
Palladium catalyzed C(sp3)–H acetoxylation of aliphatic primary amines to γ-amino alcohol derivatives†
Ding
Wang,  ‡a
Zhang-Jie
Shi
*ab
‡a
Zhang-Jie
Shi
*ab
‡a
Zhang-Jie
Shi
*ab
Abstract
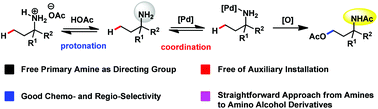
It still remains a major challenge to apply free primary amino groups as the directing group for aliphatic C–H functionalization. In this article, we used the protonation strategy to control the binding ability of primary amines and realized free amino group directed inert aliphatic C–H acetoxylation in good chemo- and regio-selectivity. This methodology provided a straightforward approach from primary amines to γ-amino alcohols.
- This article is part of the themed collection: Organic Chemistry Frontiers HOT articles for 2017
 Please wait while we load your content...
Please wait while we load your content...

