the diazonium group allows to envisage further orthogonal functionalizations of stilbene 4c under standard organometallic chemistry.
Thirdly, the methodology is remarkably efficient for synthetizing highly conjugated stilbenes that might find applications in material chemistry for the preparation of optoelectronic devices.
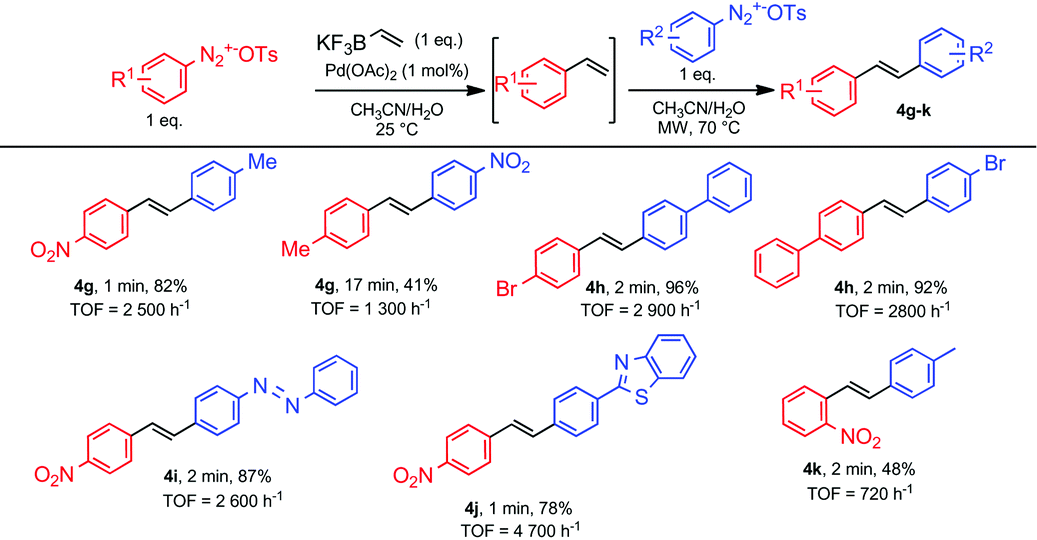
From these studies, we learned that the temperature was the only experimental parameter that differed for carrying out Suzuki and Heck couplings. Therefore, we wondered if we could prepare unsymmetrical stilbenes following sequential Suzuki–Heck reactions using two different arenediazonium salts. From a practical point of view, we reasoned that after mixing an equimolar amount of an arenediazonium salt with potassium vinyltrifluoroborate 1 for achieving ultra-fast Suzuki cross-couplings, the resulting styrene could be involved in a subsequent microwave-triggered Heck coupling upon addition of a second equivalent of another arenediazonium salt. We explored the synthetic usefulness of this promising strategy through the preparation of several representative examples depicted in Table 4. Pleasingly, unsymmetrical stilbenes could be prepared in high yields with reaction times ranging from 60 to 240 seconds corresponding to TOF ranging from 2600 to 5300 h−1. As already noticed for the preparation of symmetrical stilbenes, heterocycles and heteroatom-containing diazonium salts were tolerated for this process. As a relevant feature, we also noticed that switching the order of addition of arenediazonium salts did not affect the reaction outcome as exemplified by the preparation of compound 4h.
| a Reaction conditions: Diazonium salt (0.2 mmol), potassium vinyltrifluoroborate 1 (0.2 mmol) and Pd(OAc)2 (1 mol%) were mixed in CH3CN/H2O (1/1, v/v, 5 ml) and stirred at room temperature, then the second diazonium salt was added and the mixture was stirred at 70 °C under microwave irradiation. The reaction was stopped after full conversion of arenediazonium tosylate as determined by β-naphtol test. b Isolated yield. |
|---|
|
Conclusions
In summary, we exploited the super-electrophilic properties of arenediazonium salts for establishing ultra-fast Pd-catalyzed methodologies. Highly sensitive styrenes were prepared in seconds with TOF up to 16200 h−1 through Suzuki cross-couplings while a collection of symmetrical and unsymmetrical stilbenes were synthetized through sequential Suzuki–Heck reactions requiring only a couple of minutes to reach completion for most examples. As highlights of this work, it can be argued that these methodologies (i) were conducted under extremely simple experimental conditions without any additives such as base and ligand, (ii) allowed remarkable high TOF for such simple conditions, (iii) provided styrenes and (un)symmetrical stilbenes in high yields. We believe that this contribution, highlighting the usefulness of arenediazonium salts, could inspire chemists involved in process intensification and should be of practical interest for synthetic chemists.
Experimental
General procedure for preparation of styrenes from potassium vinyltrifluoroborate 1 and arenediazonium tosylates (ADT)
To a mixture of ADT (2a–m) (0.2 mmol) and potassium vinyltrifluoroborate (0.3 mmol, 0.040 g) in 5 ml water/acetonitrile (1/1) was added 1 mol% Pd(OAc)2 (0.002 mmol, 0.0005 g). The end of the nitrogen evolution and the formation of Pd(0) indicated the reaction completion. Brine (10 ml) was added to the reaction mixture and the organic layer was extracted with pentane (3 × 50 mL). The organic layers were dried over anhydrous MgSO4. The solvent was removed in vacuo to give the corresponding styrene without further purification (3a–m).General procedure for preparation of symmetric stilbenes from potassium vinyltrifluoroborate 1 and ADT
To a mixture of ADT (0.4 mmol) and potassium vinyltrifluoroborate (0.2 mmol, 0.027 g) in water/acetonitrile (5 mL, 1/1) was added Pd(OAc)2 (1 mol%). The reaction vessel was immediately placed into a microwave reactor (70 °C, 30 bars). After completion, brine was added, the reaction mixture was extracted with CHCl3 (3 × 50 mL) and dried over anhydrous MgSO4. The solvent was removed in vacuo to give the corresponding stilbenes without further purification.General procedure for preparation of unsymmetric stilbenes from potassium vinyltrifluoroborate 1 and ADT
To a solution of ADT (0.2 mmol) and potassium vinyltrifluoroborate (0.3 mmol, 0.040 g) water/acetonitrile (5 mL, 1/1) was added Pd(OAc)2 (1 mol%). The reaction mixture was stirred until the cessation of the nitrogen evolution, then the second ADT (0.2 mmol) was added to reaction mixture. The reaction vessel was placed into a microwave reactor (70 °C, 30 bars). After completion, brine was added, the reaction mixture was extracted with CHCl3 (3 × 50 mL) and dried over anhydrous MgSO4. The organic extracts were filtered through short silica pad. The solvent was removed in vacuo to give the corresponding stilbenes without further purification.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Tomsk Polytechnic University (project VIU-316/2017), the University of Nantes (France) and the “Centre National de la Recherche Scientifique” (CNRS). FXF is member of the “Institut Universitaire de France, IUF.” SB acknowledges funding through the DFG (SFB 1176, TRR 88). The authors greatly acknowledge the Laboratory of physical–chemical analytical methods of Tomsk State University for the NMR and HRMS analysis.References
- (a) F. Mo, G. Dong, Y. Zhang and J. Wang, Org. Biomol. Chem., 2013, 11, 1582 RSC; (b) D. M. R. Santos, R. Coriolano, M. N. Godoi, A. L. Monteiro, H. C. B. de Oliveira, M. N. Eberlin and B. A. D. Neto, New J. Chem., 2014, 38, 2958 RSC; (c) L. He, G. Qiu, Y. Gao and J. Wu, Org. Biomol. Chem., 2014, 12, 6965 RSC; (d) S. Kindt and M. R. Heinrich, Synthesis, 2016, 1597 CAS; (e) V. Dichiarante and M. Fagnoni, Synlett, 2008, 0787 CAS.
- (a) A. Roglans, A. Pla-Quintana and M. Moreno-Mañas, Chem. Rev., 2006, 106, 4622 CrossRef CAS PubMed; (b) H. Bonin, E. Fouquet and F.-X. Felpin, Adv. Synth. Catal., 2011, 353, 3063 CrossRef CAS; (c) F.-X. Felpin, L. Nassar-Hardy, F. Le Callonnec and E. Fouquet, Tetrahedron, 2011, 67, 2815 CrossRef CAS; (d) J. G. Taylor, A. V. Moro and C. R. D. Correia, Eur. J. Org. Chem., 2011, 1403 CrossRef CAS.
- F.-X. Felpin, K. Miqueu, J.-M. Sotiropoulos, E. Fouquet, O. Ibarguren and J. Laudien, Chem. – Eur. J., 2010, 16, 5191 CrossRef CAS PubMed.
- (a) F. Le Callonnec, E. Fouquet and F.-X. Felpin, Org. Lett., 2011, 13, 2646 CrossRef CAS PubMed; (b) R. Joncour, N. Susperregui, N. Pinaud, K. Miqueu, E. Fouquet, J.-M. Sotiropoulos and F.-X. Felpin, Chem. – Eur. J., 2013, 19, 9291 CrossRef CAS PubMed; (c) N. Susperregui, K. Miqueu, J.-M. Sotiropoulos, F. Le Callonnec, E. Fouquet and F.-X. Felpin, Chem. – Eur. J., 2012, 18, 7210 CrossRef CAS PubMed; (d) N. Oger, M. d'Halluin, E. Le Grognec and F.-X. Felpin, Org. Process Res. Dev., 2014, 18, 1786 CrossRef CAS; (e) N. Oger, F. Le Callonnec, D. Jacquemin, E. Fouquet, E. Le Grognec and F.-X. Felpin, Adv. Synth. Catal., 2014, 356, 1065 CrossRef CAS; (f) M. E. Trusova, K. V. Kutonova, V. V. Kurtukov, V. D. Filimonov and P. S. Postnikov, Res.-Effic. Technol., 2016, 2, 36 Search PubMed; (g) K. V. Kutonova, M. E. Trusova, A. V. Stankevich, P. S. Postnikov and V. D. Filimonov, Beilstein J. Org. Chem., 2015, 11, 358 CrossRef CAS PubMed; (h) S. Vanderheiden, N. Jung and S. Brase, Solid-Phase Org. Synth., 2012, 2, 129 CAS; (i) S. Brase and M. Schroen, Angew. Chem., Int. Ed., 1999, 38, 1071 CrossRef CAS; (j) B. Schmidt, N. Elizarov, N. Riemer and F. Hölter, Eur. J. Org. Chem., 2015, 5826 CrossRef CAS; (k) A. P. Colleville, R. A. J. Horan and N. C. O. Tomkinson, Org. Process Res. Dev., 2014, 18, 1128 CrossRef CAS; (l) B. Schmidt and F. Hölter, Org. Biomol. Chem., 2011, 9, 4914 RSC; (m) H. Bonin, D. Delbrayelle, P. Demonchaux and E. Gras, Chem. Commun., 2010, 46, 2677 RSC; (n) B. Schmidt, N. Elizarov, R. Berger and F. Hölter, Org. Biol. Chem., 2013, 11, 3674 RSC.
- (a) V. Gallo, P. Mastrorilli, C. F. Nobile, R. Paolillo and N. Taccardi, Eur. J. Inorg. Chem., 2005, 3, 582 CrossRef; (b) S. Darses, G. Michaud and J.-P. Genêt, Tetrahedron Lett., 1998, 39, 5045 CrossRef CAS; (c) S. Darses, G. Michaud and J.-P. Genêt, Eur. J. Org. Chem., 1999, 1875 CrossRef CAS.
- V. Farina, Adv. Synth. Catal., 2004, 346, 1553 CrossRef CAS.
- (a) B. P. Carrow and J. F. Hartwig, J. Am. Chem. Soc., 2011, 133, 2116 CrossRef CAS PubMed; (b) C. Amatore, A. Jutand and G. Le Duc, Chem. – Eur. J., 2011, 17, 2492 CrossRef CAS PubMed; (c) R. Joncour, N. Susperregui, N. Pinaud, K. Miqueu, E. Fouquet, J.-M. Sotiropoulos and F.-X. Felpin, Chem. – Eur. J., 2013, 19, 9291 CrossRef CAS PubMed.
- (a) F.-X. Felpin and E. Fouquet, Adv. Synth. Catal., 2008, 350, 863 CrossRef CAS; (b) F.-X. Felpin, E. Fouquet and C. Zakri, Adv. Synth. Catal., 2008, 350, 2559 CrossRef CAS; (c) F.-X. Felpin, E. Fouquet and C. Zakri, Adv. Synth. Catal., 2009, 351, 649 CrossRef CAS.
- K. V. Kutonova, N. Jung, M. E. Trusova, V. D. Filimonov, P. S. Postnikov and S. Brase, Synthesis, 2017, 1680 CAS.
- S. Fissekis, J. Org. Chem., 1973, 38, 264 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: All experimental details as well as NMR spectra for all compounds. See DOI: 10.1039/c7qo00750g |
| This journal is © the Partner Organisations 2018 |