
Gold(i)-catalyzed access to neomerane skeletons†

Abstract
The gold(I) catalyzed cycloisomerization of an enynyl propargylic ester, featuring a 1,2-acyloxy migration/intramolecular cyclopropanation sequence, opens a straightforward access to the 5,7,3-tricyclic skeleton of neomerane sesquiterpenes. The first total synthesis of 5-epi-valeneomerin B in 12 steps with an overall yield of 5.3% from the readily available hex-5-en-2-one is reported.
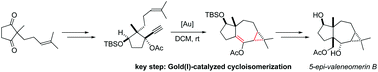
- This article is part of the themed collection: Synthetic Approaches to Natural Products via Catalytic Processes
 Please wait while we load your content...
Please wait while we load your content...

