
Thiophene-fused 1,10-phenanthroline toward a far-red emitting conjugated polymer and its polymer dots: synthesis, properties and subcellular imaging†
Chunmei
Yan‡
ab,
Zezhou
Sun‡
cd,
Hongshuang
Guo
ab,
Changfeng
Wu
cd and
Yulan
Chen
 *ab
*ab
aTianjin Key Laboratory of Molecular Optoelectronic Science, Department of Chemistry, Tianjin University, Tianjin, 300354, P. R. China. E-mail: yulan.chen@tju.edu.cn
bCollaborative Innovation Center of Chemical Science and Engineering, Tianjin, P. R. China
cState Key Laboratory on Integrated Optoelectronics, College of Electronic Science and Engineering, Jilin University, Changchun, Jilin 130012, P. R. China
dDepartment of Biomedical Engineering, Southern University of Science and Technology, Shenzhen, Guangdong 518055, P. R. China
First published on 10th October 2017
Abstract
We synthesized and characterized a new kind of far-red emitter based on a conjugated block copolymer obtained from fluorene, 4,7-di(thiophen-2-yl)benzo[c][1,2,5]thiadiazole and thiophene-fused 1,10-phenanthroline. Compact polymer dots (Pdots) that show intense fluorescence signals in the far-red and near-infrared regions were formed by nanoprecipitation. Their small particle size, high brightness, non-toxic feature, and ability to specifically target subcellular structures make them promising probes for biological imaging.
 Yulan Chen | Yulan Chen graduated from Jilin University in 2005, and obtained her PhD degree from the Institute of Chemistry, Chinese Academy of Sciences (ICCAS), in 2010. During 2010–2014, she worked as a postdoctoral researcher in the Eindhoven University of Technology and the Max Planck Institute for Polymer Research. In September 2014, she was appointed as a professor at Tianjin University. Her current research interests include functional conjugated polymers and mechano-responsive polymers. |
Introduction
Progress in understanding many biological processes has relied on applications of fluorescence microscopy, which is known as an effective and noninvasive imaging technique.1,2 With advantages such as high signal-to-noise ratios and excellent spatial and temporal resolution,3 fluorescence imaging is critically dependent on the development of fluorescent probes. Among various emissive materials, such as fluorescent proteins,4a organic fluorophores,4b and inorganic semiconductors,4c semiconducting polymer dots (Pdots)4d,e represent a promising class of fluorescent probes because of their size-tunable optical properties, robust photostability, high fluorescence quantum yields, large Stokes shifts and nontoxic features.5,6Pdots are made primarily from hydrophobic polymers and their formation results from the collapse of polymer chains due to a sudden decrease in solvent hydrophobicity. Consequently, the polymer chain morphology and packing behavior significantly affect the optical properties of the Pdots. Disturbance derived from the autofluorescence of biological samples and the bypass issues7 and the tissue damage from short-wavelength light jointly influence the spatial and temporal resolution of bioimaging.8 To this end, Pdots that have a compact size (<30 nm) and are emissive within the first optical window of biological tissues (between 650 nm and 950 nm) are a profound need in modern bioimaging and clinical diagnosis.8 Despite the field's progress, it is still highly desirable to explore new polymer species that form compact and bright far-red (FR)/near-infrared (NIR) emitting Pdots for specific subcellular imaging.9
Semiconducting polymers have emerged as attractive materials for various optoelectronic applications, including LED, OPV, and FET devices.10,11 Their appeal is based on the readily tailored electrical and optical properties of semiconductors, combined with their versatile polymer chemistry. The adoption of fluorescent conjugated polymers as fluorescent probes thus has also received considerable attention in biology and medicine.12 Various conjugated polymers can be used to prepare small Pdots as fluorescent labels.13 For instance, polyfluorene (PF) based polymers in particular exhibit great flexibility for the design of bright blue emitting materials.14 Recently, we reported a facile synthetic strategy for thiophene-fused 1,10-phenanthroline (TPhen, Scheme 1).15 This building block was used to prepare phenanthroline-alt-fluorene polymers (poly(TPhen-alt-fluorene)). We found that incorporating the π-extended phenanthroline into the polyfluorene main chain resulted in bright blue-green emission with a distinct red shift of both the absorption and PL emission bands, in comparison with polyfluorene and poly(1,10-phenanthroline-alt-fluorene). Since color tuning to the redder region in conjugated polymers can be conveniently achieved by combining a narrow-band-gap comonomer with the polyfluorene backbone,16 herein, we intended to incorporate 4,7-di(thiophen-2-yl)benzo[c][1,2,5]thiadiazole (DBT), a prototypical narrow-band-gap repeating unit, into the aforementioned poly(TPhen-alt-fluorene) main chain, so that the utility of the new monomer TPhen could be extended to fluorescence bio-imaging, particularly for the far-red fluorescence imaging technique. Such a polymer was further used to prepare compact nanoparticles by nanoprecipitation. Successful functionalization and bioconjugation produces P1 Pdot–streptavidin probes that specifically label subcellular microtubule structures. These results indicate that the highly fluorescent P1 Pdots with far-red emission are promising probes for cellular imaging and bioanalytical assays.
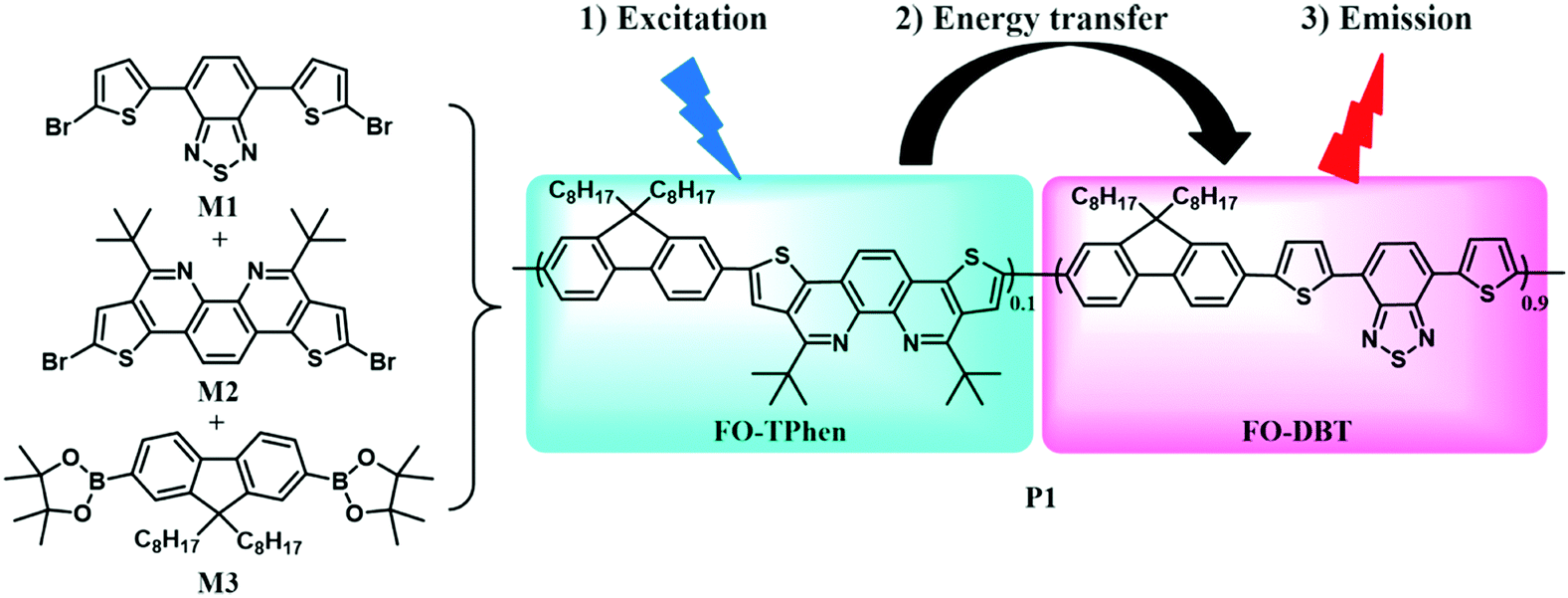 | ||
| Scheme 1 Synthetic scheme of copolymer P1 and the energy transfer process in the polymer. | ||
Results and discussion
To obtain stable Pdots that can emit bright light at far-red wavelengths, and even extending to the NIR region (650–900 nm), the conjugated copolymer was designed as an energy transfer system composed of multiple monomeric units. The chemical structures of three monomers are shown in Scheme 1, among which 4,7-di(thiophen-2-yl)benzo[c][1,2,5]thiadiazole (DBT) served as an energy acceptor owing to its high fluorescence quantum yield with light emission at long wavelengths.16a The wide band-gap monomers, fluorene and TPhen, could form light-harvesting segments. n-Octyl substituents in the 9-position of fluorene (FO) were employed to improve the solubility of the resulting copolymer, and to increase the overall hydrophobicity to stabilize the Pdots. TPhen performed as the π-bridge spacer between FO and DBT. There are three important functions taken into consideration for the selection of the π-bridge: (i) to obtain the absorption in the visible region (400–600 nm); (ii) to increase the steric effect of the semiconducting polymers and thus moderate the aggregation-caused quenching in a nanoparticle form; and (iii) to improve energy transfer from the donor segment to the acceptor moiety.8 According to our previous investigation, the thiophene-fused 1,10-phenanthroline attached with tert-butyl groups (TPhen) thus fulfills these requirements for a π-bridge spacer.15 Moreover, concerning its optophysical properties, the bright blue-emissive monomer TPhen was also expected to be a donor extender. The dibromosubstituted monomers M1 and M2 and 9,9-dioctyl-2,7-diboronic ester-fluorene M3 were synthesized according to previously reported protocols.15,17Subsequently, the polymerization was conducted via palladium-catalyzed Suzuki polycondensation. A special feature of the Suzuki coupling is that it allows an alternating type of copolymer, and each individual narrow band-gap unit is separated on both sides by wide band-gap segments, when the narrow band-gap component is less than or equal to 50% in the copolymer.16a In this case, we could expect that the narrow band-gap unit BDT functions as an exciton trap which facilitates intramolecular energy transfer from the fluorene and TPhen segments to the DBT unit. The optimized comonomer feed ratios were estimated to be 9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:10 (M1:M2:M3) to afford dark red polymer P1 in good yield (91%). P1 could be readily dissolved in common organic solvents, such as dichloromethane, chloroform, and THF. Its weight-average molecular weight (Mw), determined by GPC against polystyrene standards, was found up to 35 kg mol−1 with a polydispersity (PDI) of ∼2.1. All the starting monomers and the polymer were characterized using NMR spectroscopy.
:1:10 (M1:M2:M3) to afford dark red polymer P1 in good yield (91%). P1 could be readily dissolved in common organic solvents, such as dichloromethane, chloroform, and THF. Its weight-average molecular weight (Mw), determined by GPC against polystyrene standards, was found up to 35 kg mol−1 with a polydispersity (PDI) of ∼2.1. All the starting monomers and the polymer were characterized using NMR spectroscopy.
The thermal properties of the polymer were examined using thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC). P1 exhibited good thermal stability (Fig. S1, ESI†). It showed less than 10% weight loss up to 427 °C and a residual weight percentage of about 60% at 800 °C under a nitrogen atmosphere at a heating rate of 10 °C min−1. As shown in Fig. S1b (ESI†), no distinct glass transition of P1 was observed from 25 to 320 °C in its DSC curve of the second heating and cooling runs (10 °C min−1), suggesting that it was amorphous.16c
The photophysical properties of copolymer P1 were studied using UV-vis absorption and photoluminescence (PL) emission spectroscopy in dilute solution (1 × 10−5 mol L−1, Fig. 1). In chloroform, P1 displayed intense and broad absorption in the region ranging from 350 to 650 nm. The absorption peak at 397 nm (ε = 7.82 × 104 M−1 cm−1) originates from the π–π* transition of the FO and TPhen units, whereas the peak at 552 nm (ε = 7.91 × 104 M−1 cm−1) can be attributed to the contribution of the DBT chromophore. The two distinguished absorption features demonstrate that the electronic states of the two blocks (FO-DBT and FO-TPhen) in the copolymer are not mixed.18 The dominant visible absorption (420–600 nm) also ensured that the copolymer could be directly excited with a biologically friendly conventional laser (such as a 488 or 532 nm laser). Compared to the reported polymer that has only a polyfluorene block as the energy donor (e.g. PFO-DBT5, exhibiting a π–π* transition absorption peak at ∼380 nm16a), for P1, the π-extended phenanthroline unit performed as a donor extender and π-bridge spacer between fluorene and DBT segments, leading to a bathochromic shift of the UV-vis absorption band, while it could maintain the high-energy emissive feature of the FO-TPhen block. Photoexcitation of P1 at 397 nm results in energy transfer and emission from the FO-TPhen block to the FO-DBT part. As a result, the PL emission corresponding to the FO and TPhen segments completely disappeared, while the far-red fluorescence emission band consists exclusively of one peak at 653 nm. And the PL band edge spanned even to the NIR region (ranging from 600 to 800 nm). A relatively large Stokes shift (101 nm) and high fluorescence quantum yield (72.6%, relative to rhodamine B) for P1 are essential to increase the resolution for fluorescence imaging.
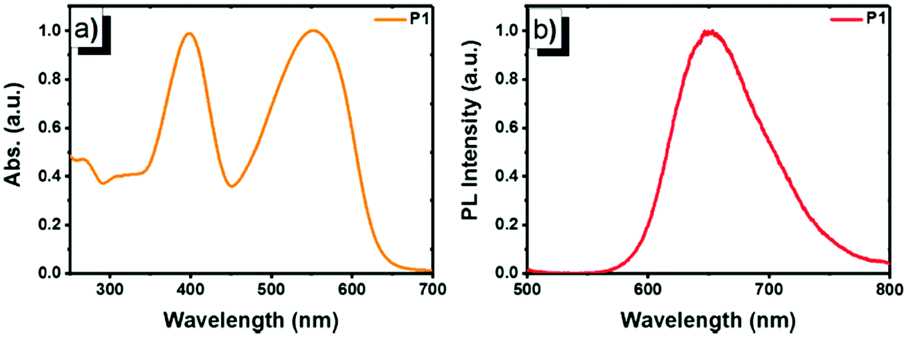 | ||
| Fig. 1 Normalized (a) UV-vis absorption and (b) PL emission spectra of P1 in chloroform (1 × 10−5 mol L−1, excited by 397 nm UV light). | ||
Next, the far-red emitting polymer P1 was used to prepare compact Pdots via the reprecipitation method as reported in our previous papers.19,20 During precipitation in water, the polymer chains folded and collapsed because of hydrophobic interactions. For further bioconjugation of the Pdots, surface functionalization was achieved by blending a small portion (20 wt%) of poly(styrene-maleic anhydride) (PSMA) followed by spontaneous hydrolyzation of maleic anhydride groups, leading to aqueous solutions of stable nanometer-sized Pdots. In contrast, the control polymer P3 (Scheme S1, ESI†) with cationic side chains attached at the fluorene moieties did not form compact nano-particles (Fig. S2 and S3, ESI†), confirming the significant role of the hydrophobicity of the polymer during the aggregation. The Pdots exhibited a spherical morphology with an average diameter of 13 nm, as determined by dynamic light scattering (DLS) and atomic force microscopy (AFM) (Fig. 2). As shown by spectroscopic measurements (Fig. 3a), which are similar to those of P1 in solution, the UV-vis absorption spectrum of the Pdots displayed a broad and dominant visible absorption band from 350 to 650 nm because of the presence of multiple monomers. Compared to the monomeric DBT, the absorption of the polymer solution and Pdots both exhibited a bathochromic shift of about 70–110 nm (Fig. S4, ESI†). This red-shift in the polymer absorption is attributed to an increase in the conjugation length compared to that of the monomer.21 Notably, the Pdots were emissive in the FR and NIR regions (580–850 nm) with a bright emission band centered at ∼677 nm. Therefore, the surface functionalization did not remarkably influence the energy transfer process. Due to the aggregation effect, the emission peak shifted bathochromically in contrast to that in solution;22 accordingly, the Stokes shift of Pdots further increased. A relatively large Stokes shift of ∼162 nm was detected for P1 in its Pdot state, which was favorable for eliminating the cross-talk between the excitation light and the Pdot emission. The absolute quantum yield of the Pdots was determined to be 6.2%, which was comparable to or higher than the values reported for ordinary FR/NIR fluorophores, such as indocyanine green (ICG, ∼2%)23 and NIR7.0 (∼2.5%).24 All the photophysical properties of P1 and its Pdots are summarized in Table S1 (ESI†).
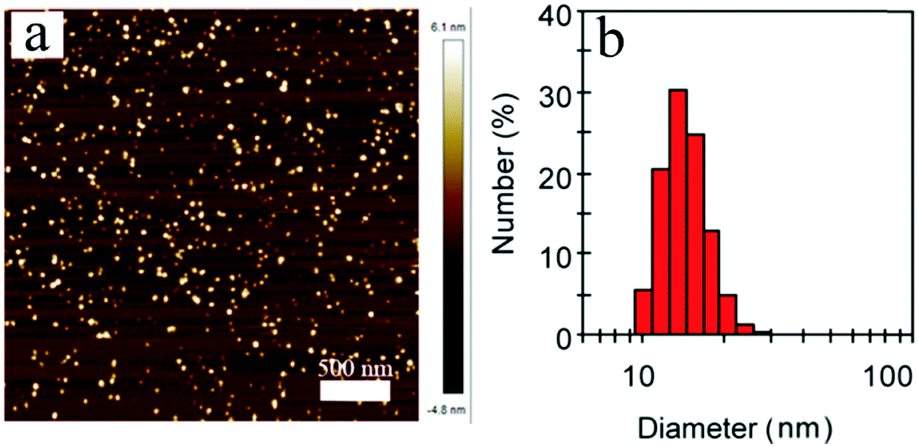 | ||
| Fig. 2 (a) Typical AFM image of P1 Pdots. (b) Hydrodynamic diameters of P1 Pdots measured by dynamic light scattering. | ||
 | ||
| Fig. 3 Normalized (a) UV-vis absorption and (b) PL emission spectra of P1 Pdots in water (excited by 515 nm UV light). | ||
These FR/NIR emissive Pdots were then used for subcellular labeling to demonstrate their applicability for biological imaging. With abundant carboxyl groups on the Pdot surface, bioconjugation to streptavidin was completed via an EDC-catalyzed coupling method to form Pdot–streptavidin bioconjugates. Subsequently, HeLa cells were incubated with the biotinylated, monoclonal anti-alpha tubulin antibody at room temperature. The cells were washed from the primary antibody with PBS followed by incubation with Pdot-bioconjugates, allowing the Pdots to target the microtubules of HeLa cells. Fig. 4a and Fig. S5 (ESI†) show the representative fluorescence images of Pdot-labeled microtubules in HeLa cells, indicating that the Pdots could be specifically targeted onto subcellular structures in the presence of the biotinylated antibody, as revealed by bright red fluorescence. Individual tubular structures were clearly seen and well-resolved in the images. Moreover, the in vitro cytotoxicity of the Pdots was evaluated using an MTT assay, as shown in Fig. 4b. The results suggested that the Pdots (10–100 ppm) had a minimal cytotoxic effect on the cells in an incubation period of 24 h testing.
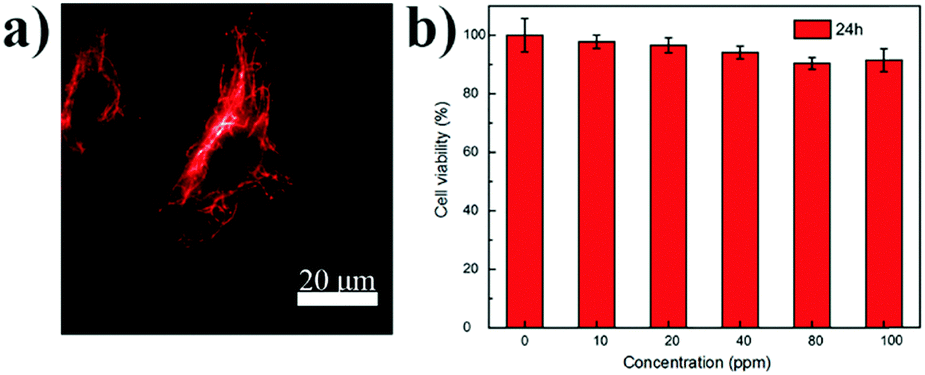 | ||
| Fig. 4 (a) Wide-field microscopy image of microtubules in HeLa cells labeled with streptavidin conjugated P1 Pdots in the presence of the biotinylated anti-alpha tubulin antibody (excited by a 405 nm laser). (b) Cell viability of HeLa cells after being incubated with the P1 Pdot solution of various concentrations for 24 h. | ||
Conclusions
In conclusion, we have synthesized a new semiconducting block copolymer based on the donor–acceptor system. For the first time, the thiophene-fused 1,10-phenanthroline has been incorporated into the bio-orientated polymer backbone that could emit far-red fluorescence with high brightness. Used as a bridge spacer and donor extender, such a repeating unit was demonstrated as a desirable visible-light-harvesting part together with the fluorene moiety. Owing to the hydrophobic feature of P1, compact FR/NIR emissive Pdots were formed, which exhibited fantastic characteristics for fluorescence probes, such as a high quantum yield (Φ = 6.2%), a large Stokes shift (162 nm) and good biocompatibility. Bioconjugation and specific subcellular labeling using streptavidin−Pdot conjugates led to successful imaging of the microtubules of HeLa cells. Our study thus brings forward a new type of highly fluorescent red emissive polymer and its nanoparticle bioconjugates that hold great promise for a wide range of fluorescence-based biological detection. Further exploration of in vivo bioimaging applications using the P1 based Pdots is now under progress.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors sincerely thank Dr Zhuping Fei for GPC measurements, and Feiyi Chen for AFM analysis. Financial support from the Ministry of Science and Technology of China (Grants 2017YFA0204503 and 2017YFA0207800), the National Natural Science Foundation of China (Grants 51503142 and 21522405), the Thousand Youth Talents Plan, and the Natural Science Foundation of Tianjin (Grant 15JCYBJC52900) is gratefully acknowledged.Notes and references
- (a) J. V. Frangioni, J. Clin. Oncol., 2008, 26, 4012 CrossRef PubMed; (b) C.-P. Chen, Y.-C. Huang, S.-Y. Liou, P.-J. Wu, S.-Y. Kuo and Y.-H. Chan, ACS Appl. Mater. Interfaces, 2014, 6, 21585 CrossRef CAS PubMed.
- (a) G. Lukinavičius, K. Umezawa, N. Olivier, A. Honigmann, G. Yang, T. Plass, V. Mueller, L. Reymond, I. R. C. Jr, Z. G. Luo, C. Schultz, E. A. Lemke, P. Heppenstall, C. Eggeling, S. Manley and K. Johnsson, Nat. Chem., 2013, 5, 132 CrossRef PubMed; (b) X. Zhang, Y. Tian, Z. Li, X. Tian, H. Sun, H. Liu, A. Moore and C. Ran, J. Am. Chem. Soc., 2013, 135, 16397 CrossRef CAS PubMed.
- (a) S. Falkner, S. Grade, L. Dimou, K. K. Conzelmann, T. Bonhoeffer, M. Götz and M. Hübener, Nature, 2016, 539, 248 CrossRef PubMed; (b) Y. Shiba, T. Gomibuchi, T. Seto, Y. Wada, H. Ichimura, Y. Tanaka, T. Ogasawara, K. Okada, N. Shiba, K. Sakamoto, D. Ido, T. Shiina, M. Ohkura, J. Nakai, N. Uno, Y. Kazuki, M. Oshimura, I. Minami and U. Ikeda, Nature, 2016, 538, 388 CrossRef CAS PubMed.
- (a) S. Xu, T. Gao, X. Feng, X. Fan, G. Liu, Y. Mao, X. Yu, J. Lin and X. Luo, Biosens. Bioelectron., 2017, 97, 203 CrossRef CAS PubMed; (b) K. T. Ly, R.-W. Chen-Cheng, H.-W. Lin, Y.-J. Shiau, S.-H. Liu, P.-T. Chou, C.-S. Tsao, Y.-C. Huang and Y. Chi, Nat. Photonics, 2017, 11, 63 Search PubMed; (c) S. W. Bae, W. Tan and J.-I. Hong, Chem. Commun., 2012, 48, 2270 RSC; (d) J. E. Lee, N. Lee, H. Kim, J. Kim, S. H. Choi, J. H. Kim, T. Kim, I. C. Song, S. P. Park, W. K. Moon and T. Hyeon, J. Am. Chem. Soc., 2010, 132, 552 CrossRef CAS PubMed; (e) Y. Yang, F. An, X. Zhang, M. Zhou, W. Li, X. Hao, C.-S. Lee and X. Zhang, Biomaterials, 2012, 33, 7803 CrossRef CAS PubMed.
- (a) M. B. Jr, M. Moronne, P. Gin, S. Weiss and A. P. Alivisatos, Science, 1998, 281, 2013 CrossRef; (b) W. C. W. Chan and S. Nie, Science, 1998, 281, 2016 CrossRef CAS PubMed; (c) X. Wu, H. Liu, J. Liu, K. N. Haley, J. A. Treadway, J. P. Larson, N. Ge, F. Peale and M. P. Bruchez, Nat. Biotechnol., 2003, 21, 41 CrossRef CAS PubMed; (d) X. Michalet, F. F. Pinaud, L. A. Bentolila, J. M. Tsay, S. Doose, J. J. Li, G. Sundaresan, A. M. Wu, S. S. Gambhir and S. Weiss, Science, 2005, 307, 538 CrossRef CAS PubMed.
- (a) C. Wu, S. J. Hansen, Q. Hou, J. Yu, M. Zeigler, Y. Jin, D. R. Burnham, J. D. McNeill, J. M. Olson and D. T. Chiu, Angew. Chem., Int. Ed., 2011, 50, 3430 CrossRef CAS PubMed; (b) D. Chen, I.-C. Wu, Z. Liu, Y. Tang, H. Chen, J. Yu, C. Wu and D. T. Chiu, Chem. Sci., 2017, 8, 3390 RSC; (c) C. Wu, Y. Jin, T. Schneider, D. R. Burnham, P. B. Smith and D. T. Chiu, Angew. Chem., Int. Ed., 2010, 49, 9436 CrossRef CAS PubMed; (d) C. Wu, B. Bull, C. Szymanski, K. Christensen and J. McNeill, ACS Nano, 2008, 2, 2415 CrossRef CAS PubMed; (e) C.-P. Chen, P.-J. Wu, S.-Y. Lioua and Y.-H. Chan, RSC Adv., 2013, 3, 17507 RSC.
- (a) E. M. Muraca, J. P. Houston and M. Gurfinkel, Curr. Opin. Chem. Biol., 2002, 6, 642 CrossRef; (b) V. Ntziachristos, C. Bremer and R. Weissleder, Eur. J. Radiol., 2003, 13, 195 Search PubMed.
- C. S. Ke, C. C. Fang, J. Y. Yan, P. J. Tseng, J. R. Pyle, C. P. Chen, S. Y. Lin, J. Chen, X. Zhang and Y. H. Chan, ACS Nano, 2017, 11, 3166 CrossRef CAS PubMed.
- H. S. Choi, S. L. Gibbs, J. H. Lee, S. H. Kim, Y. Ashitate, F. Liu, H. Hyun, G. Park, Y. Xie and S. Bae, Nat. Biotechnol., 2013, 31, 148 CrossRef CAS PubMed.
- (a) C. Wu, T. Schneider, M. Zeigler, J. Yu, P. G. Schiro, D. R. Burnham, J. D. McNeill and D. T. Chiu, J. Am. Chem. Soc., 2010, 132, 15410 CrossRef CAS PubMed; (b) S. Günes, H. Neugebauer and N. S. Sariciftci, Chem. Rev., 2007, 107, 1324 CrossRef PubMed.
- (a) X. Zhan and D. Zhu, Polym. Chem., 2010, 1, 409 RSC; (b) A. Facchetti, Chem. Mater., 2011, 23, 733 CrossRef CAS; (c) C. B. Nielsen, M. Turbiez and I. McCulloch, Adv. Mater., 2013, 25, 1859 CrossRef CAS PubMed; (d) G. Moad, M. Chen, M. Häussler, A. Postma, E. Rizzardo and S. H. Thang, Polym. Chem., 2011, 2, 492 RSC.
- (a) I.-C. Wu, J. Yu, F. Ye, Y. Rong, M. E. Gallina, B. S. Fujimoto, Y. Zhang, Y.-H. Chan, W. Sun and X.-H. Zhou, J. Am. Chem. Soc., 2014, 137, 173 CrossRef PubMed; (b) K. Li and B. Liu, Chem. Soc. Rev., 2014, 43, 6570 RSC; (c) E. Ahmed, S. W. Morton, P. T. Hammond and T. M. Swager, Adv. Mater., 2013, 25, 4504 CrossRef CAS PubMed.
- (a) R. Hu, J. W. Y. Lam, J. Liu, H. H. Y. Sung, I. D. Williams, Z. Yue, K. S. Wong, M. M. F. Yuen and B. Z. Tang, Polym. Chem., 2012, 3, 1481 RSC; (b) C. Wu and D. T. Chiu, Angew. Chem., Int. Ed., 2013, 52, 3086 CrossRef CAS PubMed.
- Q. Pei and Y. Yang, J. Am. Chem. Soc., 1996, 118, 7416 CrossRef CAS.
- T. Wang, H. Wang, G. Li, M. Li, Z. Bo and Y. Chen, Macromolecules, 2016, 49, 4088 CrossRef CAS.
- (a) Q. Hou, Y. Xu, W. Yang, M. Yuan, J. Peng and Y. Cao, J. Mater. Chem., 2002, 12, 2887 RSC; (b) M. T. Bernius, M. Inbasekaran, J. O'Brien and W. Wu, Adv. Mater., 2000, 12, 1737 CrossRef CAS; (c) S. Tokito, H. Tanaka, K. Noda, A. Okada and Y. Taga, Appl. Phys. Lett., 1997, 70, 1929 CrossRef CAS.
- (a) H. Chen, M. He, J. Pei and B. Liu, Anal. Chem., 2002, 74, 6252 CrossRef CAS PubMed; (b) W.-H. Lee, A. D. Mohanty and C. Bae, ACS Macro Lett., 2015, 4, 453 CrossRef CAS.
- R. Yang, R. Tian, J. Yan, Y. Zhang, J. Yang, Q. Hou, W. Yang, C. Zhang and Y. Cao, Macromolecules, 2005, 38, 244 CrossRef CAS.
- C. Szymanski, C. Wu, J. Hooper, M. A. Salazar, A. Perdomo, A. Dukes and J. McNeill, J. Phys. Chem. B, 2005, 109, 8543 CrossRef CAS PubMed.
- F. Ye, C. Wu, Y. Jin, M. Wang, Y.-H. Chan, J. Yu, W. Sun, S. Hayden and D. T. Chiu, Chem. Commun., 2012, 48, 1778 RSC.
- J. Zhang, Y. Huang, D. Wang, A. C. Pollard, Z. Chen and E. Egap, J. Mater. Chem. C, 2017, 5, 5685 RSC.
- C. Wu, C. Szymanski and J. McNeill, Langmuir, 2006, 22, 2956 CrossRef CAS PubMed.
- U. Resch-Genger, M. Grabolle, S. Cavaliere-Jaricot, R. Nitschke and T. Nann, Nat. Methods, 2008, 5, 763 CrossRef CAS PubMed.
- C. Bouteiller, G. Clave, A. Bernardin, B. Chipon, M. Massonneau, P. Y. Renard and A. Romieu, Bioconjugate Chem., 2007, 18, 1303 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthetic procedures, experimental details, 1H NMR and 13C NMR spectra, DSC and TGA data, and more optophysical data. See DOI: 10.1039/c7qm00379j |
| ‡ The authors contributed equally to this work. |
| This journal is © the Partner Organisations 2017 |