
Pyrolysis of metal–organic frameworks to hierarchical porous Cu/Zn-nanoparticle@carbon materials for efficient CO2 hydrogenation†
Jingzheng
Zhang‡
a,
Bing
An‡
a,
Yahui
Hong
a,
Yaping
Meng
a,
Xuefu
Hu
a,
Cheng
Wang
 *a,
Jingdong
Lin
*a,
Wenbin
Lin
ab and
Yong
Wang
ac
*a,
Jingdong
Lin
*a,
Wenbin
Lin
ab and
Yong
Wang
ac
aCollaborative Innovation Center of Chemistry for Energy Materials, State Key Laboratory of Physical Chemistry of Solid Surfaces, Department of Chemistry, College of Chemistry and Chemical Engineering, Xiamen University, Xiamen 361005, People's Republic of China. E-mail: wangchengxmu@xmu.edu.cn; jdlin@xmu.edu.cn
bDepartment of Chemistry, University of Chicago, 929 East 57th Street, Chicago, Illinois 60637, USA
cVoiland School of Chemical Engineering and Bioengineering, Washington State University, Pullman, Washington 99164, USA
First published on 11th September 2017
Abstract
Conversion of CO2 to CO via hydrogenation, also known as the reverse water-gas shift (RWGS) reaction, is an important chemical process to generate CO as a platform chemical for further conversions. Metallic Cu catalyses the RWGS reaction at a temperature of 500 °C with a high initial turnover frequency, but surface structural reorganization and particle growth at the reaction temperature deleteriously reduce its activity over time. In this work, we synthesized hierarchical structures of porous Cu@C and Cu/Zn@C materials via pyrolysis of Cu-BTC Metal–Organic Frameworks (MOFs) with or without Zn doping. Carbon encapsulation protects the Cu NPs from sintering, leading to stable catalytic activity at 500 °C under which RWGS is favored. Furthermore, the final catalyst pellet size can be controlled by tuning the crystal size of MOF precursors, eliminating the step of forming catalysts for fixed bed reactor applications.
 Cheng Wang | Dr Cheng Wang is now a professor of Chemistry at Xiamen University, China. He got his BSc in chemistry at Peking University, and PhD in inorganic chemistry at University of North Carolina at Chapel Hill in US. He worked with Professor Wenbin Lin on metal–organic frameworks for heterogeneous catalysis for his doctoral thesis. He then went to the University of Chicago as a postdoc to study photodynamics in photocatalysis using ultrafast spectroscopy. Dr Cheng Wang got the Young Investigator Award from Division of Inorganic Chemistry, American Chemical Society in 2013, and the Junior Thousand Talents Award of P. R. China in 2015. Currently Dr Cheng Wang's research focuses on metal–organic layers as solid state catalysts. His interest covers conversions of C1 molecules via solid–gas phase catalysis, electrocatalysis and photocatalysis. |
Introduction
Carbon monoxide is a widely used platform molecule that can be converted to versatile products such as methanol,1 olefin,2 and liquid hydrocarbon fuels.3 CO is also a key starting material for many important chemical processes such as the Monsanto process to synthesize acetic acid,4 hydroformylation to obtain aldehyde,5 and the synthesis of phosgene.6 Efficient hydrogenation of CO2 using hydrogen from renewable energy sources is a potentially scalable process to produce CO.Known as the reverse water-gas shift (RWGS) reaction, CO2 hydrogenation to CO is catalysed by the noble metals such as Pt7 and Rh.8 Non-precious metal catalysts for the RWGS reaction have attracted much more recent interest due to their availability for large scale applications.9 Cu is one of the promising non-precious metal catalysts for the RWGS reaction, whose activity and stability strongly depend on the metal-support interactions.10 However, Cu nanoparticles (NPs) have a strong tendency to sinter and aggregate at reaction temperatures above 300 °C,10a while RWGS with a positive entropy change is more favorable at higher temperatures in comparison to the methanol synthesis with a negative entropy change.
Metal–Organic Frameworks (MOFs) can serve as a porous support for metal NPs or clusters in these catalysts.11 Pyrolysis of MOFs can generate hierarchical structures that are stable at higher temperature with the corresponding metal, metal carbide, or metal oxide NPs embedded in a porous carbon matrix.3b,12 We hypothesized that encapsulation in a carbon matrix can prevent the growth of NPs and thus stabilize the Cu catalyst for CO2 hydrogenation.13 The porous carbon network from the pyrolysis process also enhances transport of CO2 and hydrogen to access the NP surfaces. We chose the readily available Cu-BTC MOF (HKUST-1) as the pyrolysis precursor to prepare the Cu@C material and to study its catalytic activity in CO2 hydrogenation. The feasibility of producing Cu@C via pyrolysis of Cu-BTC was previously demonstrated by Won Cheol Yoo and coworkers.14
The RWGS reaction involves both hydrogen activation and C–O bond breaking.9d A bimetallic system, comprising metals with affinity to hydrogen such as Cu and metals with affinity to oxygen such as Zn, is expected to be beneficial for the RWGS reaction.9b,15 We doped Zn into the Cu-BTC MOF for pyrolysis to prepare a Cu/Zn@C material. The Cu/Zn@C system has a much higher CO2 hydrogenation activity than Cu@C, and is selective for CO production at temperatures above 300 °C. Importantly, the carbon matrix prevents the phase separation of Cu/Zn bimetallic catalysts, which is problematic for traditional Cu/ZnO/Al2O3 catalysts at high temperatures.10a Furthermore, we also demonstrated that the sizes of the MOF crystals can be tuned from several microns to several millimeters (Zn–Cu-BTC-μm, Zn–Cu-BTC-submm and Zn–Cu-BTC-mm) to eliminate the step of forming Cu/Zn@C catalyst pellets (Scheme 1).
 | ||
| Scheme 1 Pyrolysis of MOFs to produce hierarchical Cu/Zn@C structures for RWGS. | ||
Experimental
Synthetic procedures
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of H2O and ethanol, and the solution was mixed with trimesic acid (H3BTC) and heated at 125 °C for 12 h to yield blue powders. Zn–Cu-BTC-μm crystals were synthesized in the same way by adding Zn(NO3)2·6H2O to the metal source.
:1:1:4) and placed in an oven at 55 °C for 3 days to obtain crystals.
:1 mixture of H2O and ethanol, and the solution was mixed with trimesic acid (H3BTC) and heated at 125 °C for 12 h to yield blue powders. Zn–Cu-BTC-μm crystals were synthesized in the same way by adding Zn(NO3)2·6H2O to the metal source.
:1:1:4) and placed in an oven at 55 °C for 3 days to obtain crystals.
Pyrolysis procedure
MOF crystals were put in a combustion boat inside a tube furnace under an argon flow of 80 mL min−1. The furnace temperature was programmed to increase at a rate of 0.3 °C min−1 to 80 °C, then 5 °C min−1 to 500 °C and held at 500 °C for 4 h. The pyrolyzed samples were denoted as Cu@C-μm, Cu/Zn@C-μm, Cu@C-submm, Cu/Zn@C-submm, and Cu/Zn@C-mm, respectively.Results and discussion
Preparation of MOF precursors
The sizes of the M-BTC crystals were reliably controlled by adopting different reaction temperatures, reactant concentrations, and amounts of modulators. For example, reactions at 125 °C with a metal concentration of 0.2 M led to MOF crystals several microns in size (Zn–Cu-BTC-μm, see Fig. 1a). In comparison, lowering the reaction temperature to 90 °C with similar reactant concentrations yielded crystals several hundreds of microns (sub-millimeter) in size (Zn–Cu-BTC-submm, see Fig. 1c), presumably due to lower rates of nucleation at lower temperatures. At an even lower temperature of 55 °C and a metal concentration of 0.1 M, we obtained MOF crystals several millimeters in size from the reaction mixture in the presence of acetic acid as a monocarboxylic acid modulator (Zn–Cu-BTC millimeter crystal, see Fig. 1e). The phase purities of all synthesized M-BTC crystals were confirmed by their PXRD patterns which matched with the simulated ones (Fig. S1 in the ESI†). | ||
| Fig. 1 (a) SEM image of Zn–Cu-BTC micro crystals, (b) TEM image of Cu/Zn@C-μm, (c) microscope photo of Zn–Cu-BTC submillimeter crystals, (d) microscope photo of Cu/Zn@C-submm, (e) microscope photo of Zn–Cu-BTC millimeter crystals and (f) microscope photo of Cu/Zn@C-mm. | ||
Zn was doped into the Cu-BTC framework by adding 13–17 mol% of Zn(NO3)2 with respect to the total metal (Zn/[Zn + Cu]) into the reaction mixture in the corresponding solvothermal syntheses. However, only 2.7 mol% of Zn was incorporated into the pyrolyzed Cu/Zn@C-μm, as determined by ICP-OES. Similarly, Cu/Zn@C-submm possessed only 4.6 mol% of Zn and Cu/Zn@C-mm possessed 3.6 mol% of Zn. Zn2+ ions may not fit into the Cu-BTC structure like Cu2+ ions due to the slightly larger ionic radius of Zn2+ (0.68 Å) than Cu2+ (0.65 Å), which can account for the lower amount of Zn incorporated into the MOFs than the amount added into the solvothermal syntheses.
Characterization of the pyrolyzed catalysts
After pyrolysis at 500 °C for 4 h, the samples all changed from blue to black as a result of carbonization of the organic linkers. All of the pyrolyzed samples maintained the sizes and morphologies of the corresponding original MOF crystals as shown in Fig. 1b, d and f and Fig. S8 (ESI†). TEM further revealed that the pyrolyzed particles contain numerous small NPs around 20 ± 5 nm in size that are uniformly distributed in the carbon matrix. Fig. 2a shows a zoomed-in view of the Cu/Zn@C-μm TEM image, while Fig. 2c and e show the TEM images of pieces stripped off the Cu/Zn@C-submm and Cu/Zn@C-mm catalyst pellets by vigorous sonication.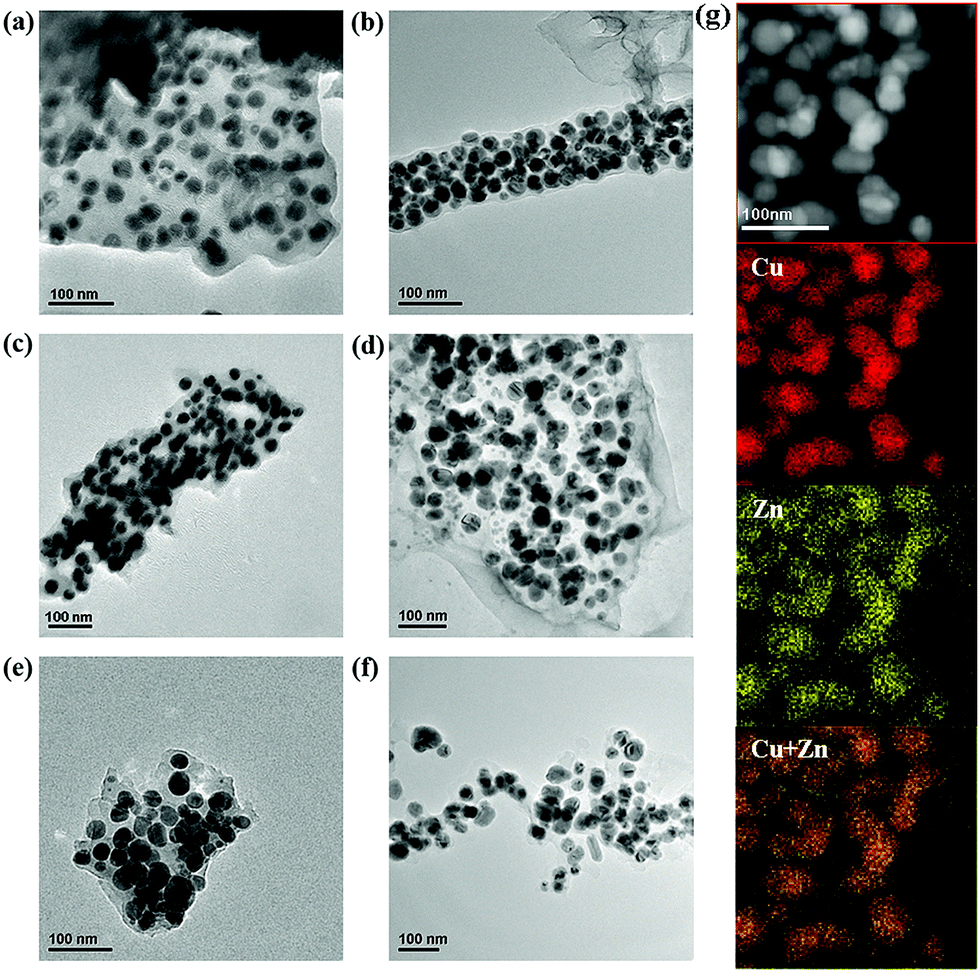 | ||
| Fig. 2 TEM images of Cu/Zn@C materials before and after reaction at 500 °C and 1 bar. (a) Cu/Zn@C-μm as synthesized, (b) Cu/Zn@C-μm after the reaction, (c) Cu/Zn@C-submm as synthesized, (d) Cu/Zn@C-submm after the reaction, (e) Cu/Zn@C-mm as synthesized, (f) Cu/Zn@C-mm after the reaction and (g) EDX mapping of fresh Cu/Zn@C-submm, showing well mixed Cu and Zn. | ||
The NPs are mostly Cu or Cu/Zn(OH)2 as shown by the PXRD patterns of the pyrolyzed samples (Fig. S2, ESI†). A Cu/Zn alloy is also a possible phase in this system. These NPs account for 70.5 wt% of Cu/Zn@C-submm as shown by TGA, and 75.8 wt% of Cu/Zn@C-μm (Fig. S3–S5, ESI†). The complete conversion of the MOFs was also verified by fourier-transform infrared (FT-IR) spectroscopy (Fig. S6, ESI†). EDX mapping showed the overlap of the Cu and Zn distributions in both Cu/Zn@C-submm and Cu/Zn@C-μm, consistent with finely mixed Zn and Cu (Fig. 2g and Fig. S9, S10, ESI†). XPS analysis of the pyrolyzed samples after reduction with 5% H2 in Ar at 250 °C showed Cu(0) with peaks at 951.8 eV and 931.8 eV for Cu 2p1/2 and 2p3/2, respectively (Fig. S11 and S12, ESI†). These two peaks do not change positions after treating the catalysts with the reaction gas at 260 °C, suggesting that Cu is in the Cu(0) valence state under the reaction conditions. The peaks at 1021.2 eV and 1044.5 eV in the XPS spectra belong to Zn 2p3/2 and Zn 2p1/2 and are consistent with Zn(II) both after reduction with hydrogen at 250 °C and after treatment of the reaction gas at 260 °C.
It is worth noting that Cu NPs are easily oxidized upon exposure to air. However, the Cu NPs encapsulated by a carbon matrix in the pyrolyzed MOFs are much more resistant to oxidation, as shown by the retention of the PXRD patterns of metallic copper after the samples were kept in air for weeks. A similar phenomenon was observed for Cu NPs encapsulated by graphene or Ni.13 Consistent with this, treatment of these catalysts with N2O showed a negligible amount of Cu oxidation in Cu/Zn@C-submm (0.011% of the total Cu or 2.9% of surface Cu for NPs of 20 nm) or Cu/Zn@C-μm (0.077% of the total Cu or 0.16% of surface Cu for NPs of 20 nm) as determined by subsequent temperature programmed reduction with hydrogen (H2-TPR) measurements.10b
The porous nature of the carbon matrix in these samples was confirmed by N2 adsorption measurements, with BET surface areas of 95.85 m2 g−1, 125.03 m2 g−1 and 123.09 m2 g−1 for Cu/Zn@C-mm, Cu/Zn@C-submm and Cu/Zn@C-μm, respectively. The mesopore volumes were 0.043 cm3 g−1 for Cu/Zn@C-mm, 0.125 cm3 g−1 for Cu/Zn@C-submm and 0.123 cm3 g−1 for Cu/Zn@C-μm (see Fig. S13–S15 and Table S1, ESI†).
Catalytic activities of Cu/Zn@C-submm and Cu@C-submm
Hydrogenation of CO2 was investigated in a fixed bed flow reactor. We have listed the catalytic results together with those of representative catalysts in the literature for comparison (Table S2, ESI†). The Cu/Zn@C-submm exhibited 5.0% CO2 conversion and 100% CO selectivity at 500 °C and a total pressure of 1 bar, comparable to that of Cu/SiO2 (1.8% conversion at 500 °C/1 bar)19 and Cu–Zn–Al (100% CO selectivity at 500 °C/1 bar).10a The activities of the Cu/Zn@C-submm are higher than that of the Cu@C-submm (entries 1–4 vs. entries 5–8 in Table S2, ESI†). For example, Cu/Zn@C-submm gave a conversion of 5.0% at 500 °C and 1 bar, while Cu@C-submm only gave a conversion of 1.9%. These observations demonstrate the beneficial synergistic effects between Cu and Zn in RWGS. The pyrolysis of bimetallic MOFs achieves a maximal mixing of Cu and Zn in the NPs and a good dispersion of them in a porous carbon matrix, both of which help to obtain high catalytic activities.Catalyst stability
The stability of the Cu/Zn@C-submm catalyst was evaluated over 20 hours under several conditions. Strikingly, the activity of the catalyst remained steady during the entire test at a rather high temperature of 500 °C (1 bar H2/CO2 = 3/1, GHSV = 18000 h−1). The selectivity of CO was 100% in the test. For comparison, Cu–Zn/AC (AC = activated carbon) (Cu 56.3 wt% and Zn 7.4 wt%) and Cu–Zn/OMC (OMC = ordered mesoporous carbon) (Cu/Zn row ratio = 5:1, the same as Cu–Zn/AC) catalysts were synthesized according to a modified literature procedure (the detailed synthetic procedure can be found in the ESI†).20 Cu–Zn/AC showed a 10-fold decrease of CO2 conversion activity after 11 hours on stream, however Cu–Zn/OMC with a low CO2 conversion did not show apparent deactivation on stream under the same reaction conditions (H2/CO2 = 3/1, temperature = 500 °C, pressure = 1 bar, GHSV = 18000 h−1). This result is consistent with the hypothesis that mesopores play key roles in stabilizing the catalyst (Fig. 3).
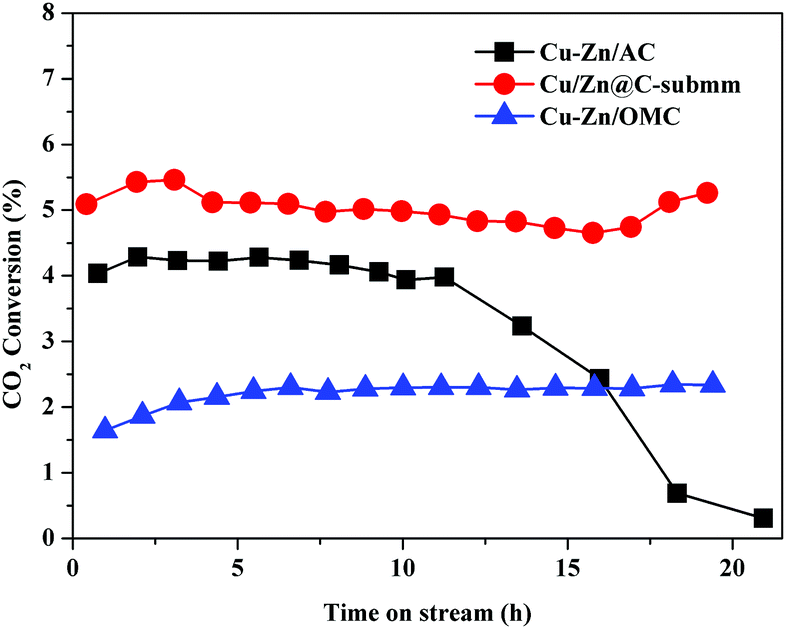 | ||
| Fig. 3 The CO2 conversion over Cu/Zn@C-submm, Cu–Zn/AC and Cu–Zn/OMC as a function of time on stream. Reaction conditions: 500 °C, 1 bar H2/CO2 = 3/1, GHSV = 18000 h−1. | ||
We recovered the Cu/Zn@C-submm catalysts after the reaction of 20 hours at 500 °C at a pressure of 1 bar (H2/CO2 = 3/1), and compared the sizes of the Cu/Zn NPs in the carbon matrix with the fresh catalysts using TEM. The size of the NPs before and after the catalyst testing only changed slightly (Fig. 2c, d and Table S3 (ESI†), 20 ± 5 nm before the reaction vs. 31 ± 8 nm after reaction; see Fig. S19–S24 (ESI†) for statistics of size distribution). In contrast, the size of the nanoparticles in Cu–Zn/AC increased significantly (33 ± 9 nm before reaction vs. 75 ± 9 nm after reaction), agreeing with the declining activity due to particle aggregation (Fig. S26 and S27, ESI†).
Control of pellet sizes
The pellet size of the MOF derived catalysts can be tuned by controlling the crystal size of MOFs via crystal engineering. We can thus control the pellet size of the catalysts without the additional step of catalyst forming for fixed bed reactor applications. As expected, pellet size affects the internal mass transfer limitation (Table S4, ESI†). The catalytic activity of Cu/Zn@C-submm is higher than that of Cu/Zn@C-μm and Cu/Zn@C-mm (Table S5, ESI†), suggesting the presence of internal mass transfer limitation within pellets in this size range (further discussions of pellet size are included in the ESI†).Conclusions
We have synthesized porous hierarchical structures of Cu/Zn@C via pyrolysis of MOF crystals of Zn-doped Cu-BTC. The encapsulation of the resultant Cu/Zn NPs in the porous carbon matrix protects the NPs from sintering, leading to superior stability of the catalysts in the RWGS reaction. We also confirmed the synergistic effects between Cu and Zn to increase catalytic activity owing to the well mixed Cu and Zn from pyrolysis. A study of the dependence of the catalytic activity on pellet size reveals the importance of fine-tuning the crystal sizes of the pyrolysis precursors, which eliminates an additional step of catalyst forming. This work highlights the opportunities in strategically converting MOFs into high performance catalysts by combining techniques of crystal engineering and requirements from chemical engineering.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Junhu Lei, Zi Wang, Zhe Li and Lingzhen Zeng for experimental help. We acknowledge funding support from the National Natural Science Foundation and the Ministry of Science and Technology of the P. R. China (NSFC21471126, NSFC21671162, NSFC21673189, and 2016YFA0200702), the National Thousand Talents Program of the P. R. China, and the Fundamental Research Funds for the Central Universities (No. 20720160032).Notes and references
- (a) S. Felix, A.-P. Frank, W. Qiongxiao, D. J. Anker, T. Burcin, G. Jan-Dierk and K. N. Jens, J. Catal., 2012, 293, 51–60 CrossRef; (b) A. P. Konstantin, D. R. Michael and T. B. Alexis, J. Catal., 2005, 368–377 Search PubMed; (c) S. Kuld, M. Thorhauge, H. Falsig, C. F. Elkjær, S. Helveg, I. Chorkendorff and J. Sehested, Science, 2016, 352, 969–974 CrossRef CAS PubMed.
- (a) K. Cheng, B. Gu, X. Liu, J. Kang, Q. Zhang and Y. Wang, Angew. Chem., Int. Ed., 2016, 55, 4725–4728 CrossRef CAS PubMed; (b) H. Mistry, A. S. Varela, C. S. Bonifacio, I. Zegkinoglou, I. Sinev, Y.-W. Choi, K. Kisslinger, E. A. Stach, J. C. Yang, P. Strasser and B. R. Cuenya, Nat. Commun., 2016, 7, 12123 CrossRef PubMed; (c) F. Jiao, J. Li, X. Pan, J. Xiao, H. Li, H. Ma, M. Wei, Y. Pan, Z. Zhou, M. Li, S. Miao, J. Li, Y. Zhu, D. Xiao, T. He, J. Yang, F. Qi, Q. Fu and X. Bao, Science, 2016, 351, 1065–1068 CrossRef CAS PubMed; (d) H. M. Torres Galvis, J. H. Bitter, C. B. Khare, M. Ruitenbeek, A. I. Dugulan and K. P. de Jong, Science, 2012, 335, 835–838 CrossRef CAS PubMed; (e) P. Zhai, C. Xu, R. Gao, X. Liu, M. Li, W. Li, X. Fu, C. Jia, J. Xie, M. Zhao, X. Wang, Y.-W. Li, Q. Zhang, X.-D. Wen and D. Ma, Angew. Chem., Int. Ed., 2016, 55, 9902–9907 CrossRef CAS PubMed.
- (a) Z. Yang, S. Guo, X. Pan, J. Wang and X. Bao, Energy Environ. Sci., 2011, 4, 4500–4503 RSC; (b) B. Qiu, C. Yang, W. Guo, Y. Xu, Z. Liang, D. Ma and R. Zou, J. Mater. Chem. A, 2017, 5, 8081–8086 RSC; (c) C.-x. Xiao, Z.-p. Cai, T. Wang, Y. Kou and N. Yan, Angew. Chem., Int. Ed., 2008, 47, 746–749 CrossRef CAS PubMed; (d) X. Peng, K. Cheng, J. Kang, B. Gu, X. Yu, Q. Zhang and Y. Wang, Angew. Chem., Int. Ed., 2015, 54, 4553–4556 CrossRef CAS PubMed.
- H. Cheung, R. S. Tanke and G. P. Torrence, Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH Verlag GmbH & Co. KGaA, 2000 Search PubMed.
- K. Weissermel and H.-J. Arpe, Industrial Organic Chemistry, Wiley-VCH Verlag GmbH, 2008, ch. 6, pp. 127–144 Search PubMed.
- W. Schneider and W. Diller, Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH Verlag GmbH & Co. KGaA, 2000 Search PubMed.
- (a) S. G. Jadhav, P. D. Vaidya, B. M. Bhanage and J. B. Joshi, Can. J. Chem. Eng., 2016, 94, 101–106 CrossRef CAS; (b) S. Kattel, B. Yan, J. G. Chen and P. Liu, J. Catal., 2016, 343, 115–126 CrossRef CAS; (c) A. Goguet, F. Meunier, J. P. Breen, R. Burch, M. I. Petch and A. Faur Ghenciu, J. Catal., 2004, 226, 382–392 CrossRef CAS.
- (a) K. Kitamura Bando, K. Soga, K. Kunimori and H. Arakawa, Appl. Catal., A, 1998, 175, 67–81 CrossRef CAS; (b) H. Kusama, K. K. Bando, K. Okabe and H. Arakawa, Appl. Catal., A, 2001, 205, 285–294 CrossRef CAS.
- (a) D. H. Kim, J. L. Park, E. J. Park, Y. D. Kim and S. Uhm, ACS Catal., 2014, 4, 3117–3122 CrossRef CAS; (b) C. Alvarez Galvan, J. Schumann, M. Behrens, J. L. G. Fierro, R. Schlogl and E. Frei, Appl. Catal., B, 2016, 195, 104–111 CrossRef CAS; (c) A. Wolf, A. Jess and C. Kern, Chem. Eng. Technol., 2016, 39, 1040–1048 CrossRef CAS; (d) M. D. Porosoff, B. Yan and J. G. Chen, Energy Environ. Sci., 2016, 9, 62–73 RSC.
- (a) X. Zhang, X. Zhu, L. Lin, S. Yao, M. Zhang, X. Liu, X. Wang, Y.-W. Li, C. Shi and D. Ma, ACS Catal., 2017, 7, 912–918 CrossRef CAS; (b) C. S. Chen, J. H. Wu and T. W. Lai, J. Phys. Chem. C, 2010, 114, 15021–15028 CrossRef CAS; (c) J. A. Rodriguez, P. Liu, D. J. Stacchiola, S. D. Senanayake, M. G. White and J. G. Chen, ACS Catal., 2015, 5, 6696–6706 CrossRef CAS.
- (a) Z. Li, A. W. Peters, V. Bernales, M. A. Ortuño, N. M. Schweitzer, M. R. DeStefano, L. C. Gallington, A. E. Platero-Prats, K. W. Chapman, C. J. Cramer, L. Gagliardi, J. T. Hupp and O. K. Farha, ACS Cent. Sci., 2017, 3, 31–38 CrossRef CAS PubMed; (b) E. D. Metzger, C. K. Brozek, R. J. Comito and M. Dincă, ACS Cent. Sci., 2016, 2, 148–153 CrossRef CAS PubMed; (c) R. J. Comito, K. J. Fritzsching, B. J. Sundell, K. Schmidt-Rohr and M. Dincă, J. Am. Chem. Soc., 2016, 138, 10232–10237 CrossRef CAS PubMed; (d) D. Yang, S. O. Odoh, T. C. Wang, O. K. Farha, J. T. Hupp, C. J. Cramer, L. Gagliardi and B. C. Gates, J. Am. Chem. Soc., 2015, 137, 7391–7396 CrossRef CAS PubMed; (e) K. Na, K. M. Choi, O. M. Yaghi and G. A. Somorjai, Nano Lett., 2014, 14, 5979–5983 CrossRef CAS PubMed; (f) A. N. Mlinar, B. K. Keitz, D. Gygi, E. D. Bloch, J. R. Long and A. T. Bell, ACS Catal., 2014, 4, 717–721 CrossRef CAS.
- (a) B. An, K. Cheng, C. Wang, Y. Wang and W. B. Lin, ACS Catal., 2016, 6, 3610–3618 CrossRef CAS; (b) T. A. Wezendonk, V. P. Santos, M. A. Nasalevich, Q. S. E. Warringa, A. I. Dugulan, A. Chojecki, A. C. J. Koeken, M. Ruitenbeek, G. Meima, H.-U. Islam, G. Sankar, M. Makkee, F. Kapteijn and J. Gascon, ACS Catal., 2016, 6, 3236–3247 CrossRef CAS; (c) B. Liu, H. Shioyama, H. Jiang, X. Zhang and Q. Xu, Carbon, 2010, 48, 456–463 CrossRef CAS; (d) M. Ding, S. Chen, X.-Q. Liu, L.-B. Sun, J. Lu and H.-L. Jiang, ChemSusChem, 2017, 10, 1898–1903 CrossRef CAS PubMed; (e) B. Liu, H. Shioyama, T. Akita and Q. Xu, J. Am. Chem. Soc., 2008, 130, 5390–5391 CrossRef CAS PubMed; (f) V. P. Santos, T. A. Wezendonk, J. J. D. Jaén, A. I. Dugulan, M. A. Nasalevich, H.-U. Islam, A. Chojecki, S. Sartipi, X. Sun, A. A. Hakeem, A. C. J. Koeken, M. Ruitenbeek, T. Davidian, G. R. Meima, G. Sankar, F. Kapteijn, M. Makkee and J. Gascon, Nat. Commun., 2015, 6, 6451 CrossRef CAS PubMed; (g) L. Oar-Arteta, T. Wezendonk, X. Sun, F. Kapteijn and J. Gascon, Mater. Chem. Front., 2017, 1, 1709–1745 RSC.
- (a) S. Wang, X. Huang, Y. He, H. Huang, Y. Wu, L. Hou, X. Liu, T. Yang, J. Zou and B. Huang, Carbon, 2012, 50, 2119–2125 CrossRef CAS; (b) F. Muench, L. Sun, T. Kottakkat, M. Antoni, S. Schaefer, U. Kunz, L. Molina-Luna, M. Duerrschnabel, H.-J. Kleebe, S. Ayata, C. Roth and W. Ensinger, ACS Appl. Mater. Interfaces, 2017, 9, 771–781 CrossRef CAS PubMed.
- W. Bak, H. S. Kim, H. Chun and W. C. Yoo, Chem. Commun., 2015, 51, 7238–7241 RSC.
- (a) Y. Bu, C. J. Weststrate, J. W. Niemantsverdriet and H. O. A. Fredriksson, ACS Catal., 2016, 6, 7994–8003 CrossRef CAS; (b) J. Schumann, M. Eichelbaum, T. Lunkenbein, N. Thomas, M. C. Álvarez Galván, R. Schlögl and M. Behrens, ACS Catal., 2015, 5, 3260–3270 CrossRef CAS; (c) S.-I. Fujita, M. Usui and N. Takezawa, J. Catal., 1992, 134, 220–225 CrossRef CAS.
- B. Jee, K. Eisinger, F. Gul-E-Noor, M. Bertmer, M. Hartmann, D. Himsl and A. Pöppl, J. Phys. Chem. C, 2010, 114, 16630–16639 CAS.
- M. K. Bhunia, J. T. Hughes, J. C. Fettinger and A. Navrotsky, Langmuir, 2013, 29, 8140–8145 CrossRef CAS PubMed.
- T. M. Tovar, J. Zhao, W. T. Nunn, H. F. Barton, G. W. Peterson, G. N. Parsons and M. D. LeVan, J. Am. Chem. Soc., 2016, 138, 11449–11452 CrossRef CAS PubMed.
- C.-S. Chen, W.-H. Cheng and S.-S. Lin, Appl. Catal., A, 2003, 238, 55–67 CrossRef CAS.
- O. Arbeláez, A. Orrego, F. Bustamante and A. L. Villa, Top. Catal., 2012, 55, 668–672 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7qm00328e |
| ‡ These authors contributed equally. |
| This journal is © the Partner Organisations 2017 |