
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBinding of ion pairs in a thiourea-functionalized self-folding cavitand†
A.
Lledó
 * and
A.
Soler
* and
A.
Soler
Institute of Computational Chemistry and Catalysis and Department of Chemistry, Universitat de Girona, Maria Aurelia de Campmany 69, 17003 Girona, Spain. E-mail: agusti.lledo@udg.edu
First published on 21st March 2017
Abstract
We report the synthesis of a new synthetic cavitand receptor with an embedded thiourea moiety directed at effective molecular recognition of ion pairs. Initial prospects towards the use of cavitands in biomimetic carbocationic cyclizations have been carried out, employing ionic guests as stable surrogates for putative substrates and intermediates. The cavitand displays tight binding with tetramethylammonium salts, with unforeseen slow exchange kinetics of both cation and anion components.
Introduction
Supramolecular catalysis constitutes a promising approach for streamlining the synthesis of complex molecules by promoting selective transformations of organic substrates.1 This concept is inspired mostly by nature, and hence the most genuine examples are arguably those which rely on the encapsulation of substrates in confined spaces akin to buried enzymatic pockets.2 Imitation of the natural terpene cyclization machinery (Scheme 1a) is one of the most sought goals in this area, and a variety of encapsulation strategies have been described to carry out analogous carbocationic cyclizations in the confined spaces of π-rich synthetic receptors.3 However, none of the previously reported examples feature specific activation of the cyclization precursors by purpose-built functionalities. Given the existing parallels with anion binding catalysis,4 developed mainly by the group of Jacobsen (Scheme 1b), we envisaged melding the two concepts by installing a thiourea activating handle to a supramolecular receptor, with the aim of encapsulating, isolating and modulating the reactivity of carbocationic intermediates derived from linear polyene precursors (Scheme 1c).5 As long term goals of this research, we envisage promoting non-stop propagation of carbocationic cascades and overriding the innate selectivity of cyclizations carried in the bulk. | ||
| Scheme 1 Carbocationic cyclization processes (a) in the buried active site of a terpene cyclase, triggered by specific interactions with neighboring residues, (b) through small molecule anion-binding with a thiourea catalyst, and (c) proposed bioinspired strategy with a thiourea-appended synthetic receptor. | ||
We describe herein our latest efforts in this area, including the preparation of a new thiourea-functionalized self-folding cavitand,6 the study of its binding abilities and initial attempts to bind substrates relevant to enzymatic carbocationic cyclization processes.
Results and discussion
Inspiration for this work came from a recent report describing a hitherto unknown cyclization mechanism in bacterial sesquiterpene cyclases, whereby ionization of farnesyl pyrophosphate is triggered by the synchronized effect of a guanidinium group hydrogen bonding to the pyrophosphate anionic head, and a neighbouring carbonyl providing nucleophilic anchimeric assistance (Scheme 1a).7Capitalizing on our experience, we envisaged the preparation of a resorcin[4]arene derived, amide stabilized deep cavitand 1,8 bearing an attached thiourea group (Fig. 1). We sought to use a 3,5-bis(trifluoromethyl)phenyl thiourea to provide a receptor with improved anion binding ability with respect to our previously reported octa(ureido) cavitand 3.9 Central to our design is the ability of resorcin[4]arene-derived cavitands to stabilize positively charged species through cation–π interactions,10 adding up to the thiourea motif to provide an effective ion-pair receptor.11 Preliminary modelling studies revealed that a para-substituted phenyl spacer between the thiourea group and the customary benzimidazole attaching moiety sufficed to prevent interference with the stabilizing amide hydrogen bond seam, while providing enough space to bind a monoterpene precursor such as a geraniol derivative, its polar end interacting with the thiourea's NHs and the aliphatic portion being buried in the deep aromatic pocket (Fig. S1†).
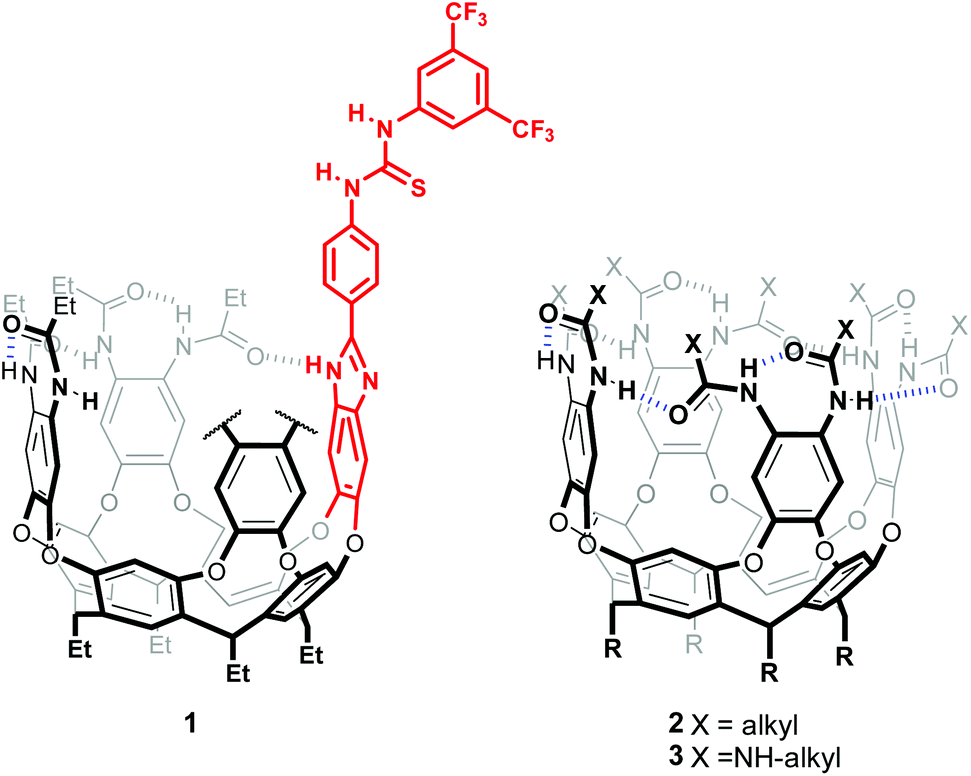 | ||
| Fig. 1 Structure of hexaamide-thiourea self-folding cavitand 1, and previously reported octa(amido)- and octa(ureido)- cavitands, 2 and 3. | ||
Cavitand 1 was prepared from the known diamino platform10 which has been used for the construction of other monofunctionalized self-folding cavitands (Scheme 2).12 After some experimentation, we found that reduction of dinitro cavitand 4a followed by FeCl3 catalysed oxidative condensation with aldehyde 5 delivered the desired thiourea-appended cavitand in satisfactory yields.13 We prepared model compound 6 in an analogous manner, while 5 was obtained in two steps from commercial materials (Schemes S1 and S2†).
 | ||
| Scheme 2 Synthesis of hexaamide-thiourea cavitand 1, and model compound 6. | ||
Cavitand 1 was characterized by 1H NMR, 13C NMR and HRMS. The 1H NMR spectra of 1 in acetone-d6 or CDCl3 (Fig. 2) display the distinct features of hexaamide-benzimidazole cavitands, namely, a far downfield shift at δ 11.8 ppm corresponding to the hydrogen-bonded benzimidazole NH, six separate resonances for the amide NH's indicative of slow rotation around the aryl-N bond relative to the NMR time-scale, and four overlapping triplets around δ 5.65 ppm diagnostic for the cavitand adopting a folded, chiral “vase” conformation.14
 | ||
| Fig. 2 Downfield regions of the 1H NMR spectra (400 MHz) of 1 in acetone-d6 (top) and CDCl3 (bottom). Blue squares indicate the NH resonances. | ||
Effective substrate binding is a requisite for supramolecular catalysis and we hypothesized that species already bearing an anionic portion such as polyisoprene pyrophosphates would benefit from enhanced binding in 1, with respect to analogues bearing neutral leaving groups (e.g. halides, acetates). The preparation of organic pyrophosphates is cumbersome, requiring tedious purifications which limit its practicability.15 We reasoned that analogous alkyl sulphates, which can be easily prepared by reaction of an alcohol with sulphur trioxide,16 would serve our purpose just as well. Eventually, we found that reaction of the requisite alcohol with SO3·pyridine, followed by cation exchange with tetra(n-butylammonium) bromide furnished the corresponding tetra(n-butylammonium) sulphates 7a–h in multigram quantities and excellent yields, without the need for chromatographic purification (Scheme 3). Most conveniently, the salts prepared herein are soluble in both water and polar organic solvents. Very much like polyisoprenyl pyrophosphates, allylic sulphates 7a–b hydrolyse in neutral or mildly acidic aqueous solution, making them promising ionization precursors for bioinspired carbocationic cyclizations.
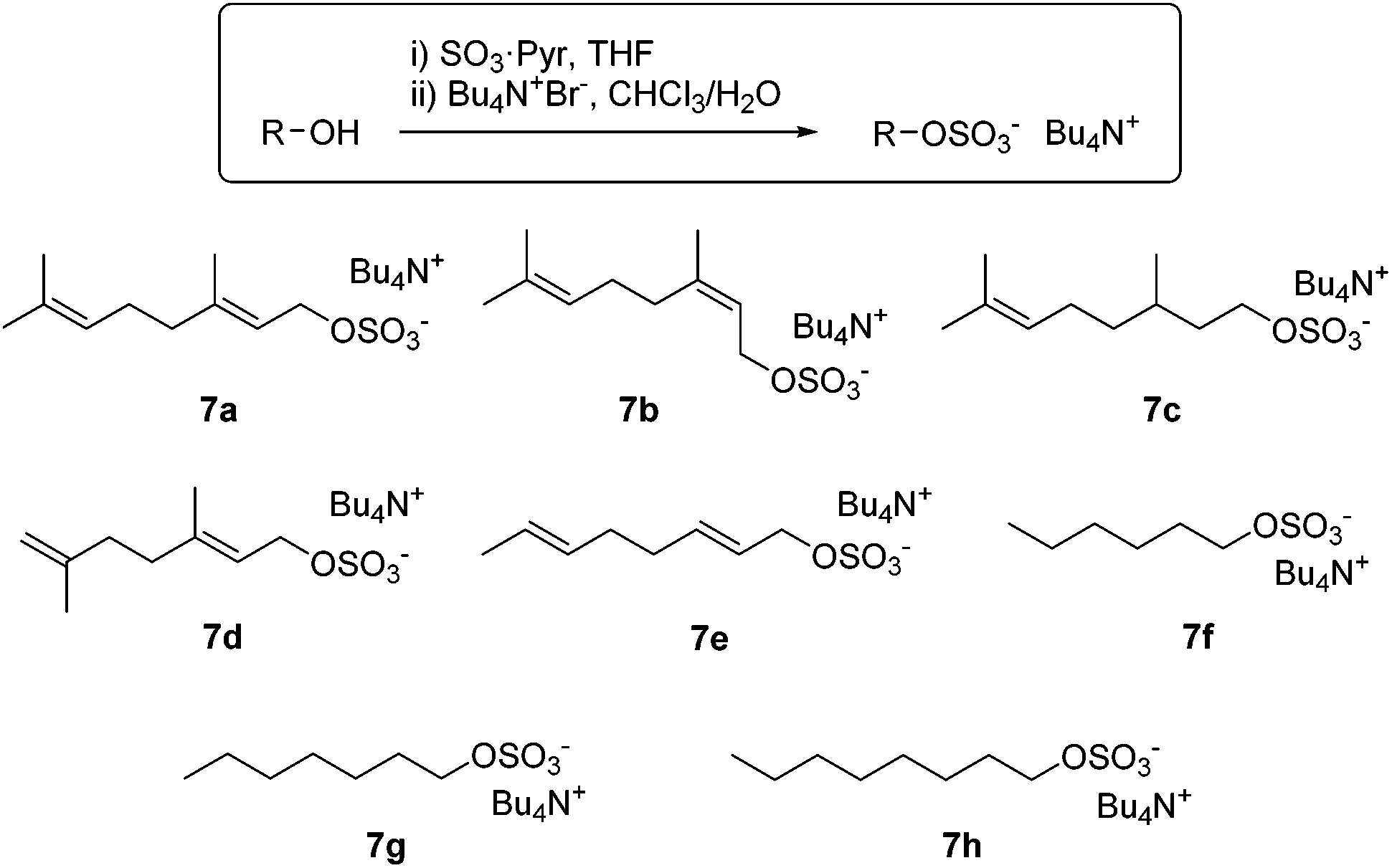 | ||
| Scheme 3 Synthesis of tetrabutylammonium monoterpene sulphates and non-biological analogues. | ||
We then proceeded to test the binding of 7a–h with 1 in CDCl3 solution. Host–guest complexes of amide-stabilized self-folding cavitands are kinetically stable in the NMR time scale and molecules buried in the cavity experience strong upfield shifts in the 1H NMR spectra caused by the anisotropic shielding of the multiple aromatic rings.17 For guests with aliphatic portions, this feature facilitates the monitoring of complex formation by the appearance of the bound guest's resonances in the far upfield region of the spectrum (δ 0 to −4 ppm) which is devoid of any interference from other species present in solution. Much to our dismay, no upfield resonances were observed at all upon treatment of 1 with excess quantities of monoterpene sulphates 7a–c. Smaller terpene analogues (7d–e) or n-alkyl sulphates (7f–h) – with increased conformational flexibility – did not provide evidence of internalization in the receptor's cavity either. In spite of this, a downfield shift of the urea's NHs is apparent in all these cases (Fig. S2†), suggesting that a significant interaction between the thiourea and the sulphate moiety is taking place. This is corroborated by the downfield shift of the singlet observed in the 19F NMR spectra of 1 in CDCl3 upon titration with increasing amounts of 7c. Non-linear fit analysis of the obtained binding isotherm revealed a binding constant Ka of 1.04 × 104 M−1. The presence of a single resonance throughout the titration indicates a binding event in the fast exchange regime. We also tested the analogue acetates of 7a–h as potential neutral substrates with hydrogen bonding leaving groups, but no binding was observed either. These results illustrate the challenge of displacing highly competitive solvents – such as CDCl3 – by attempting to couple the effect of separate binding motifs in a cooperative manner.‡
We next examined the binding of ionic substrates which could mimic carbocationic intermediates along a putative cyclization process inside 1. We chose to use ammonium cations, which are typical inhibitors of terpene cyclases and work as static surrogates of a reacting carbocationic species.18 Cognizant of the challenge of binding linear aliphatic substrates, we set out to explore the binding of cyclic ammonium salts as surrogates for carbocationic intermediates folded in compact but energetically disfavoured contorted conformations. Different ring sizes and counter anions were assayed with the aim of probing the binding space of 1 (Fig. 3). Gratifyingly, upon addition of various 6-, 7- and 8-membered cyclic ammonium salts to 1, the appearance of new resonances in the far upfield region confirmed the formation of new encapsulation species –now in the slow exchange regime (Fig. 4). Binding constants were estimated by direct integration of the NMR spectra (Fig. 3). The low values extracted from our data prevent an accurate quantitative analysis but some trends can be inferred. The cyclooctylammonium ion seems to be close to the upper size limit for the number of atoms that can be accommodated in the aromatic pocket of 1. Interestingly, while cyclohexylammonium ion pairs (8) display a reasonable affinity, the addition of an extra cyclohexyl group to the ammonium moiety (11) virtually depletes binding, despite the fact that it can protrude out of the cavity through the upper rim. We interpret this as a result of the second cyclohexyl ring interfering with the anion binding site. For cyclic ammonium salts, no general trends can be observed upon variation of the counter-anion. Again, the low magnitude of the binding constants precludes any analysis invoking subtle effects of basicity, size, and shape.19 The signature of the upfield peaks in the 1H NMR spectra reveals that these species bind with the ammonium group close to the polar rim defined by the hydrogen bonded amide groups, and therefore charge–dipole interactions with the amide carbonyl groups must significantly contribute to the overall binding stabilization.
 | ||
Fig. 3 Ammonium guests used in this study and binding constants with 1 (Ka) estimated by 1H NMR. a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Binding is too weak to accurately integrate the bound guest's resonances. Binding is too weak to accurately integrate the bound guest's resonances. | ||
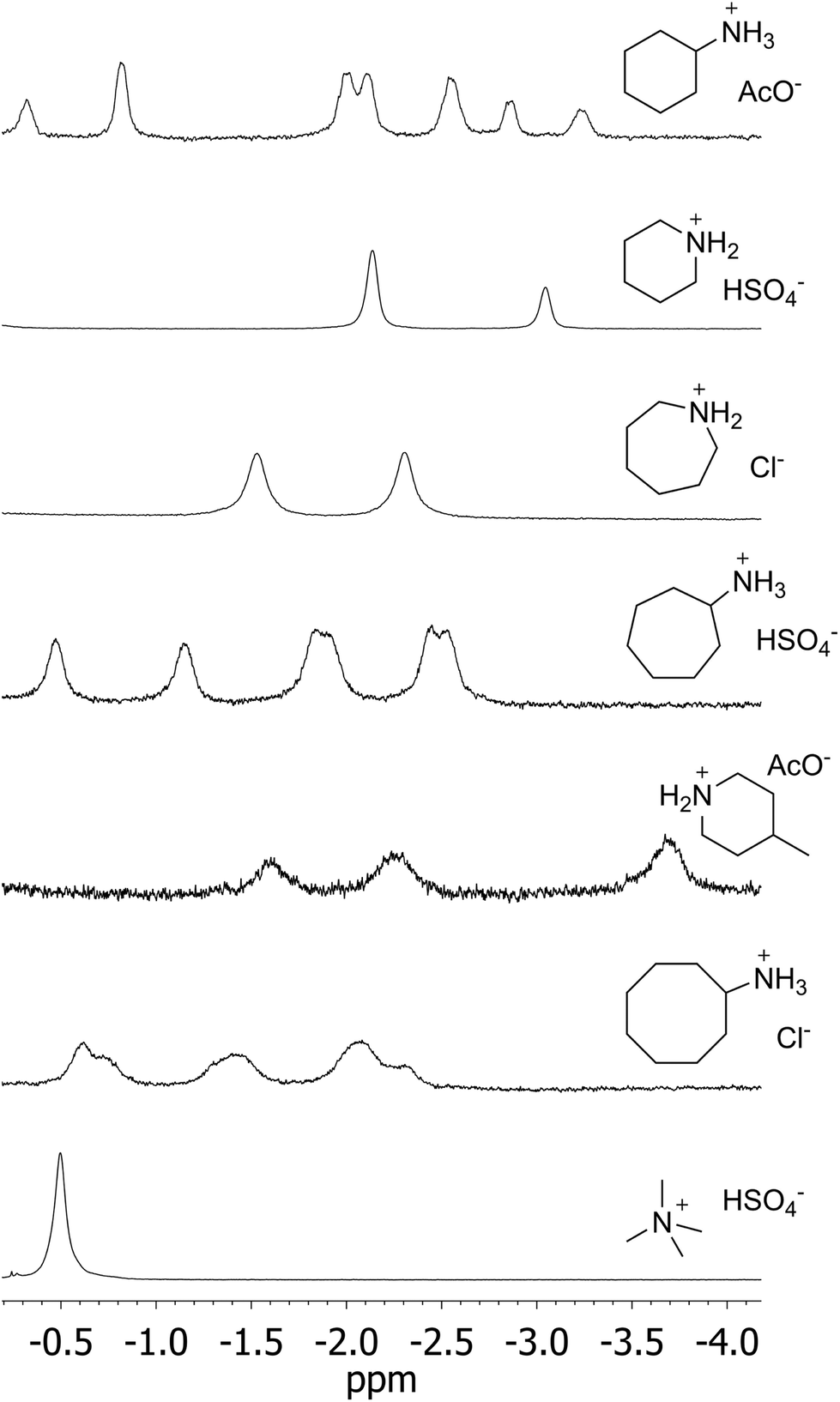 | ||
| Fig. 4 Far upfield region of the 1H NMR spectra of 1 (CDCl3) with excess of various ammonium salts, showing the resonances of aliphatic portions of the guests buried deep in the cavity. | ||
Finally, much increased binding was observed when we employed tetramethylammonium salts 17a–e as guests.§ Even with sub-stoichiometric amounts of the guest, tetramethylammonium fluoride, chloride, acetate or hydrogensulphate were totally encapsulated within 1 as ascertained by 1H and 19F NMR (vide infra). Only traces of the free ammonium species can hardly be made out in the 1H NMR spectra – beyond the integration threshold. For these ion pairs the magnitude of the association constant is thus too large to be measured by NMR techniques. On the other hand, weaker binding is observed for tetramethylammonium bromide, in good agreement with the well-established lower affinity of thioureas for anions with larger polarizability.20 Since previously reported cavitands devoid of specific anion binding motifs bind tetraalkylammonium salts with reasonable affinities,21 we set out to clarify the role of the thiourea moiety with additional experiments. First, the parent octaamide cavitand 2a (Fig. 1, X = R = Et) showed an association constant for Me4N+AcO− (17e, Ka = 7.5 × 102 M−1) which is at least one order of magnitude weaker than that with 1. Model compound 6, lacking a specific anchor for the tetramethylammonium cation, displays also much reduced affinity for tetramethylammonium ion pairs (including acetate 17e, Ka = 4.9 × 102 M−1), as ascertained by non-linear fit analysis of 19F NMR titration data (Table 1). Taken together, these data support a cooperative effect of both binding motifs in 1. Most interestingly, 19F NMR titration experiments with 6 always show a fast exchange scenario, while titration of 1 with tetramethylammonium salts reveals slow exchange dynamics (Fig. 5). While this is expected when looking at the bound ammonium ion resonances in the 1H NMR spectra, it is not immediately apparent that the anion binding site – which is exposed to the solvent – would behave similarly. This is a promising feature for future developments in catalysis because the combination of both anion and cation specific binding sites in the cavitand enables not only improved binding but also effective confinement of the cation from bulk medium by temporally blocking the cavitand's opening with the anion.
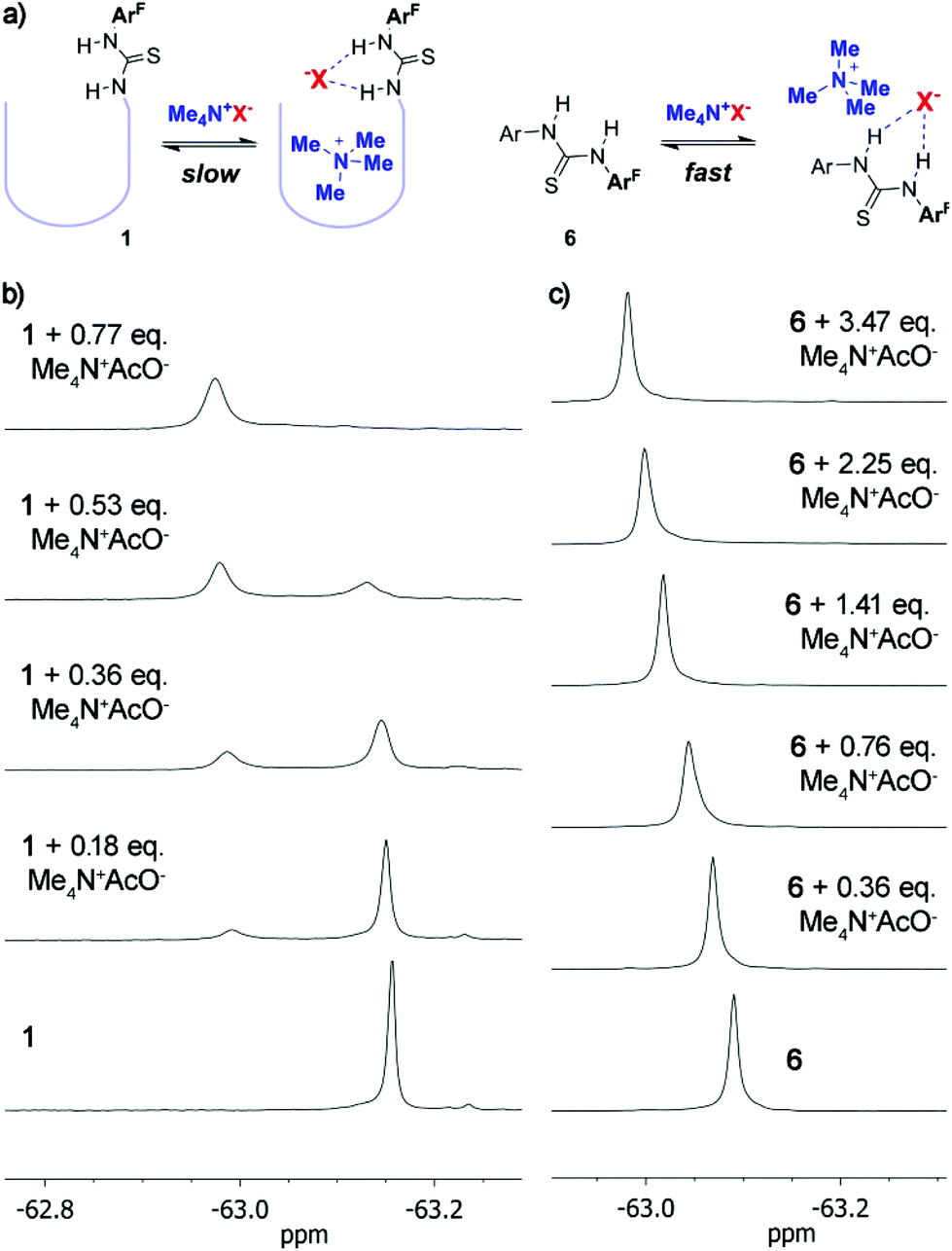 | ||
| Fig. 5 (a) Different exchange regimes with tetramethylammonium salts and evolution of the 19F NMR spectra (CDCl3/CD3OD 95:5) upon titration with Me4N+AcO− of (b) 1 and (c) 6. | ||
Despite the reluctance of 1 to form kinetically stable complexes with linear substrates buried in the hydrophobic pocket, we hypothesized whether ionization of allylic sulphates could take place by specific activation with the thiourea function outside the cavity, followed by rapid internalization of the nascent carbocation in the aromatic pocket. We therefore monitored solutions of 7a–b/7d–e and 1 in CDCl3 over time to check for the formation of new compounds. As ascertained by 1H NMR, the solutions remain basically unaltered at room temperature. When heated to 40 °C the substrates remain stable and no trace of new compounds is observed, but decomposition of 1 is quite apparent.
Conclusions
In summary, we have prepared a new self-folding cavitand receptor with two complementary binding sites: an electron-rich hydrophobic pocket – suitable for binding of hydrocarbons and cationic species – and a thiourea motif directed to anion binding. The two sites work in concert to tightly bind tetramethylammonium ion pairs in CDCl3, resulting in slow exchange of both components relative to the NMR time scale. Salts of cyclic ammonium ions – used as stable models for reactive carbocationic species – were also bound in 1 albeit with much reduced affinity. The lack of binding for linear substrates prevented the use of 1 in developing a biomimetic carbocationic cyclization of terpene-like polyene substrates. Future developments in this area will address the main limitations of the current design, namely (a) the reduced buried space in 1, which makes it unlikely to bind precursors and/or reactive intermediates of 8 carbon atoms or more, and (b) the low binding affinities resulting from the use of a competitive solvent. For the latter, the preparation of water soluble equivalents of 1 is underway in order to exploit hydrophobic effects, which should facilitate folding of linear substrates into energetically disfavoured more compact conformations.22Acknowledgements
We thank Prof. Christopher A. Hunter (U. of Cambridge) for kindly providing a purpose written software for non-linear fitting of titration curves. Assistance with the software from Dr Rafel Cabot (U. of Cambridge) is gratefully acknowledged. We thank MINECO – Spanish Government (“Ramón y Cajal” grant to A. L. RYC2012-11112, CTQ2014-54306-P, and CTQ2014-61629-EXP), Generalitat de Catalunya, 2014SGR931, and Universitat de Girona (MPCUdG2016/096) for financial support.Notes and references
- M. Raynal, P. Ballester, A. Vidal-Ferran and P. W. N. M. van Leeuwen, Chem. Soc. Rev., 2014, 43, 1660–1733 RSC.
- (a) L. Catti, Q. Zhang and K. Tiefenbacher, Chem. – Eur. J., 2016, 22, 9060–9066 CrossRef CAS PubMed; (b) M. Raynal, P. Ballester, A. Vidal-Ferran and P. W. N. M. van Leeuwen, Chem. Soc. Rev., 2014, 43, 1734–1787 RSC.
- (a) Q. Zhang and K. Tiefenbacher, Nat. Chem., 2015, 7, 197–202 CrossRef CAS PubMed; (b) W. M. Hart-Cooper, K. N. Clary, F. D. Toste, R. G. Bergman and K. N. Raymond, J. Am. Chem. Soc., 2012, 134, 17873–17876 CrossRef CAS PubMed.
- (a) R. R. Knowles, S. Lin and E. N. Jacobsen, J. Am. Chem. Soc., 2010, 132, 5030–5032 CrossRef CAS PubMed; (b) R. R. Knowles and E. N. Jacobsen, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 20678–20685 CrossRef CAS PubMed.
- A. Galan and P. Ballester, Chem. Soc. Rev., 2016, 45, 1720–1737 RSC.
- R. J. Hooley and J. Rebek Jr., Chem. Biol., 2009, 16, 255–264 CrossRef CAS PubMed.
- P. Baer, P. Rabe, K. Fischer, C. A. Citron, T. A. Klapschinski, M. Groll and J. S. Dickschat, Angew. Chem., Int. Ed., 2014, 53, 7652–7656 CrossRef CAS PubMed.
- D. M. Rudkevich, G. Hilmersson and J. Rebek, J. Am. Chem. Soc., 1998, 120, 12216–12225 CrossRef CAS.
- A. Lledó, Org. Lett., 2015, 17, 3770–3773 CrossRef PubMed.
- P. Amrhein, A. Shivanyuk, D. W. Johnson and J. Rebek Jr., J. Am. Chem. Soc., 2002, 124, 10349–10358 CrossRef CAS PubMed.
- S. K. Kim and J. L. Sessler, Chem. Soc. Rev., 2010, 39, 3784–3809 RSC.
- (a) F. Durola and J. Rebek Jr., Angew. Chem., Int. Ed., 2010, 49, 3189–3191 CrossRef CAS PubMed; (b) T. Iwasawa, R. J. Hooley and J. Rebek Jr., Science, 2007, 317, 493–496 CrossRef CAS PubMed; (c) U. Luecking, J. Chen, D. M. Rudkevich and J. Rebek Jr., J. Am. Chem. Soc., 2001, 123, 9929–9934 CrossRef CAS.
- O. Dumele, N. Trapp and F. Diederich, Angew. Chem., Int. Ed., 2015, 54, 12339–12344 CrossRef CAS PubMed.
- U. Lücking, F. C. Tucci, D. M. Rudkevich and J. Rebek, J. Am. Chem. Soc., 2000, 122, 8880–8889 CrossRef.
- V. Jo Davisson, A. B. Woodside and C. Dale Poulter, Methods Enzymol., 1985, 110, 130–144 Search PubMed.
- S. Maltsev, O. Sizova, N. Utkina, V. Shibaev, T. Chojnacki, W. Jankowski and E. Swiezewska, Acta Biochim. Pol., 2008, 55, 807–813 CAS.
- D. Ajami, T. Iwasawa and J. Rebek, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 8934–8936 CrossRef CAS PubMed.
- (a) J. A. Faraldos, B. Kariuki and R. K. Allemann, J. Org. Chem., 2010, 75, 1119–1125 CrossRef CAS PubMed; (b) F. Karp, Y. Zhao, B. Santhamma, B. Assink, R. M. Coates and R. B. Croteau, Arch. Biochem. Biophys., 2007, 468, 140–146 CrossRef CAS PubMed.
- S. Kaabel, J. Adamson, F. Topic, A. Kiesila, E. Kalenius, M. Oeren, M. Reimund, E. Prigorchenko, A. Lookene, H. J. Reich, K. Rissanen and R. Aav, Chem. Sci., 2017, 8, 2184–2190 RSC.
- V. Blažek Bregović, N. Basarić and K. Mlinarić-Majerski, Coord. Chem. Rev., 2015, 295, 80–124 CrossRef.
- (a) O. B. Berryman, A. C. Sather and J. Rebek, Org. Lett., 2011, 13, 5232–5235 CrossRef CAS PubMed; (b) M. Kvasnica and B. W. Purse, New J. Chem., 2010, 34, 1097–1099 RSC.
- (a) S. Mosca, Y. Yu and J. Rebek Jr., Nat. Protocols, 2016, 11, 1371–1387 CrossRef CAS PubMed; (b) A. Lledo and J. Rebek Jr., Chem. Commun., 2010, 46, 8630–8632 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: 1H and 13C NMR spectra of new compounds, HRMS spectrum of 1, 1H NMR spectra showing thiourea–alkyl sulphate interactions, 19F NMR titration data for 1 and 6, synthetic schemes not detailed in the main text, molecular model of 1 with geranyl sulphate. See DOI: 10.1039/c7qo00191f |
| ‡ Analysis in less competitive solvents such as mesitylene or tetrachloromethane was prevented by the low solubility of 1 and the ionic guests in these compounds. |
| § CDCl3/CD3OD 95:5 was used as a solvent to ensure full solubility of the salts. The vase conformation of 1 remains unaltered in this solvent mixture. |
| This journal is © the Partner Organisations 2017 |