
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The impact of interplay between electronic and steric effects on the synthesis and the linear and non-linear optical properties of diketopyrrolopyrrole bearing benzofuran moieties†
Anna
Purc
a,
Beata
Koszarna
a,
Irina
Iachina
b,
Daniel H.
Friese
c,
Mariusz
Tasior
a,
Krzysztof
Sobczyk
a,
Tomasz
Pędziński
d,
Jonathan
Brewer
*b and
Daniel T.
Gryko
 *a
*a
aInstitute of Organic Chemistry, Polish Academy of Sciences, Kasprzaka 44/52, 01-224 Warsaw, Poland. E-mail: dtgryko@icho.edu.pl
bDepartment of Biochemistry and Molecular Biology, University of Southern Denmark, Odense 200-701, Denmark. E-mail: brewer@memphys.sdu.dk
cUniversitetet i Tromsø - Norges Arktiske Universitet, Centre for Theoretical and Computational Chemistry, Tromsø, Norway
dAdam Mickiewicz University, Department of Chemistry, Poznań, Poland
First published on 7th February 2017
Abstract
An in-depth investigation of the reaction of substituted salicylaldehydes with chloroacetonitrile led to the development of new conditions for the synthesis of 2-cyanobenzofurans. The crucial improvement lies in the use of phase-transfer catalysis in the second step, i.e., intramolecular aldol type condensation. In a two-step process, the reactants were transformed into a library of 3,6-bis(benzofuran-2-yl)diketopyrrolopyrroles. We show that the presence of a methyl group in a position adjacent to the cyano functionality only slightly decreased the yield of diketopyrrolopyrroles (to 30–57%). An analysis of the relationship between the degree of polarization/planarization of aryl-diketopyrrolopyrroles and their one- and two-photon spectroscopic properties is reported. Careful design of the desired dyes and enhanced control of their ability to assume a planar molecular structure resulted in interesting photophysical properties, such as absorption and emission in the so-called biological window. Despite having less promising linear spectroscopic properties, the deplanarized molecules possess pretty strong two-photon absorbing properties. Placing methyl groups at adjacent positions to the linkage between benzofuran and the DPP core caused the formation of yellow-emitting dyes with almost quantitative fluorescence quantum yield, moderate Stokes shift and reasonable two-photon absorption cross-sections.
Introduction
The optical properties of complex organic functional dyes depend, to a large extent, not only on the particular core, but also on the character of electron-donating and electron-withdrawing substituents.1 Previously, strongly coupled dyes linked by carbon–carbon double or triple bonds were a prevailing area of research,2 but recently, weakly coupled chromophores containing biaryl linkages have drawn greater attention.3 In these cases, the actual dihedral angle between various moieties of the molecule is more important. Based on the investigations by Matile and co-workers, modulation of the planarization and polarization of such push–pull chromophores has become a conceptually new approach for preparing modern fluorescent probes, which have been shown to respond to changes in the fluidity of lipid bilayer membranes.4 Interestingly, this planarization process takes place in the ground state, allowing these novel mechanoprobes to report the changes in their environment by shifts in their absorption and excitation maxima. These mechanoprobes stand in striking contrast to the conventional planar probes, which report the viscosity of their environment by changes in their emission spectra due to deplanarization of the excited state.5Diketopyrrolopyrroles (DPPs),6 which have been used as high-quality pigments, have been repositioned in research related to organic electronics over the last two decades.7 Recently, however, a shift of paradigm has focused attention on the application of DPPs in fluorescence imaging.8 In numerous studies, the relationship between the structure of DPPs and their linear and non-linear optical properties has been examined in great detail.9–11 The replacement of benzene with aromatic five-membered rings at positions 3 and 6 of N,N′-dialkylated DPPs decreased the dihedral angle from ca. 30 to 7 degrees.6a This smaller angle led to significant changes in the optical properties, including a bathochromic shift of absorption,6a,12 better packing in the crystalline state, stronger charge transfer, and larger two-photon absorption cross-sections.13 In this context, thiophene-functionalized DPPs have attracted particular attention due to the high electron density of the thiophene ring, which induces strong intramolecular charge transfer with the electron poor DPP core.14
We have chosen the well-known diketopyrrolopyrrole's core as the electron-withdrawing central moiety and the electron-rich benzofuran moiety to design a library of quadrupolar dyes. We have modified the type and strength of the electron-donating substituents, the geometry of the donor, and the dihedral angle. Although replacing phenyl substituents with five-membered heterocycles and their π-expanded analogues offers some opportunities to modulate the absorption and emission properties, the most significant change in the optical properties was achieved by expanding the DPP-core via fusion with other aromatics/dyes. Four strategies for making such compounds have been recently revealed by Zumbusch,15 Shimizu,16 Würthner,17 and the Gryko group.18 Taking advantage of Matile's concept of planarization, the diaryl-DPPs would perfectly complement these previously reported approaches. Thus far, DPPs decorated with a benzofuran ring have not been extensively investigated, except for some photoinitiators19 and our preliminary studies on two-photon absorbing materials.20 We are interested in this particular substitution pattern because the furan- and benzofuran-derived DPPs have higher fluorescence quantum yields (Φfl) than the corresponding derivatives of thiophene and benzothiophene.20 Also, the literature data suggest that the dihedral angle between the furan and the DPP core is as low as 0.9 degree.6a,21 The goal of this work was to elaborate the synthesis of diketopyrrolopyrrole containing benzofuran moieties at positions 3 and 6 from the corresponding 2-cyanobenzofurans and to establish a relationship between their structural and optical properties. We aimed to identify the molecule(s) possessing both appreciable linear (i.e., high fluorescence quantum yield) and non-linear (i.e., large two-photon absorption cross-section) properties. The two-photon absorbing properties of DPPs have rarely been studied despite the fact that their electron-poor core seems to be an excellent starting point for the construction of D–A–D or A–D architectures, which are important for generating large two-photon absorption cross-sections.20,22 Therefore, we have also investigated how the structure of the benzofuran moiety and its ability for planarization will affect the nonlinear properties of DPPs.
Results and discussion
Design and synthesis
Our design of N,N′-dialkyldiketopyrrolopyrroles was based on enhancing the push–pull effect within the DPP molecules by locating two electron-donating groups, the MeO and amino groups, on benzofuran moieties. Two different modifications have been introduced. Since it is well known that the position of the electron-donating groups has a paramount influence on the absorption and, especially, emission of various dyes,23 our first modification was to locate the methoxy substituent at different positions around the benzene ring of benzofuran. We also modified the character of the dialkylamino group via elongating the alkyl chains (to achieve good solubility of the pigment itself) and via bridging, which is known to cause bathochromic shifts24 and enhance the fluorescence quantum yields.25 Previous studies, with mostly coumarin and xanthene dyes, have indicated that bridging had an immense influence on the optical properties.24,25 Heavy atoms also often influence the properties of organic fluorophores, and therefore bromine was also used. The most important element of our design, however, was the introduction of methyl groups at positions adjacent to the linkage between the benzofuran moieties and the DPP core. We expected the presence of the methyl group to significantly influence both the linear and non-linear optical properties via an increase of the dihedral angle between the benzofuran moieties and the DPP core.Our synthetic strategy required access to 2-cyanobenzofurans and 2-cyano-3-methylbenzofurans. Although the literature26 stated that 2-cyanobenzofurans could be synthesized in a one-pot process directly from salicylaldehydes and chloroacetonitrile, the reality was more complex. The procedure consisting of a Williamson etherification followed by an intramolecular aldol type reaction did not always work efficiently. Indeed, only in the case of salicylaldehydes 1h20b and 1i containing a dialkylamino group at position 6 did this literature procedure (Method C, Table 1) work efficiently as a one-pot process giving rise directly to nitriles 3h and 3i. In almost all other cases the original methodology failed (Table 1). The notable exceptions were aldehydes 1e and 1f which were also successfully transformed into the corresponding benzofurans 3e and 3f in moderate yields (Table 1). In the remaining cases, the yields were below the detection limit or at best below 5%.
|
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1a–l | R1 | R2 | R3 | R4 | R5 | 2a–l (yield) | 3a–l (yield, method) | 4a–l (yield) | 5a–l (yield) |
| 1a | H | Br | H | H | H | 2a (90%) | 3a (52%, A) | 4a (52%) | 5a (60%) |
| 1b | H | H | H | Br | H | 2b (93%) | 3b (33%, A; 11%, B) | 4b (33%) | 5b (70%) |
| 1c | H | OMe | H | Br | H | 2c (86%) | 3c (24%, A; 45%, B) | 4c (57%) | 5c (90%) |
| 1d | H | OMe | H | H | H | 2d (85%) | 3d (48%, A; 72%, B) | 4d (36%) | 5d (61%) |
| 1e | H |
|
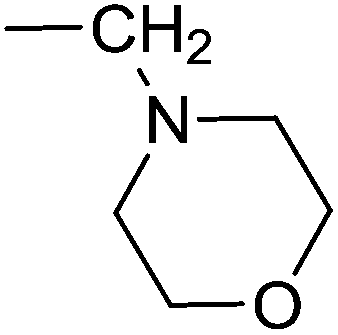
H | OMe | H | — | 3e (51%, C) | 4e (42%) | 5e (79%) |
| 1f | H | H | H |
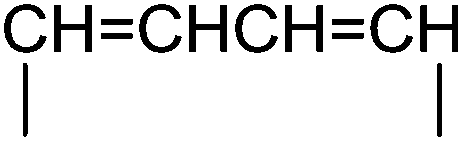
|
— | 3f (32%, C) | 4f (72%) | 5f (46%) | |
| 1g | H | H | OMe | H | H | 2g (78%) | 3g (48%, B) | 4g (28%) | 5g (75%) |
| 1h | H | H | N(C12H25)2 | H | H | — | 3h (62%, C) | 4h (15%) | — |
| 1i | H |
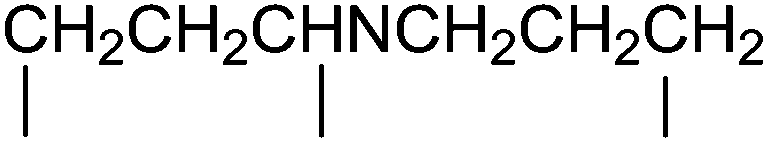
|
H | 2i (49%) | 3i (59%, A) | 4i (69%) | 5i (71%) | ||
| 1j | CH3 | H | H | H | H | 2j (89%) | 3j (97%, A) | 4j (57%) | 5j (24%) |
| 1k | CH3 | H | H | H | H | 2k (63%) | 3k (57%, A) | 4k (53%) | 5k (36%) |
| 1l | CH3 | H | H | OMe | H | 2l (75%) | 3l (89%, A) | 4l (30%) | 5l (21%) |
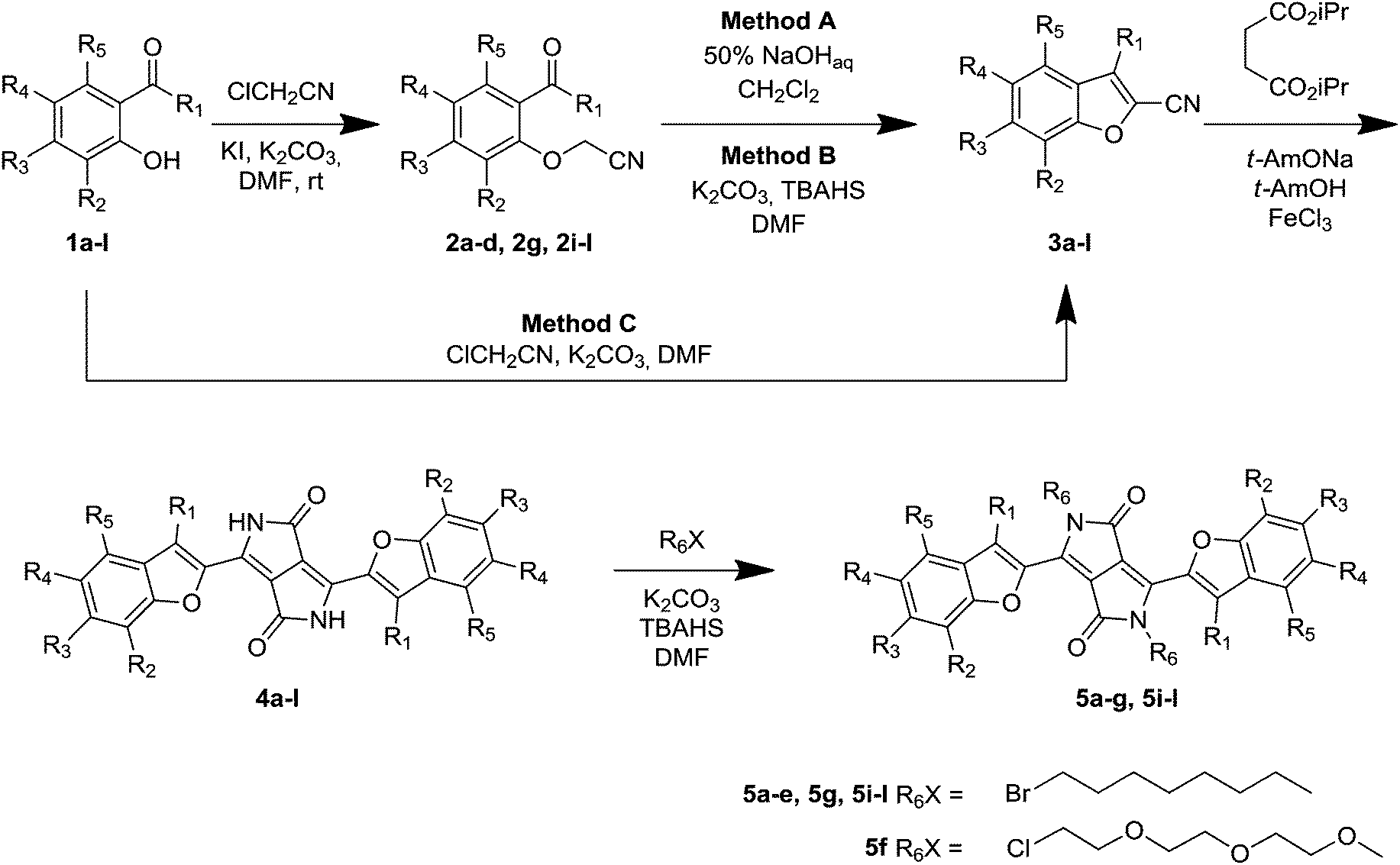
We therefore opted to perform both steps under different conditions. The replacement of ClCH2CN with BrCH2CN and/or replacing DMF with other solvents as well as modifying the reaction temperature did not provide satisfactory yields of the 2-cyanobenzofurans. To address this problem, we prepared the target building blocks in two separate steps. The optimization process using aldehyde 1b as a model system revealed that the best conditions for the formation of O–C bonds are K2CO3/KI in DMF, at room temperature (Table 1). Aldehydes 1a–d and 1g were successfully transformed into aldehydes 2a–d and 2g using this procedure in 78–93% yields. The great limitation in this reaction sequence was the second step, which gave very poor yields due to unidentified side-reactions. Therefore, after investigating various conditions we identified two complementary procedures: method A, in which the aldol reaction takes place in a 50% NaOHaq/DCM mixture; and method B, utilizing K2CO3 and tetrabutylammonium hydrogen sulfate as a phase transfer catalyst (Table 1). These methods allowed us to synthesize a library of 2-cyanobenzofurans 3a–d and 3g in moderate to high yields. The same procedure has been subsequently utilized in the two-step synthesis of nitriles 3j–l from acetophenones 1j–l in overall yields of 36–86% (Table 1).
All substituted 2-cyanobenzofurans 3a–l were subsequently reacted with di-isopropyl succinate to give rise to desired diketopyrrolopyrroles 4a–l in 15–69% yields (Table 1). To our delight, reactions of nitriles 3j–l led to the formation of DPPs 4j–l in 30–57% yield, which is only slightly lower than the average yield of DPP from 2-cyanobenzofurans lacking a substituent at position 3. At the same time, these yields are an order of magnitude higher than the yield of di(2-tolyl)diketopyrrolopyrrole prepared by Langhals from 2-tolunitrile.27 All pigments, except compound 4h, in which long alkyl chains placed on amino groups secured good solubility, were transformed into N-alkylated derivatives 5a–g and 5i–l (Table 1).
To gain insights into the structures of compounds 5j–5l bearing sterically demanding methyl groups, we undertook X-ray analysis. Although for the alkylated 5a–g and 5i the angle between the DPP core and the five membered aromatic ring was expected to be less than 10 degrees, the repulsion between the carbonyl and the methyl group in 5j–5l should lead to substantially larger angles. Indeed, X-ray analysis of crystals of compound 5j obtained by the slow evaporation of a solution of 5j in a dichloromethane/cyclohexane mixture showed that the 3-methylbenzofuran rings were twisted by 41.8 degrees with respect to the DPP mean plane, while both fragments were perfectly planar (Fig. 1).
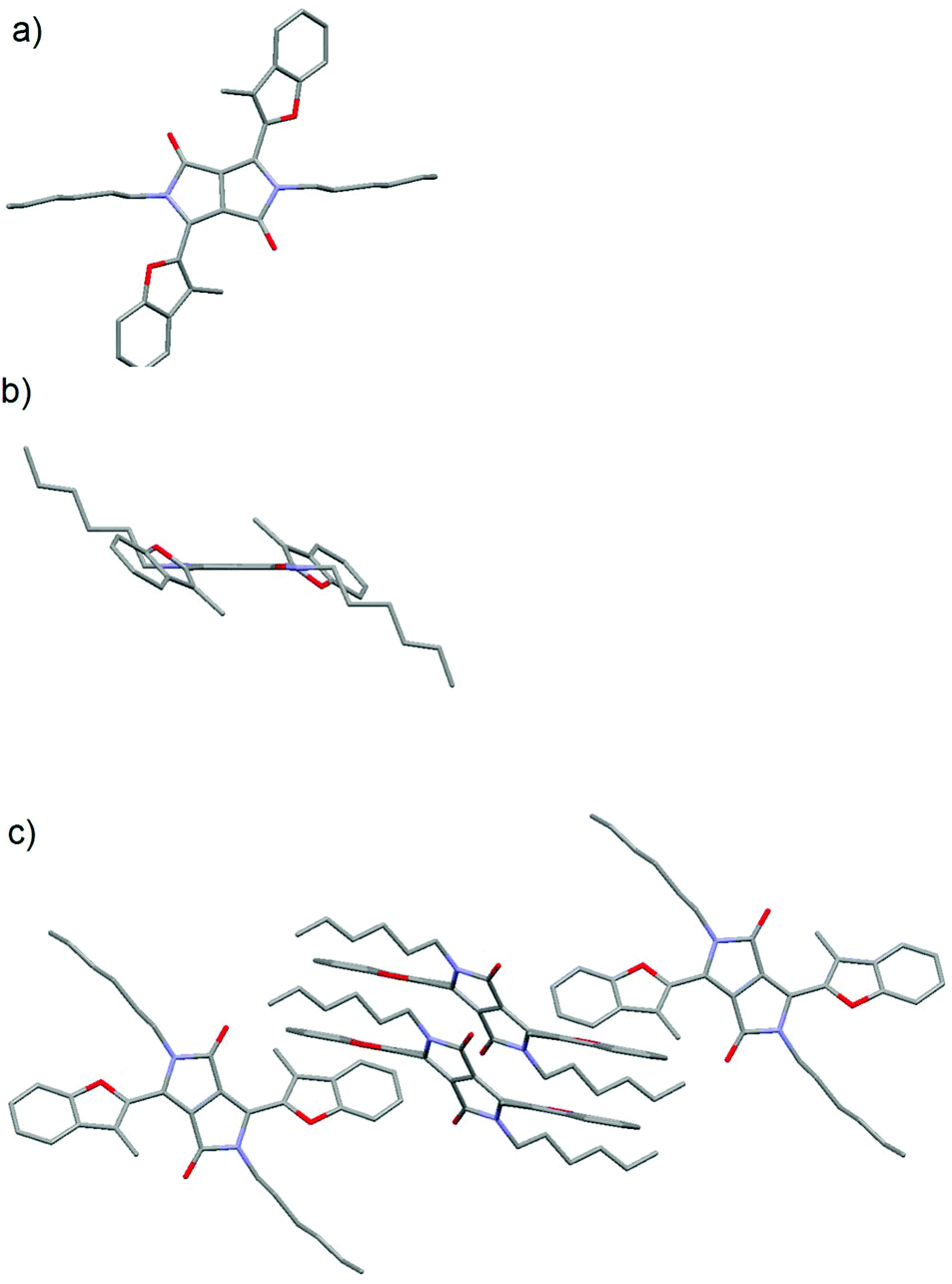 | ||
| Fig. 1 (a) Top view, (b) side view and (c) packing of 5j in the crystal as determined by X-ray analysis (CCDC 1515317). Hydrogens are omitted for clarity. | ||
Photophysical properties
With a library of N,N′-bisalkylated benzofuran-DPPs available, we systematically studied their spectroscopic properties. There was a clear relationship between the absorption and emission maxima and the substitution pattern of the prepared dyes, which can be divided into three categories (Table 2, Fig. 2–4). DPPs that belong to the first group (compounds 5a–5g) are characterized by absorption and emission maxima in the 574–600 nm and 586–611 nm regions, respectively, with molar absorption coefficients of 54![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000–93000 M−1 cm−1 and Φfl ranging from 0.76 to 0.97, when measured in DCM. Interestingly, the presence of a moderately electron-donating OMe group at position 6 of the benzofuran ring in dye 5g did not shift its absorption and emission maxima significantly, although a small hyperchromic effect was easily observed. The substitution of a chromophore with a heavy atom usually resulted in a dramatic decrease of fluorescence quantum yield as the heavy atom activates additional radiationless deactivation channels by favoring intersystem crossing within the molecule.24 Contrary to our expectations, compounds 5a–5c bearing a Br at various locations displayed very high fluorescence quantum yields, reaching 0.97 for 5c in DCM.
000–93000 M−1 cm−1 and Φfl ranging from 0.76 to 0.97, when measured in DCM. Interestingly, the presence of a moderately electron-donating OMe group at position 6 of the benzofuran ring in dye 5g did not shift its absorption and emission maxima significantly, although a small hyperchromic effect was easily observed. The substitution of a chromophore with a heavy atom usually resulted in a dramatic decrease of fluorescence quantum yield as the heavy atom activates additional radiationless deactivation channels by favoring intersystem crossing within the molecule.24 Contrary to our expectations, compounds 5a–5c bearing a Br at various locations displayed very high fluorescence quantum yields, reaching 0.97 for 5c in DCM.
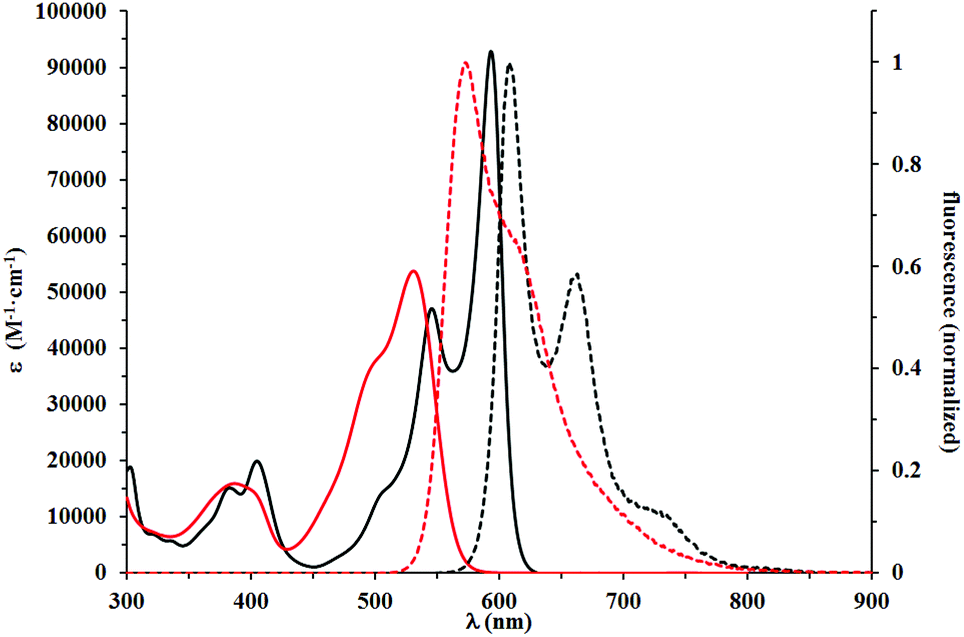 | ||
| Fig. 2 Absorption (solid lines) and normalized emission (dashed lines) spectra of 5j (black) and 5k (red). All spectra were measured in DCM. Compound 5k was characterized by a lower degree of conjugation between the benzofuran moiety and the DPP core. | ||
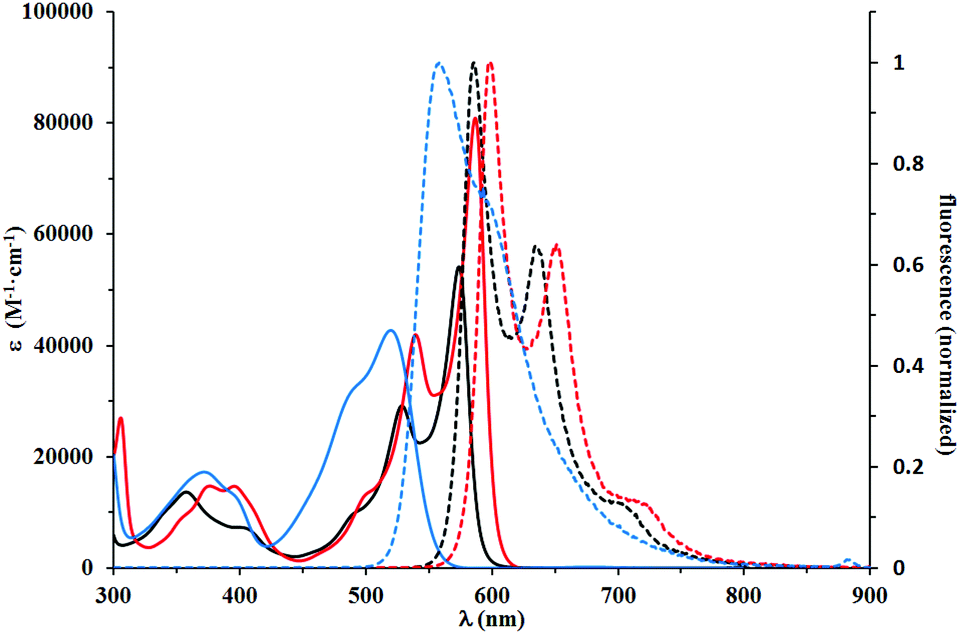 | ||
| Fig. 3 Absorption (solid lines) and normalized emission (dashed lines) spectra of 5d (black)and 5e (red), which have electron-donating substituents on the benzofuran ring, and 5l (blue), which was modified with only one OMe group. Spectra were measured in DCM. | ||
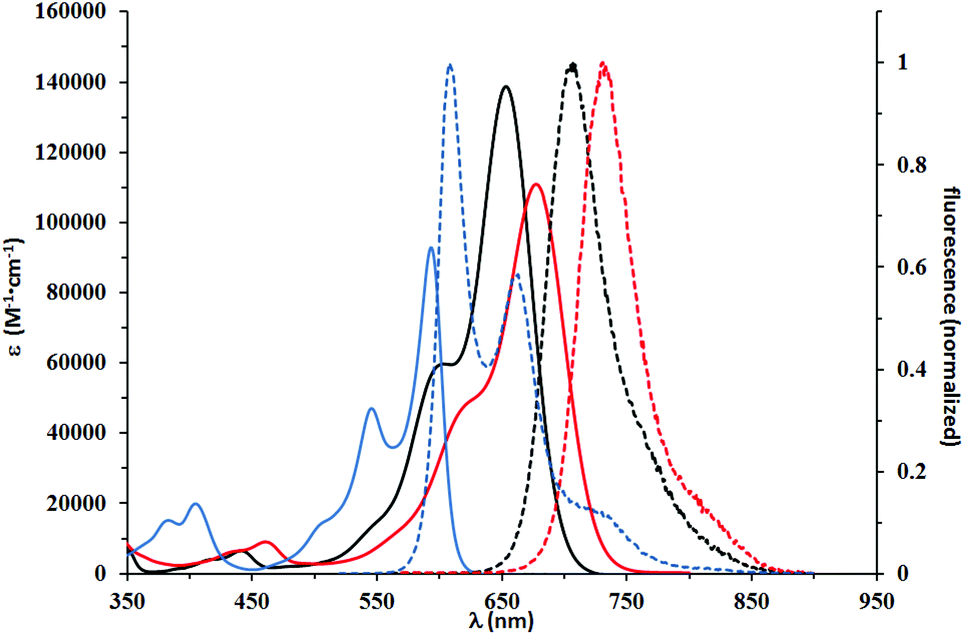 | ||
| Fig. 4 Absorption (solid lines) and normalized emission (dashed lines) spectra of 4h (black)and 5i (red), which have strong electron-donating substituents on the benzofuran ring and 5g (blue), which was modified with only one OMe group. Spectra were measured in DCM. | ||
| Compound | Solvent | λ abs [nm] | ε max [M−1 cm−1] | λ em [nm] | Stokes shift [cm−1] | Φ fl |
|---|---|---|---|---|---|---|
| a Fluorescence quantum yield. b Measured with rhodamine 6G as a reference. c Measured with cresyl violet as a reference. d Measured with 1,1′,3,3,3′,3′-hexamethylindocarbocyanine iodide (HITC) as a reference. | ||||||
| 5a | DCM | 580 | 71000 |
592 | 350 | 0.77b |
| THF | 579 | 68000 |
591 | 350 | 0.77b | |
| Toluene | 583 | 69000 |
596 | 370 | 0.70b | |
| 5b | DCM | 582 | 69000 |
594 | 350 | 0.92b |
| THF | 581 | 66000 |
591 | 290 | 0.87b | |
| 5c | DCM | 577 | 68000 |
589 | 350 | 0.97b |
| THF | 575 | 69000 |
587 | 360 | 0.94b | |
| 5d | DCM | 574 | 54000 |
586 | 360 | 0.80b |
| THF | 574 | 69000 |
582 | 240 | 0.70b | |
| Toluene | 575 | 71000 |
587 | 360 | 0.57b | |
| 5e | DCM | 587 | 81000 |
597 | 290 | 0.82b |
| THF | 585 | 83000 |
595 | 290 | 0.77b | |
| 5f | DCM | 600 | 92800 |
611 | 300 | 0.81c |
| 5g | DCM | 593 | 93000 |
607 | 390 | 0.76b |
| THF | 593 | 100000 |
603 | 280 | 0.62b | |
| Toluene | 594 | 98000 |
607 | 360 | 0.66b | |
| 4h | DCM | 653 | 139000 |
707 | 1200 | 0.70c |
| Toluene | 643 | 148000 |
664 | 490 | 0.72c | |
| Cyclohexane | 620 | 150000 |
635 | 380 | 0.89c | |
| THF | 665 | 127000 |
707 | 890 | 0.49c | |
| 5i | DCM | 677 | 111000 |
731 | 1100 | 0.77d |
| Toluene | 665 | 126000 |
701 | 770 | — | |
| Cyclohexane | 644 | 128000 |
659 | 350 | — | |
| THF | 665 | 79000 |
707 | 890 | — | |
| 5j | DCM | 515 | 40000 |
555 | 1400 | 0.95b |
| THF | 513 | 41000 |
558 | 1600 | 0.95b | |
| Toluene | 517 | 37000 |
564 | 1600 | 0.85b | |
| iPrOH | 513 | 43000 |
554 | 1400 | 0.90b | |
| DMSO | 517 | 39000 |
560 | 1500 | 0.90b | |
| Cyclohexane | 513 | 36000 |
557 | 1500 | 0.89b | |
| 5k | DCM | 531 | 54000 |
573 | 1400 | 0.94b |
| THF | 528 | 54000 |
569 | 1400 | 0.89b | |
| 5l | DCM | 520 | 43000 |
558 | 1300 | 0.92b |
| THF | 518 | 44000 |
563 | 1500 | 0.93b | |
| Toluene | 522 | 46000 |
568 | 1600 | 0.73b | |
The optical properties of dye 5g are nearly identical with its analog possessing decyloxy groups instead of MeO groups at position 6 of benzofuran.18 Indeed, the methoxy group at position 7 even has a hypsochromic effect on both absorption and emission maxima when compared to DPP derived from benzofuran bearing only two tert-butyl groups at positions 5 and 7,18 or dyes 5a and 5b possessing Br atoms. A slight bathochromic shift (10 nm) is caused by placing MeO groups at position 5. Angular attachment of an additional ring (dye 5f) caused only ∼20 nm bathochromic shift of absorption and emission. All these quadrupolar diketopyrrolopyrroles possess very strong orange fluorescence, which is only slightly affected by the type and position of substituents.
The second group consisted of compounds 4h and 5i, bearing strongly donating amino groups at position 6 of the benzofuran. Their absorption and emission maxima were bathochromically shifted by ca. 100 nm in DCM, as compared to compounds 5a and 5b, which lacked the electron-donating substituents; the absorption coefficients of 4h and 5i were also significantly higher. Both 4h and 5i exhibited solvatochromism and strong solvatofluorochromism, which possibly originated from the highly polarized character of both molecules in the ground state and stabilization of a charge-separated excited state by polar solvents. The influence of strong electron-donating substituents was well visualized by direct comparison of the absorption maxima for 4h and 5i (λmax in DCM of 653 and 677 nm, respectively) with DPPs bearing unsubstituted benzofuran and benzothiophene,28 which showed absorption maxima at 616 and 620 nm, respectively, in DCM. Bridging gives rise to ∼20 nm bathochromic shift of absorption and even stronger 40 nm shift of emission. Emission in DCM beyond 700 nm has been reached for the first time for non-fused DPP. The presence of long alkyl chains made it possible to study the influence of lack of N-alkyl substituent. One clear difference is that compound 4h is much more sensitive to solvent polarity. This is especially true for emission, which shifts from 636 nm in cyclohexane to 707 nm in THF, while at the same time Φfl decreases substantially. This type of behavior has been much less pronounced for analogous DPP with two Et2N substituents.13 Another explanation would be the possibility of hydrogen bond formation for 4h, leading to aggregation processes in solvents of low polarity and alteration of spectroscopic properties. The structurally related 5i, however, for which the ability for hydrogen bond formation is blocked, display a similar albeit less pronounced tendency. Aggregation involving H-bond formation has been confirmed for non-alkylated diketopyrrolopyrroles and through exciton delocalization (excited state energy dissipation) it leads to increased stability of these pigments.6 The fact that there is an increase of fluorescence quantum yield for solutions of dye 4i in non-polar solvents further supports our assumption of charge-transfer character of the excited state observed for both 4h and 5i. Symmetry breaking in the excited state leading to strong solvatofluorochromism has been observed for many centrosymmetric dyes29,30 including diketopyrrolopyrrole derivatives.31
This red shift in the absorption spectra of DPPs bearing R2N groups stands in striking contrast with optical properties recorded for the third group of chromophores, to which compounds 5j–5l belong. These compounds displayed a lower degree of conjugation between the DPP core and the benzofuran moiety. The decreased conjugation is attributed to the almost 42 degrees twist of the mean plane of the latter subunit. This led to the strong blue shift of their absorption and emission maxima, which were in the 515–531 nm and 555–573 nm regions, respectively. Their molar absorption coefficients barely reached 40000 M−1 cm−1, considered the lowest among all tested compounds. The Stokes shifts measured for compounds 5j–5l are in the range of 1300–1400 cm−1, up to three times higher than those observed for 5a–5g, which suggested that the geometry of the S1 state of 5j–5l was more distorted than that of 5a–5g. Direct comparison of the absorption and emission of 5k and 5g both possessing electron-donating MeO groups shows that the presence of the Me group causes ∼60 nm blue-shift of absorption and only ∼35 nm shift of emission. The fundamental differences in the conformation in the ground state (dihedral angle ∼40 degrees) and partial planarization in the excited state are responsible for this result. A similar observation can be drawn when compounds 5e and 5l are compared. At the same time the absorption and emission of DPPs 5j–l are strongly bathochromically shifted vs. that of di(2-tolyl)diketopyrrolopyrrole (∼80 nm).27 The Stokes shift remains at the same level. Fig. 2–4 illustrate the differences between the selected compounds that belong to each category.
Two-photon absorption spectra were recorded using two-photon excited fluorescence measurements (Table 3, Fig. 4–6). The highest value of the cross-section of the two-photon absorption of 1547 GM was measured for compound 5i at 780 nm (Fig. 5). Similarly, very high values were measured for other dyes possessing electron-donating groups in the 6-position of the benzofuran (5e and 5g); however, for compound 5g bearing a methoxy substituent, the highest value of two-photon absorption was reached at a lower wavelength (730 nm). The constrained structure of dye 5e only slightly increased the cross-section value. Given the large quantum yield of fluorescence, the two-photon brightness of all three dyes is very high and in the preferred range for biological studies.
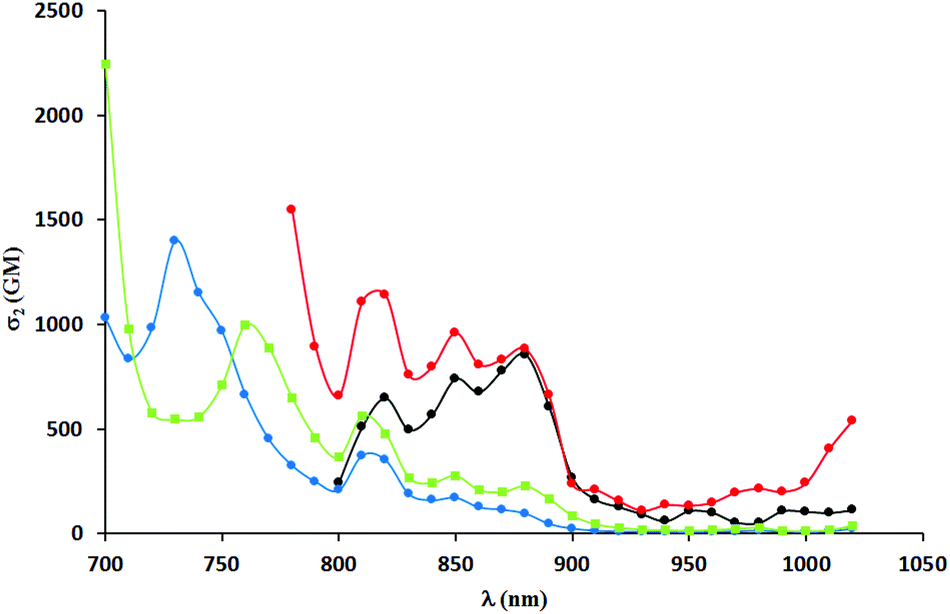 | ||
| Fig. 5 TPA spectra of compounds 4h (black), 5i (red), 5g (blue) and 5f (green) measured in DCM. | ||
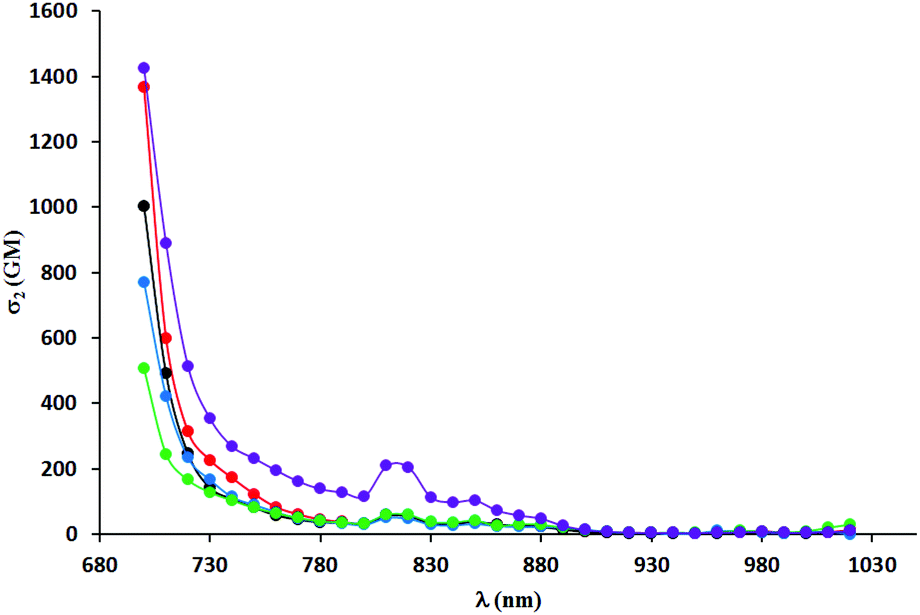 | ||
| Fig. 6 TPA spectra of compounds 5a (black), 5b (red) and 5c (blue), 5d (green) and 5e (violet) measured in DCM. | ||
| Compound | Φ fl | 2·λOPA [nm] | λ max1TPA [nm] | σ max12 [GM] | σ max12·Φ [GM] |
|---|---|---|---|---|---|
| 5a | 0.77 | 1160 | ≤700 | ≥1004 | 770 |
| 5b | 0.92 | 1164 | ≤700 | ≥1365 | 1260 |
| 5c | 0.97 | 1154 | ≤700 | ≥771 | 750 |
| 5d | 0.80 | 1148 | ≤700 | ≥506 | 400 |
| 5e | 0.82 | 1174 | ≤700 | ≥1426 | 1170 |
| 5f | 0.81 | 1200 | 760 | 997 | 810 |
| 5g | 0.76 | 1186 | 730 | 1400 | 1060 |
| 4h | 0.70 | 1306 | 880 | 855 | 600 |
| 5i | 0.77 | 1354 | ≤780 | ≥1547 | 1190 |
| 5j | 0.95 | 1030 | 740 | 342 | 320 |
| 5k | 0.94 | 1062 | 760 | 737 | 690 |
| 5l | 0.92 | 1040 | 740 | 292 | 270 |
A significant decrease in two-photon absorption at wavelengths longer than 700 nm was observed for all the synthesized compounds having bromine atoms, and for compounds with methoxy groups placed in positions other than position 6 (Fig. 6). The similarity of the overall shape of the spectra obtained for those dyes in comparison with a strong absorption of the compounds described in the previous paragraph showed the importance of the presence of an electron-donating substituent in the 6-position of the benzofuran. The direct comparison of TPA spectra of dyes 5j–l with their analogs lacking Me groups reveals significant differences. The values of TPA cross-sections for the latter one are smaller but they are located at higher wavelength, which is a rather unexpected. The presence of a methyl group in the 3-position of the benzofuran led to a significant increase of angle between the DPP core and the aromatic substituents, but did not result in the expected decrease in two-photon absorption cross-section (Fig. 7). The maximum values were found at wavelengths of 740–760 nm and within this range were even higher than those of the dyes with less steric hindrance (except those having electron-donating substituents at position 6 of benzofuran). One has to emphasize however that TPA has been measured only from 700 nm due to the instrument limitations, and it is possible that TPA maxima are situated below 700 nm.
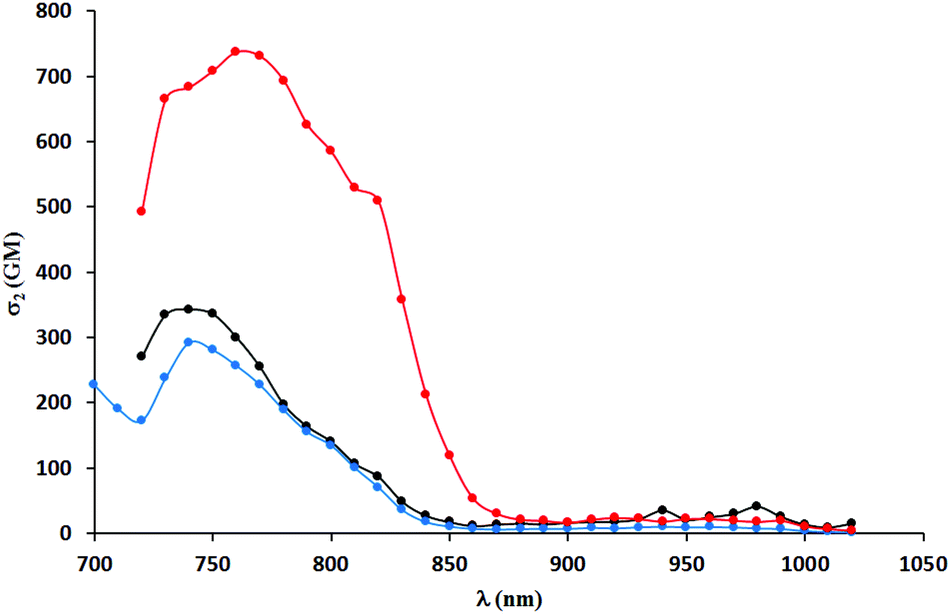 | ||
| Fig. 7 TPA spectra of compounds 5j (black), 5k (red) and 5l (blue) measured in DCM. | ||
The results clearly confirm the centrosymmetric nature of the presented diketopyrrolopyrrole derivatives. Quantum selection rules state that for the centrosymmetrical chromophores, one-photon permitted transitions are not allowed for two-photon transitions and vice versa. This means that the energy level of the excited state achieved in a two-photon absorption process is different from the ‘classical’ (i.e., achieved by single-photon excitation) energy of the S1. Therefore, the two-photon absorption maximum measured for, e.g., compound 4h is located at 880 nm and not at 1PA × 2 (i.e., 653 nm × 2 = 1306 nm) (Table 3).
Computational studies
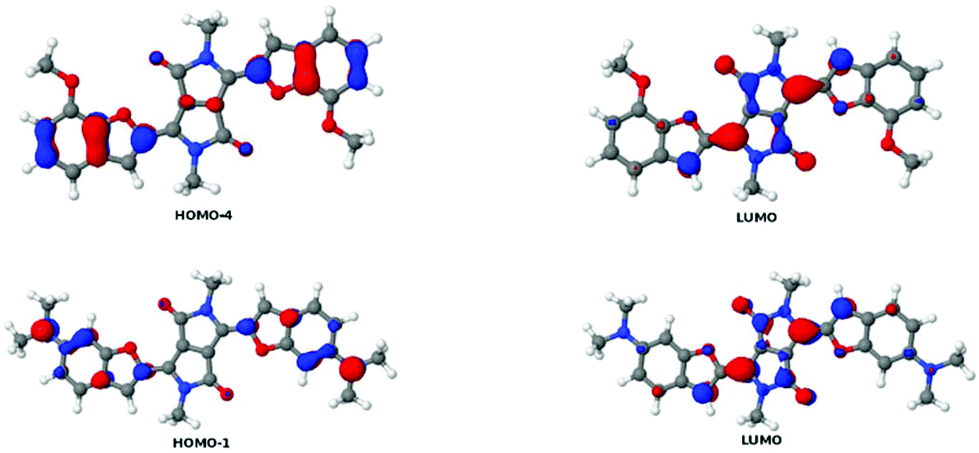
To extend our understanding, we compared the experimental results for two-photon absorption with computational methods. Evaluating the excited states of the molecules revealed that the different systems showed great variation. Only the lowest excited state, which is always dark in TPA, was found in all molecules. However, several states were identified in several molecules that could be compared with each other. Excited states were assigned to each other by comparison of the involved orbital transitions. As both the involved orbitals and the states differ in order, we have named the states with capital letters A–G. Naming either by state number or orbital transition is not possible under these circumstances. However, each state can be correlated with a certain dominant orbital transition. While the involved virtual orbitals are the LUMO and the LUMO+1 in most cases, the occupied orbitals involved differ a lot in order. In Fig. 8 we give an overview of the orbital transitions involved in the excitation to states A, B, C, D and E for dye 5b. In Fig. 9 the dominant orbital transitions for states F and G are shown exemplified on 5d and 5h, respectively. In the other molecules the orbitals look very similar but the occupied ones are in a different order. States which have been identified in several molecules are named with capital letters A–G and listed in Table 4. The results of our calculations are listed in Table 5.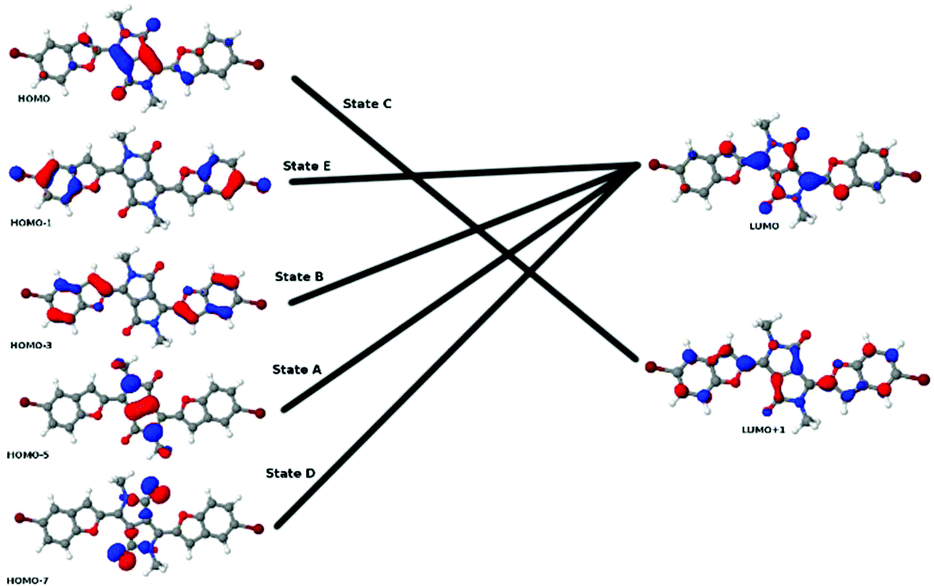 | ||
| Fig. 8 Overview of the orbital transitions which dominate the five lowest TPA-active states in 5b. Note that the LUMO+1 and the HOMO are symmetric while all other orbitals are antisymmetric with respect to inversion. The state with the lowest energy corresponds to the HOMO–LUMO transition. Note that this state is not indicated in the picture as it is dark in TPA. | ||
 | ||
| Fig. 9 Orbital transitions dominating the states F in 5d (top line) and G in 5h. Note that the LUMO is qualitatively the same as the one in 5b. | ||
| Compound | A | B | C | D | E | F | G |
|---|---|---|---|---|---|---|---|
| 5a | 1 | 3 | 4 | 5 | |||
| 5b | 1 | 4 | 2 | 5 | 3 | ||
| 5c | 1 | 4 | 5 | ||||
| 5d | 1 | 3 | 4 | ||||
| 5e | 2 | 8 | 5 | 3 | |||
| 5f | 2 | ||||||
| 5g | 1 | 2 | 3 | ||||
| 4h | 3 | 4 | 1 | ||||
| 5i | 3 | 1 | |||||
| 5j | 1 | 2 | 3 | 4 | |||
| 5k | 1 | 2 | 3 | 4 | |||
| 5l | 1 | 3 | 4 | 2 |
The most abundant states were A and C which were found in nearly all molecules. State A, which is always the lowest TPA-active state when present, is not found in 5h and 5i, the two molecules containing amino groups bound to the benzofuran moieties. These molecules have also shown remarkable differences in their measured TPA spectra compared to the other ones. In molecules 5i and 5h we observe two well-separated, low-lying states of G and C type. In all other molecules, the lowest TPA-active state is of A-type. This shows that the amino groups have a strong influence on the chromophore and induce remarkable changes in the excited state manifold of the molecules. The most remarkable indication for this is the occurrence of the G-type states, which have only been found for 5h and 5i. From Fig. 9 we also note that the occupied orbital involved in this transition has a strong coefficient at the nitrogen of the amino group. Similar observations have been made on 5f where the annelated ring also induces a strong modification of the chromophore. For this reason there is only one state left in 5i which is comparable to the ones of the other molecules. In the other molecules the lowest TPA-active states are much more comparable as can be seen from Table 4. State B was not found in molecules with inductive donors in 7-position. We also note that in some cases the TPA-active states are very close to each other. Therefore, it is likely that they were not well separable in some cases. This outcome was observed for 5a (states D and E), 5b (states E and B), 5d (state F and another non-comparable one), 5e (states C and B) and 5f (state D with two non-comparable ones). State A was of valence character. The most relevant involved orbitals were located at the diketopyrrolopyrrole backbone of the molecules. State B has partial charge transfer character. It corresponded to charge transfer from the benzofuran substituents to the diketopyrrolopyrrole backbone. States C and D show slight charge transfer in the opposite direction.
Overall, the strongest TPA was found for dye 5i. However, this TPA is of no experimental relevance as it is caused by a resonance effect with a very strong OPA-active state (not listed in the table). For this reason, this strong TPA will not be visible but hidden behind the OPA of the lowest OPA-active state. Therefore, this state is irrelevant experimentally and will not be further discussed here. The strongest TPA below possible resonances is found for 5e with 9075 GM at 627 nm. It is of B-character and corresponds to the transition between the highest occupied and the lowest unoccupied ungerade molecular orbitals. In the other cases this state shows TPAs in the range between 1000 and 3000 GM. The strong boost of this state in 5e cannot be interpreted without taking state mixing into account since the B-state is found very close to the C-state, which shows a much smaller TPA. This state mixing is reflected in the involved orbital transitions. The B and C states in 5e were dominated by the same orbital transitions as in the other molecules (see Fig. 8). However, in 5e there are relevant contributions to state C also from the transition dominating state B and vice versa, which is not the case in the other molecules.
Comparing the C states in the different molecules, we note that they showed a large variety of TPAs that are extremely strong in the two amino-substituted molecules 5h and 5i. This state is of charge transfer character as the corresponding occupied orbital is mostly located at the benzofuran moieties while the virtual orbital is located at the diketopyrrolopyrrole backbone (see Fig. 8).
The B and C states were investigated using a few-state model analysis in an effort to reproduce the dominating element of the TPA tensor M (yy in most cases, xx for 5g) using an approximation to the sum-over-states approach according to eqn (1):
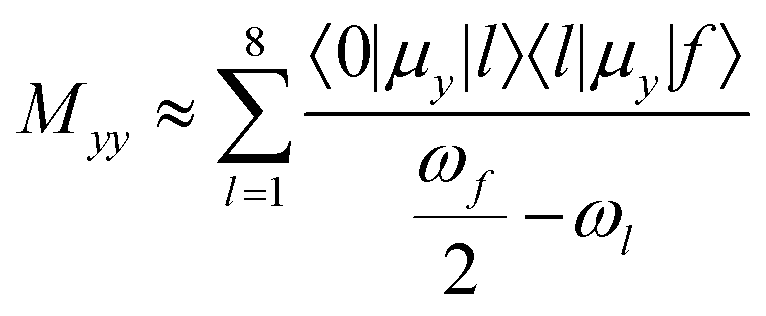 | (1) |
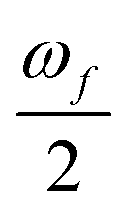 is half the excitation energy of the final state, which is identical to the energy of the incident photons. The summation runs over the lowest eight excited states. This limit is due to computational restrictions as transition moments between a large number of excited states are extremely difficult to compute.
is half the excitation energy of the final state, which is identical to the energy of the incident photons. The summation runs over the lowest eight excited states. This limit is due to computational restrictions as transition moments between a large number of excited states are extremely difficult to compute.
The following discussion is based on an investigation of 5d, 5e, 5g, 5j, 5k and 5l. This means that for this part of the study only molecules which contain neither bromine nor amino groups nor annelated rings on the aromatic moiety are taken into account. Using eqn (1) we found that for all the states that have been considered, by far the most dominant contribution came from the intermediate state l that is the lowest excited state. This state is always TPA-dark for symmetry reasons but the brightest state in one-photon absorption (OPA). This state can be considered a hub with respect to the non-linear optical properties.
Evaluating the one-photon transition moments we note that there are correlations between 〈0|μy|1〉 and 〈1|μy|B〉, and between 〈1|μy|A〉 and 〈1|μy|C〉: 〈0|μy|1〉 and 〈1|μy|B〉 have been found to be strong in 5d, 5e and 5g while 〈1|μy|A〉 and 〈1|μy|C〉 are stronger in 5j, 5k and 5l. An explanation for this different behaviour is the shape of the involved orbitals. 〈0|μy|1〉 is always dominated by the HOMO–LUMO transition. Fig. 8 reveals that these orbitals are mainly located at the diketopyrrolopyrrole backbone and also that they have small but reasonable coefficients at the benzofuran. This is the same for the orbitals involved in the transition to the B-type state. Here the involved occupied orbital is, to a large extent, located at the benzofurans. Therefore, these transition moments and especially the ones to the B-state should strongly depend on the coupling between the backbone and the substituent, which is the strongest for planar structures. The CH3 groups in 5j, 5k and 5l enforce a stronger torsion between these moieties. This means that a large TPA for the B state can be correlated with a large OPA of the lowest state.
By contrast, the transitions to states A and C are, to a larger extent, dominated by orbitals which are mostly located at the backbone. Therefore they are less dependent on the torsion between the backbone and benzofuran rings. Here the prediction is more difficult than for the B state as 〈0|μy|1〉 on the one side and 〈1|μy|A〉 and 〈1|μy|C〉 on the other side behave differently.
Table 6 provides an overview of the calculated partial charges for six of the molecules in the named positions. In most cases, we noted that substituents (all substituents under consideration can be taken as mesomeric or inductive donors) decreased the partial charge of the atoms to which they were bound. Comparing the TPA for the B and C states (which have the strongest TPA and also occur in enough molecules to enable comparisons) we noted that the strongest values were found for systems with donors located in 5- or 6-position (values for 5 being stronger than those for 6) while a methyl group in 3-position leads to a lower TPA. Molecules with a CH3 group in 3-position showed lower TPA than the ones without this substitution. At the same time, the numbers in Table 6 indicated that especially in this position, the change of partial charge by substitution is enormous. One might therefore argue that a substitution in 3-position hampers the formation of a transition dipole moment between the electron-rich substituent and the electron-poor backbone. While this factor may provide a partial explanation, the answer it provides is quite qualitative and reveals just a part of the puzzle, especially because the torsion between the different moieties has to be taken into account, which is known to have a strong influence on the TPA (Table 6).
Experimental
Materials and methods
All chemicals were used as received unless otherwise noted. Reagent grade solvents (MeCN, DCM, hexane, and toluene) were distilled prior to use. All reported NMR spectra were recorded on a 500 MHz spectrometer unless otherwise noted. Chemical shifts (δ ppm) for 1H and 13C NMR were determined with TMS as the internal reference. J values are given in Hz. Chromatography was performed on silica (Kieselgel 60, 200–400 mesh). Mass spectra were obtained with an EI ion source and an EBE double focusing geometry mass analyzer or spectrometer equipped with an electrospray ion source with a q-TOF type mass analyzer.Synthesis
All experimental details, including synthetic procedures, spectroscopic characterization of new compounds and X-ray analysis, are given in the ESI.†Two-photon excited fluorescence (TPEF) measurements
Dichloromethane (CHROMASOLV, for HPLC, ≥99.9%), methanol (CHROMASOLV, for HPLC, ≥99.9), 5-carboxyfluorescein (99% (HPLC)) and Rhodamine B were purchased from Sigma-Aldrich, Denmark.Solutions of the samples 4h, 5a–g and 5i–l (2 μM in dichloromethane) were prepared and used for the experiments. Solutions of Rhodamine B, 2 μM in methanol, and fluorescein, 2 μM in a CAPS buffer (N-cyclohexyl-3-aminopropanesulfonic acid), pH 11, were also prepared.
The concentrations of the samples and references were measured using a dilution series in a spectrophotometer (PerkinElmer lambda 35). Single photon fluorescence emission spectra were recorded using a spectrofluorometer (ChronosFD from ISS, Champaign, IL USA). Two photon absorption measurements were performed on a custom built multiphoton excitation spectrofluorometer described previously.32 The absolute 2-photon absorption cross sections were calculated by comparing the relative fluorescence intensity of the sample to a well-known reference. The references used were fluorescein in a CAPS buffer (pH 11) and Rhodamine B in methanol, using a references absorption spectrum as described previously.33 The measurements for the two photon cross-sections were carried out at least five times for the different samples. To remove artifacts due to photobleaching or linear absorption, the samples were measured at different excitation intensities to ensure that the fluorescence signal increased as the square of the excitation intensity at different excitation wavelengths for different samples.
The excitation wavelength ranged from 700 nm to 1020 nm. However, due to the linear absorption of some of the samples, the lower wavelengths were excluded.
Calculations
All molecules were optimized using TURBOMOLE,34 the B3LYP functional35 and the TZVP basis sets.36 One- and two-photon absorption cross-sections were calculated using the DALTON program,37 the CAM-B3LYP density functional38 and the aug-cc-pVDZ basis set.39 Scalar relativistic effects of the bromine atoms were taken into account using effective core potentials.40 Apart from 5e and 5f, which have been calculated without point group symmetry, all molecules were treated using Ci point group symmetry containing an inversion point. In the molecules with Ci symmetry, only the totally symmetric states are taken into account since for the antisymmetric state transitions, TPA is symmetry-forbidden. However, also in the two molecules treated without point group symmetry, an alternation of TPA-dark and bright states was found due to the quasi-centrosymmetry of these molecules. Derivations from centrosymmetry are mainly caused by conformations of the substituents, not by the chromophore. In all calculations, the pyrrole-N-atoms were capped with CH3 groups instead of the long side chains of the synthesized molecules for ease of calculation. This variation had special implications for the 5h molecule. Of the h-type structures, an alkyl substituted molecule was not synthesized, however it was treated computationally. We furthermore note that in the calculations, methyl substituents are used instead of octyl chains (5a–5e, 5g, 5i–5l) or polyether chains (5f) for ease of calculation and especially for geometry optimization. In 5e and 5i the substituents at the amino groups have been replaced by methyl groups for the ease of both optimization and property calculation.Conclusions
The direct, one-pot transformation of substituted salicylaldehydes into 2-cyanobenzofurans is capricious, most probably due to numerous side reactions influenced by the reactivity of the formyl group. In turn, the reactivity of the formyl group depends on the particular arrangement of substituents on the benzene ring. Dividing the whole process into two steps, i.e., Williamson etherification followed by intramolecular aldol type condensation in combination with the use of phase-transfer catalysts for the second step, allowed a more general and reliable procedure which enabled the straightforward preparation of 2-cyanobenzofurans from a range of salicylaldehydes. Due to the wider angle between the CN and the methyl group in 2-cyano-3-methylbenzofurans (compared to the 2-tolunitrile), the formation of diketopyrrolopyrroles occurred in good yields (30–57%). Polarization and planarization of the aryl-diketopyrrolopyrroles is a powerful tool for fine-tuning their one- and two-photon spectroscopic properties. By taking advantage of the natural tendency of benzofuran-substituted DPPs to adopt a planar conformation, we were able to shift their absorption maxima up to 677 nm. This shift to longer wavelengths is of considerable importance because of the interest in far-red absorbing dyes for deep tissue imaging and reduced photodamage. Thus, this procedure places these compounds perfectly in the spectral region suitable for bio-imaging applications. In contrast, analogous compounds bearing methyl substituents in the furan ring exhibited highly twisted conformations that hampered π-conjugation along the molecule and shifted the absorption and emission maxima to much shorter wavelengths. All the tested DPPs exhibited strong two-photon absorption (TPA) and centrosymmetrical behavior, with good two-photon brightness. Despite the interruption of the π-conjugation, the twisted DPPs display considerably promising TPA.Acknowledgements
The authors would like to thank the National Science Centre, Poland (Grant MAESTRO-2012/06/A/ST5/00216), the Foundation for Polish Science (TEAM/2009-4/3) and the Global Research Laboratory Program (2014 K1A1A2064569) through the National Research Foundation (NRF) funded by the Ministry of Science, ICT & Future Planning (Korea), for financial support.Notes and references
- (a) L. Zhua and Y. Zhao, J. Mater. Chem. C, 2013, 1, 1059–1065 RSC; (b) A. Goel, A. Sharma, M. Kathuria, A. Bhattacharjee, A. Verma, P. R. Mishra, A. Nazir and K. Mitra, Org. Lett., 2014, 16, 756–759 CrossRef CAS PubMed; (c) J. Sun, X. Lv, P. Wang, Y. Zhang, Y. Dai, Q. Wu, M. Ouyang and C. Zhang, J. Mater. Chem. C, 2014, 2, 5365–5371 RSC; (d) L. S. Kocsis, K. M. Elbel, B. A. Hardigree, K. M. Brummond, M. A. Haidekker and E. A. Theodorakis, Org. Biomol. Chem., 2015, 13, 2965–2973 RSC; (e) L. Kong, Y.-P. Tian, Q.-Y. Chen, Q. Zhang, H. Wang, D.-Q. Tan, Z.-M. Xue, J.-Y. Wu, H.-P. Zhou and J.-X. Yang, J. Mater. Chem. C, 2015, 3, 570–581 RSC; (f) L. Ji, A. Lorbach, R. M. Edkins and T. B. Marder, J. Org. Chem., 2015, 80, 5658–5665 CrossRef CAS PubMed; (g) C. Hu, F. Liu, H. Zhang, F. Huo, Y. Yang, H. Wang, H. Xiao, Z. Chen, J. Liu, L. Qiu, Z. Zhen, X. Liua and S. Bo, J. Mater. Chem. C, 2015, 3, 11595–11604 RSC; (h) M. Ipuy, Y.-Y. Liao, E. Jeanneau, P. L. Baldeck, Y. Bretonnière and C. Andraud, J. Mater. Chem. C, 2016, 4, 766–779 RSC; (i) G. Haberhauer, R. Gleiter and C. Burkhart, Chem. – Eur. J., 2016, 22, 971–978 CrossRef CAS PubMed.
- (a) M. Barzoukas and M. Blanchard-Desce, J. Chem. Phys., 2000, 113, 3951–3959 CrossRef CAS; (b) M. Drobizhev, A. Karotki, Y. Dzenis, A. Rebane, Z. Suo and C. W. Spangler, J. Phys. Chem. B, 2003, 107, 7540–7543 CrossRef CAS; (c) H. Meier, Angew. Chem., Int. Ed., 2005, 44, 2482–2506 CrossRef CAS PubMed; (d) M. Charlot, L. Porrès, C. D. Entwistle, A. Beeby, T. B. Marder and M. Blanchard-Desce, Phys. Chem. Chem. Phys., 2005, 7, 600–606 RSC; (e) M. Charlot, N. Izard, O. Mongin, D. Riehl and M. Blanchard-Desce, Chem. Phys. Lett., 2006, 417, 297–302 CrossRef CAS; (f) Y. Niko, H. Moritomo, H. Sugihara, Y. Suzuki, J. Kawamata and G.-I. Konishi, J. Mater. Chem. B, 2015, 3, 184–190 RSC; (g) M. Tasior, I. Bald, I. Deperasińska, P. J. Cywiński and D. T. Gryko, Org. Biomol. Chem., 2015, 13, 11714–11720 RSC; (h) B. Dereka, A. Rosspeintner, Z. Li, R. Liska and E. Vauthey, J. Am. Chem. Soc., 2016, 138, 4643–4649 CrossRef CAS PubMed.
- (a) Z.-Q. Liu, Q. Fang, D.-X. Cao, D. Wang and G.-B. Xu, Org. Lett., 2004, 6, 2933–2936 CrossRef CAS PubMed; (b) M. Parent, O. Mongin, K. Kamada, C. Katana and M. Blanchard-Desce, Chem. Commun., 2005, 2029–2031 RSC; (c) H. M. Kim, W. J. Yang, C. H. Kim, W.-H. Park, S.-J. Jeon and B. R. Cho, Chem. – Eur. J., 2005, 11, 6386–6391 CrossRef CAS PubMed; (d) L. Porrès, O. Mongin and M. Blanchard-Desce, Tetrahedron Lett., 2006, 47, 1913–1917 CrossRef; (e) T.-C. Lin, Y.-Y. Liu, M.-H. Li, C.-Yu. Liu, S.-Y. Tseng, Y.-T. Wang, Y.-H. Tseng, H.-H. Chu and C.-W. Luo, Chem. – Asian J., 2014, 9, 1601–1610 CrossRef CAS PubMed; (f) M. Krzeszewski, B. Thorsted, J. Brewer and D. T. Gryko, J. Org. Chem., 2014, 79, 3119–3128 CrossRef CAS PubMed; (g) M. Murai, S.-Y. Ku, N. D. Treat, M. J. Robb, M. L. Chabinyc and C. J. Hawker, Chem. Sci., 2014, 5, 3753–3760 RSC; (h) R. Orłowski, M. Banasiewicz, G. Clermont, F. Castet, R. Nazir, M. Blanchard-Desce and D. T. Gryko, Phys. Chem. Chem. Phys., 2015, 17, 23724–23731 RSC.
- (a) D. Alonso Doval, M. Dal Molin, S. Ward, A. Fin, N. Sakai and S. Matile, Chem. Sci., 2014, 5, 2819–2825 RSC; (b) A. Fin, A. Vargas-Jentzsch, N. Sakai and S. Matile, Angew. Chem., Int. Ed., 2012, 51, 12736–12739 CrossRef CAS PubMed; (c) M. Dal Molin and S. Matile, Org. Biomol. Chem., 2013, 11, 1952–1957 RSC; (d) D. Alonso Doval and S. Matile, Org. Biomol. Chem., 2013, 11, 7467–7471 RSC.
- (a) M. Dal Molin, Q. Verolet, S. Soleimanpour and S. Matile, Chem. – Eur. J., 2015, 21, 6012–6021 CrossRef CAS PubMed; (b) M. Dal Molin, Q. Verolet, A. Colom, R. Letrun, E. Derivery, M. Gonzalez-Gaitan, E. Vauthey, A. Roux, N. Sakai and S. Matile, J. Am. Chem. Soc., 2015, 137, 568–571 CrossRef CAS PubMed; (c) Q. Verolet, S. Soleimanpour, K. Fujisawa, M. Dal Molin, N. Sakai and S. Matile, ChemistryOpen, 2015, 4, 264–267 CrossRef CAS PubMed.
- (a) M. Grzybowski and D. T. Gryko, Adv. Opt. Mater., 2015, 3, 280 CrossRef CAS; (b) A. C. Rochat, L. Cassar and A. Iqbal, Eur. Pat. Appl, 94911, 1983 Search PubMed; (c) A. Iqbal, M. Jost, R. Kirchmayr, J. Pfenninger, A. Rochat and O. Wallquist, Bull. Soc. Chim. Belg., 1988, 97, 615 CrossRef CAS; (d) D. G. Farnum, G. Mehta, G. G. I. Moore and F. P. Siegal, Tetrahedron Lett., 1974, 29, 2549 CrossRef; (e) Z. M. Hao and A. Iqbal, Chem. Soc. Rev., 1997, 26, 203 RSC; (f) J. S. Zambounis, Z. Hao and A. Iqbal, Nature, 1997, 388, 131 CrossRef CAS; (g) O. Wallquist and R. Lenz, Macromol. Symp., 2002, 187, 617 CrossRef CAS.
- (a) Y. Li, P. Sonar, L. Murphy and W. Hong, Energy Environ. Sci., 2013, 6, 1684 RSC; (b) L. Burgi, M. Turbiez, R. Pfeiffer, F. Bienewald, H. J. Kirner and C. Winnewisser, Adv. Mater., 2008, 20, 2217 CrossRef CAS; (c) M. Tantiwiwat, A. Tamayo, N. Luu, X. D. Dang and T. Q. Nguyen, J. Phys. Chem. C, 2008, 112, 17402 CrossRef CAS; (d) S. L. Suraru, U. Zschieschang, H. Klauk and F. Würthner, Chem. Commun., 2011, 47, 1767 RSC; (e) W. S. Yoon, S. K. Park, I. Cho, J.-A. Oh, J. H. Kim and S. Y. Park, Adv. Funct. Mater., 2013, 23, 3519 CrossRef CAS; (f) B. Tieke, A. R. Rabindranath, K. Zhang and Y. Zhu, Beilstein J. Org. Chem., 2010, 6, 830 CrossRef PubMed; (g) M. M. Wienk, M. Turbiez, J. Gilot and R. A. J. Janssen, Adv. Mater., 2008, 20, 2556 CrossRef CAS; (h) P. Sonar, S. P. Singh, E. van L. Williams, Y. Li, M. S. Soh and A. Dodabalapur, J. Mater. Chem., 2012, 22, 4425 RSC; (i) O. Kwon, J. Jo, B. Walker, G. C. Bazan and J. H. Seo, J. Mater. Chem. A, 2013, 1, 7118 RSC; (j) J. Kuwabara, N. Takase, T. Yasuda and T. Kanbara, J. Polym. Sci., Part A: Polym. Chem., 2016, 54, 2337–2345 CrossRef CAS.
- (a) H. Ftouni, F. Bolze and J.-F. Nicoud, Dyes Pigm., 2013, 97, 77 CrossRef CAS; (b) H. Ftouni, F. Bolze, H. de Rocquigny and J.-F. Nicoud, Bioconjugate Chem., 2013, 24, 942 CrossRef CAS PubMed.
- M. Grzybowski, E. Glodkowska-Mrowka, V. Hugues, W. Brutkowski, M. Blanchard-Desce and D. T. Gryko, Chem. – Eur. J., 2015, 21, 9101 CrossRef CAS PubMed.
- (a) J. Yu, N. Li, D.-F. Chen and S.-W. Luo, Tetrahedron Lett., 2014, 55, 2859 CrossRef CAS; (b) S. Luňák Jr., J. Vyňuchal, M. Vala, L. Havel and R. Hrdina, Dyes Pigm., 2009, 82, 102 CrossRef; (c) M. Vala, J. Krajčovič, S. Luňák Jr., I. Ouzzane, J.-P. Bouillon and M. Weiter, Dyes Pigm., 2014, 106, 136 CrossRef CAS; (d) M. Vala, M. Weiter, J. Vyňuchal, P. Toman and S. Luňák Jr., J. Fluoresc., 2008, 18, 1181 CrossRef CAS PubMed; (e) S. Luňák Jr., M. Vala, J. Vyňuchal, I. Ouzzane, P. Horáková, P. Možíšková, Z. Eliáš and M. Weiter, Dyes Pigm., 2011, 91, 269 CrossRef; (f) T. Yamagata, J. Kuwabara and T. Kanbara, Heterocycles, 2014, 89, 1173–1181 CrossRef CAS.
- (a) L. Zhang, L.-Y. Zou, J.-F. Guo and A.-M. Ren, New J. Chem., 2016, 40, 4899–4910 RSC; (b) T. He, Y. Gao, S. Sreejith, X. Tian, L. Liu, Y. Wang, H. Joshi, S. Z. F. Phua, S. Yao, X. Lin, Y. Zhao, A. C. Grimsdale and H. Sun, Adv. Opt. Mater., 2016, 4, 746–755 CrossRef CAS.
- (a) J. Dhar, N. Venkatramaiah, A. Anitha and S. Patil, J. Mater. Chem. C, 2014, 2, 3457 RSC; (b) H. Bürckstümmer, A. Weissenstein, D. Bialas and F. Würthner, J. Org. Chem., 2011, 76, 2426–2432 CrossRef PubMed.
- A. Purc, K. Sobczyk, Y. Sakagami, A. Ando, K. Kamada and D. T. Gryko, J. Mater. Chem. C, 2015, 3, 742–749 RSC.
- (a) S. Qu, C. Qin, A. Islam, Y. Wu, W. Zhu, J. Hua, H. Tian and L. Han, Chem. Commun., 2012, 48, 6972–6974 RSC; (b) S. Qu and H. Tian, Chem. Commun., 2012, 48, 3039–3051 RSC and references cited therein; (c) C. B. Nielsen, M. Turbiez and I. McCulloch, Adv. Mater., 2013, 13, 1859–1880 CrossRef PubMed and references cited therein; (d) S.-Y. Liu, M.-M. Shi, J.-C. Huang, Z.-N. Jin, X.-L. Hu, J.-Y. Pan, H.-Y. Li, A. K.-Y. Jen and H.-Z. Chen, J. Mater. Chem. A, 2013, 1, 2795–2805 RSC; (e) Y.-H. Jeong, C.-H. Lee and W.-D. Jang, Chem. – Asian J., 2012, 7, 1562 CrossRef PubMed; (f) C. Lu and W.-C. Chen, Chem. – Asian J., 2013, 8, 2813–2821 CrossRef CAS PubMed; (g) M. Kaur and D. H. Choi, Chem. Soc. Rev., 2015, 44, 58 RSC; (h) E. Q. Guo, P. H. Ren, Y. L. Zhang, H. C. Zhang and W. J. Yang, Chem. Commun., 2009, 39, 5859 RSC; (i) T. Yamagata, J. Kuwabara and T. Kanbara, Tetrahedron, 2014, 70, 1451–1457 CrossRef CAS; (j) J. Kuwabara, T. Yamagata and T. Kanbara, Tetrahedron, 2010, 66, 3736 CrossRef CAS; (k) T. Yamagata, J. Kuwabara and T. Kanbara, Tetrahedron Lett., 2010, 51, 159 CrossRef.
- (a) G. M. Fischer, M. Isomaki-Krondahl, I. Gottker-Schnetmann, E. Daltrozzo and A. Zumbusch, Chem. – Eur. J., 2009, 15, 4857 CrossRef CAS PubMed; (b) G. M. Fischer, M. K. Klein, E. Daltrozzo and A. Zumbusch, Eur. J. Org. Chem., 2011, 3421 CrossRef CAS.
- (a) S. Shimizu, T. Iino, Y. Araki and N. Kobayashi, Chem. Commun., 2013, 49, 1621 RSC; (b) S. Shimizu, T. Iino, A. Saeki, S. Seki and N. Kobayashi, Chem. – Eur. J., 2015, 21, 2893–2904 CrossRef CAS PubMed.
- W. Yue, S.-L. Suraru, D. Bialas, M. Müller and F. Würthner, Angew. Chem., Int. Ed., 2014, 53, 6159–6162 CrossRef CAS PubMed.
- (a) M. Grzybowski, E. Glodkowska-Mrowka, T. Stoklosa and D. T. Gryko, Org. Lett., 2012, 14, 2670 CrossRef CAS PubMed; (b) D. T. Gryko, M. Grzybowski, P. Hayoz and A. Jeżewski, BASF SE, Patent Appl, PCT/EP2014/054060, 2013 Search PubMed.
- J. Zhang, N. Zivic, F. Dumur, C. Guo, Y. Li, P. Xiao, B. Graff, D. Gigmes, J. P. Fouassier and J. Lalevée, Mater. Today Commun., 2015, 4, 101–108 CrossRef CAS.
- (a) M. Grzybowski, V. Hugues, M. Blanchard-Desce and D. T. Gryko, Chem. – Eur. J., 2014, 20, 12493–12501 CrossRef CAS PubMed; (b) R. Nazir, P. Danilevicius, A. I. Ciuciu, M. Chatzinikolaidou, D. Gray, L. Flamigni, M. Farsari and D. T. Gryko, Chem. Mater., 2014, 26, 3175–3184 CrossRef CAS.
- C. J. H. Morton, R. Gilmour, D. M. Smith, P. Lightfoot, A. M. Z. Slawin and E. J. MacLean, Tetrahedron, 2002, 58, 5547 CrossRef CAS.
- (a) E. Q. Guo, P. H. Ren, Y. L. Zhang, H. C. Zhang and W. J. Yang, Chem. Commun., 2009, 5859 RSC; (b) C. Yang, M. Zheng, Y. Li, B. Zhang, J. Li, L. Bu, W. Liu, M. Sun, H. Zhang, Y. Tao, S. Xue and W. Yang, J. Mater. Chem. A, 2013, 1, 5172 RSC.
- X. Liu, J. M. Cole, P. G. Waddell, T.-C. Lin, J. Radia and A. Zeidler, J. Phys. Chem. A, 2012, 116, 727–737 CrossRef CAS PubMed.
- B. Valeur and M. N. Berberan-Santos, Molecular Fluorescence, Wiley, Weinheim, 2012 Search PubMed.
- D. T. Gryko, J. Piechowska, M. Tasior, J. Waluk and G. Orzanowska, Org. Lett., 2006, 8, 4747–4750 CrossRef CAS PubMed.
- R. Chinchilla and C. Nájera, Chem. Rev., 2007, 107, 874 CrossRef CAS PubMed.
- T. Potrawa and H. Langhals, Chem. Ber., 1987, 120, 1075–1078 CrossRef CAS.
- E. Ripaud, D. Demeter, T. Rousseau, E. Boucard-Cétol, M. Allain, R. Po, P. Leriche and J. Roncali, Dyes Pigm., 2012, 95, 126 CrossRef CAS.
- (a) B. Strehmel, A. M. Sarker and H. Detert, ChemPhysChem, 2003, 4, 249 CrossRef CAS PubMed; (b) S. Amthor, C. Lambert, S. Dümmler, I. Fischer and J. Schelter, J. Phys. Chem. A, 2006, 110, 5204 CrossRef CAS PubMed; (c) G. Verbeek, S. Depaemelaere, M. Van der Auweraer, F. C. De Schryver, A. Vaes, D. Terrell and S. De Meutter, Chem. Phys., 1993, 176, 195 CrossRef CAS; (d) H. Detert, E. Sugiono and G. Kruse, J. Phys. Org. Chem., 2002, 15, 638 CrossRef CAS; (e) C. Le Droumaguet, O. Mongin, M. H. V. Werts and M. Blanchard-Desce, Chem. Commun., 2005, 2802 RSC.
- (a) F. Terenziani, A. Painelli, C. Katan, M. Charlot and M. Blanchard-Desce, J. Am. Chem. Soc., 2006, 128, 15742–15755 CrossRef CAS PubMed; (b) D. H. Friese, A. Mikhaylov, M. Krzeszewski, Y. M. Poronik, A. Rebane, K. Ruud and D. T. Gryko, Chem. – Eur. J., 2015, 21, 18364 CrossRef CAS PubMed; (c) A. Rebane, M. Drobizhev, N. S. Makarov, G. Wicks, G. P. Wnuk, Y. Stepanenko, J. E. Haley, D. M. Krein, J. L. Fore, A. R. Burke, J. E. Slagle, D. G. McLean and T. M. Cooper, J. Phys. Chem. A, 2014, 118, 3749–3759 CrossRef CAS PubMed; (d) B. Dereka, A. Rosspeintner, Z. Li, R. Liska and E. Vauthey, J. Am. Chem. Soc., 2016, 138, 4643 CrossRef CAS PubMed; (e) H. Y. Woo, B. Liu, B. Kohler, D. Korystov, A. Mikhailovsky and G. C. Bazan, J. Am. Chem. Soc., 2005, 127, 14721 CrossRef CAS PubMed.
- W. Kim, J. Sung, M. Grzybowski, D. T. Gryko and D. Kim, J. Phys. Chem. Lett., 2016, 7, 3060–3066 CrossRef PubMed.
- J. R. Brewer, M. Schiek, I. Wallmann and H.-G. Rubahn, Opt. Commun., 2008, 281, 3892–3896 CrossRef CAS.
- N. S. Makarov, M. Drobizhev and A. Rebane, Opt. Express, 2008, 16, 4029–4047 CrossRef CAS PubMed.
- TURBOMOLE V6.6 2014, a development of University of Karlsruhe and Forschungszentrum Karlsruhe GmbH, 1989–2007, TURBOMOLE GmbH, since 2007; available from http: //www.turbomole.com.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- A. Schäfer, C. Huber and R. Ahlrichs, J. Chem. Phys., 1989, 100, 5829–5835 CrossRef.
- K. Aidas, C. Angeli, K. L. Bak, V. Bakken, R. Bast, L. Boman, O. Christiansen, R. Cimiraglia, S. Coriani, P. Dahle, E. K. Dalskov, U. Ekström, T. Enevoldsen, J. J. Eriksen, P. Ettenhuber, B. Fernández, L. Ferrighi, H. Fliegl, L. Frediani, K. Hald, A. Halkier, C. Hättig, H. Heiberg, T. Helgaker, A. C. Hennum, H. Hettema, E. Hjertenæs, S. Høst, I.-M. Høyvik, M. F. Iozzi, B. Jansík, H. J. Aa. Jensen, D. Jonsson, P. Jørgensen, J. Kauczor, S. Kirpekar, T. Kjærgaard, W. Klopper, S. Knecht, R. Kobayashi, H. Koch, J. Kongsted, A. Krapp, K. Kristensen, A. Ligabue, O. B. Lutnæs, J. I. Melo, K. V. Mikkelsen, R. H. Myhre, C. Neiss, C. B. Nielsen, P. Norman, J. Olsen, J. M. H. Olsen, A. Osted, M. J. Packer, F. Pawlowski, T. B. Pedersen, P. F. Provasi, S. Reine, Z. Rinkevicius, T. A. Ruden, K. Ruud, V. V. Rybkin, P. Salek, C. C. M. Samson, A. Sánchez de Merás, T. Saue, S. P. A. Sauer, B. Schimmelpfennig, K. Sneskov, A. H. Steindal, K. O. Sylvester-Hvid, P. R. Taylor, A. M. Teale, E. I. Tellgren, D. P. Tew, A. J. Thorvaldsen, L. Thøgersen, O. Vahtras, M. A. Watson, D. J. D. Wilson, M. Ziolkowski and H. Ågren, The Dalton quantum chemistry program system, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2013, 4, 269, DOI:10.1002/wcms.1172.
- T. Yanai, D. P. Tew and N. C. Handy, Chem. Phys. Lett., 2004, 393, 51–57 CrossRef CAS.
- T. H. Dunning, J. Chem. Phys., 1989, 90, 1007–1023 CrossRef CAS.
- K. A. Peterson, D. Figgen, E. Goll, H. Stoll and M. Dolg, J. Chem. Phys., 2003, 1, 21 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Full synthetic and analytical data of the prepared compounds as well as copies of 1H NMR and 13C NMR spectra. CCDC 1515317. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6qo00869k |
| This journal is © the Partner Organisations 2017 |