
Catalytic approaches to assemble cyclobutane motifs in natural product synthesis

Abstract
Cyclobutanes, as the second most strained monocyclic all-carbon rings, are found in many complex natural products. Although recent methodological developments in the formation of cyclobutanes have been reviewed, few have focused on the topic of total synthesis of cylcobutane-containing natural products. Here we summarize recent advances in natural product synthesis, featuring late-stage functionalization of four-membered rings and novel strategies for the stereocontrolled assembly of cyclobutanes or cyclobutenes.
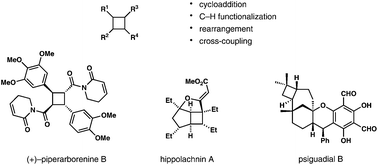
- This article is part of the themed collections: 2018 Organic Chemistry Frontiers Review-type Articles and Synthetic Approaches to Natural Products via Catalytic Processes
 Please wait while we load your content...
Please wait while we load your content...

