DOI:
10.1039/C7QM00291B
(Research Article)
Mater. Chem. Front., 2017,
1, 2349-2355
A thieno[3,4-b]thiophene-based small-molecule donor with a π-extended dithienobenzodithiophene core for efficient solution-processed organic solar cells†
Received
29th June 2017
, Accepted 18th August 2017
First published on 18th August 2017
Abstract
A small molecule donor STB-4 with dithieno[2,3-d′:2′,3′-d′]benzo[1,2-b:4′,5′-b′]dithiophene (DTBDT) as the central moiety was designed and synthesized for solution-processed bulk-heterojunction solar cells. An optimized power conversion efficiency of 8.17% with an open-circuit voltage of 0.904 V, a short-circuit current of 13.33 mA cm−2, and a fill factor FF of 0.67 was achieved after solvent annealing (SVA). According to the detailed morphology investigations, we found that SVA refined the phase-separated morphologies of the blends, allowing the domains to become well defined with anappropriate size that is beneficial for device performance.
1. Introduction
Bulk-heterojunction organic solar cells have drawn considerable attention in both academia and industry because of their unique features of being light-weight, flexible, and low-cost.1–3 Recently, the power conversion efficiencies (PCEs) of polymer solar cells (PSCs) have exceeded 11% with tremendous efforts based on material and device engineering.4–10 Compared with polymers, small-molecule donor materials possess distinct advantages such as a well-defined molecular structure, low batch-to-batch variation and feasible tunability toward their electronic structure. Significant progress has been made for small-molecule solar cells (SMSCs) with high PCEs over 9%.11–22
Some approaches have been successfully adopted in molecular design in order to optimize BHJ-OSC performances, among which introducing heteroatoms and adopting two-dimensional (2D) conjugation are two well-recognized methods. Bazan et al. reported a small-molecule donor, DTS(PTTh2)2, by replacing the benzene rings of DTS(BTTh2)2 with pyridine ones and achieved a higher PCE of 6.7%.23 Further investigations indicated that this improvement can be attributed to its wider absorption range, better crystallinity and higher carrier mobility. By applying a dialkylthiol-substituted benzodithiophene (BDT) unit, Chen et al. designed a small-molecule donor, DR3TSBDT, and achieved a very high PCE of 9.95% attributed to the enhanced absorption and the preferable morphology of the optimized blend.24 Meanwhile, introducing conjugated groups in the orthogonal direction of the conjugated framework with π-electrons being delocalized to the conjugated side groups may increase inter-chain π–π stacking and benefit exciton diffusion and charge transport.25 By altering alkyloxy side chains to thienyl ones, a small-molecule donor, DR3TBDTT, showed a high PCE of 9.58% with a significant thirty percent improvement.26 Quite recently, we developed a new category of small-molecule donors (STB-n) based on electron-rich 2-alkyl-substituted thieno[3,4-b]thiophenes (TbT). By molecular optimizations with heteroatoms and 2D-BDT, high PCEs of 8.68% and 9.26% were obtained.22,27 To achieve better photovoltaic performance, further optimizations should be performed. For D (donor)–A (acceptor) polymer materials, expanding the π-conjugated surface of electron-rich or electron-deficient moieties is another effective design strategy, which can further delocalize the holes and thus lower the local positive charge density. With reduced Coulombic interactions between the hole and the electron (exciton binding energy), charge separation at the donor–acceptor interfaces may become easier, leading to higher device performance.28 Dithieno[2,3-d′:2′,3′-d′]benzo[1,2-b:4′,5′-b′]dithiophene (DTBDT), which can be viewed as a π-expanded BDT, is widely utilized as an electron-rich moiety in PSCs.29–42 Through alkyl-chain optimizations, PTDBD3, a DTBDT-based polymer, realized higher PCEs (7.6%) than that of PTB-7 with a BDT unit as shown by Yu et al.30 Another DTBDT-based polymer, PDT-S-T, was reported by Hou et al., showing PCEs as high as 7.79% in PSCs with a PC61BM acceptor.31 Subsequently, Sun et al. copolymerized DTBDT with an electron-deficient moiety (1,3-bis(5-bromo-thiophen-2-yl)-5,7-bis(2-ethylhexyl)-4H,8H-benzo[1,2-c:4,5-c′]dithi-ophene-4,8-dione) to synthesize a wide-bandgap polymer PBDT-T1 and achieved a high PCE of 9.7% after using a solvent additive, DIO.37 However, DTBDT is rarely utilized for the design of small-molecule materials.43
In this work, we introduced DTBDT into our thieno[3,4-b]thiophene-based molecular framework and synthesized a new small-molecule donor, STB-4 (Fig. 1), in which electron-rich DTBDT and electron-deficient rhodanine44 were bridged by thieno[3,4-b]thiophene with a significant quinoid-resonance effect.45STB-4-based SMSCs exhibited a high PCE of 8.17%, with a Voc of 0.904 V and Jsc of 13.33 mA cm−2 after careful device optimization.
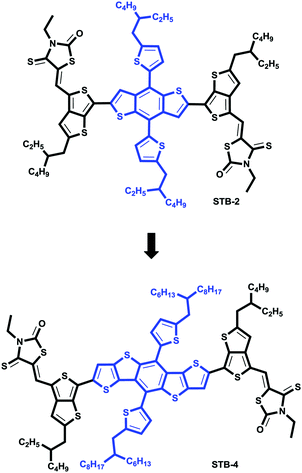 |
| | Fig. 1 Molecular structures of STB-4 and STB-2. | |
2. Results and discussion
Material preparation
The synthetic route of STB-4 is depicted in Scheme 1. Diiodide 3 was synthesized according to the literature methods.28,34 Compound 4 was synthesized via Stille coupling reaction between compound 3 and tributyl(2-(2-ethylhexyl)thieno[3,4-b]thiophen-6-yl)stannane. Compound 4 was rapidly flushed through an Alumina column and used for the next step without further purification because of its low ambient stability. Dialdehyde 5 was synthesized in 30% yield by quick deprotonation followed by the reaction with DMF. Finally, STB-4 was synthesized via the Knoevenagel reaction between compound 5 and 3-ethylrhodanine in 84% yield. STB-4 is a black solid and shows good solubility in common organic solvents, so it is suitable for fabricating solution-processed OPVs. High-purity STB-4 for device fabrication was further recrystallized in DCE/EtOH three times. As shown in Fig. S1 (ESI†), STB-4 exhibits good thermal stability with a high decomposition temperature (5 wt% weight loss) of 349 °C under a nitrogen atmosphere.
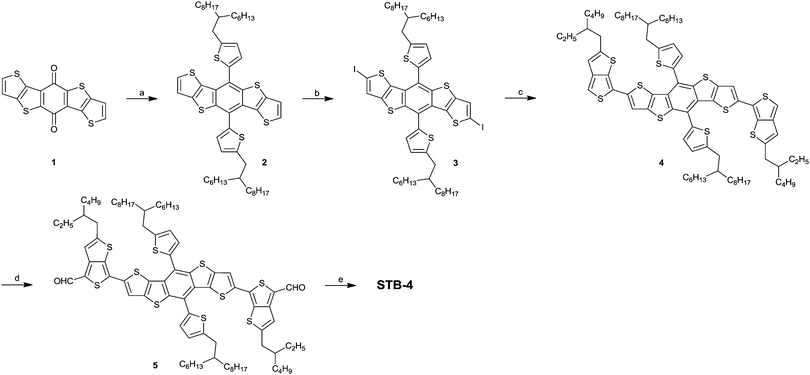 |
| | Scheme 1 Synthetic procedures for STB-4. (a) (i) (5-(2-Hexyldecyl)thiophen-2-yl)lithium, tetrahedrofuran (THF); (ii) SnCl2, aqueous HCl (10%); (b) n-BuLi, diiodoethane, THF; (c) Pd(PPh3)4, tributyl(2-(2-ethylhexyl)thieno[3,4-b]thiophen-6-yl)stannane, N,N-dimethylformamide (DMF)/toluene; (d) n-BuLi, THF, DMF; (e) 3-ethyl-rhodanine, β-alanine, dichloroethane (DCE)/ethanol (EtOH). | |
Absorption and electrochemical properties
The UV-vis absorption spectra of STB-4 in diluted dichloromethane solution (1.0 × 10−5 M−1 L−1) and in a thin film are shown in Fig. 2a, and the corresponding data are summarized in Table 1. STB-4 displayed a maximum absorption at 620 nm with a high absorption coefficient (ε) of 1.05 × 105 M−1 cm−1 in dichloromethane. In a thin film, STB-4 showed a new and intense peak at approximately 680 nm, which may be attributed to intermolecular π–π stacking. The absorption onset of the STB-4 film is 732 nm, corresponding to an optical band gap of 1.69 eV. Cyclic voltammetry measurement was applied to evaluate the frontier orbital energy levels of STB-4 (Fig. 2b). The potentials were internally calibrated using the ferrocene/ferrocenium (Fc/Fc+) redox couple (4.8 eV below the vacuum level), and the energy levels were calculated from the onset oxidation potential (0.26 eV) and reduction potential (−1.35 eV). The energy levels of the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) were estimated to be −5.06 and −3.45 eV, respectively, which matched well with the LUMO energy level of PC71BM for efficient charge separation. Compared with STB-2, STB-4 shows slightly elevated HOMO/LUMO energy levels and an increased optical bandgap.
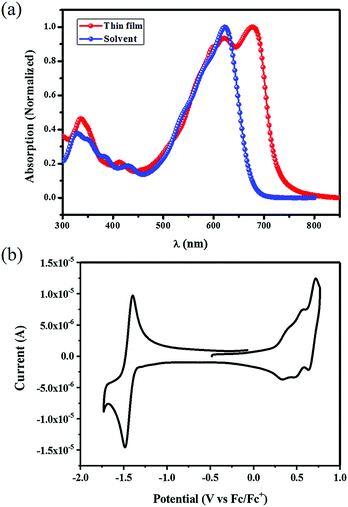 |
| | Fig. 2 (a) UV-vis absorption spectra of STB-4 in dichloromethane solution and in a thin film. (b) Cyclic voltammogram of STB-4 in dichloromethane solution (0.1 M Bu4NPF6). | |
Table 1 Photophysical and electrochemical data for STB-4 and STB-2
| Cpd.a |
λ
absmax (sol, nm) |
ε (×105 M−1 cm−1) |
λ
absmax (film, nm) |
λ
absonset (film, nm) |
E
onsetox
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) (V)
(V) |
E
HOMO
(eV) |
E
onsetred
(V) |
E
LUMO
(eV) |
E
CVg (eV) |
E
optg
(eV) |
|
Measured in dichloromethane.
CV on a carbon electrode with n-Bu4NClO4 in dichloromethane (0.1 M, vs. Fc+/Fc).
The HOMO and LUMO energy levels were determined by EHOMO/LUMO = −(4.80 + Eonsetox/red) (eV).
E
optg = 1240/λabsonset (eV).
|
|
STB-4
|
620 |
1.05 |
620, 680 |
732 |
0.26 |
−5.06 |
−1.35 |
−3.45 |
1.61 |
1.69 |
|
STB-2
|
625 |
1.12 |
620, 678 |
740 |
0.29 |
−5.09 |
−1.30 |
−3.50 |
1.59 |
1.68 |
Photovoltaic performance
BHJ organic solar cells based on STB-4 were fabricated and optimized with a conventional device structure of ITO/PEDOT:PSS/STB-4:PC71BM/Ca/Al and examined under AM 1.5G solar illumination (100 mW cm−2). Chloroform was used as the processing solvent. Fig. 3a shows the current density versus voltage (J–V) curves of solar cells before and after solvent vapor annealing (SVA) with chloroform. The results are summarized in Table 2. For the as-cast devices, STB-4 showed a poor PCE of 2.57% with a Voc of 1.04 V, a Jsc of 7.27 mA cm−2 and a low fill factor (FF) of 0.34. By contrast, the active layer processed after SVA delivered a significantly improved PCE of 8.17%, with a Voc of 0.904 V, Jsc of 13.33 mA cm−2 and FF of 0.67. External quantum efficiency (EQE) curves of the optimal STB-4-based devices covered a wide wavelength range from 300 to 750 nm (Fig. 3b), which agrees well with its absorption spectrum. In STB-4-based devices after SVA processing, high EQE values over 60% were observed in a wide range of 550–700 nm, with a peak value of 69% at 620 nm. The Jsc values calculated by integrating the EQE curve of the STB-4-based device were consistent with those from photovoltaic tests (3% error). As indicated in Fig. S3 (ESI†), the absorption of the blend film was enhanced after SVA treatment. Obviously, STB-4-based devices are more sensitive to SVA that STB-2-based ones delivering relatively high PCEs.
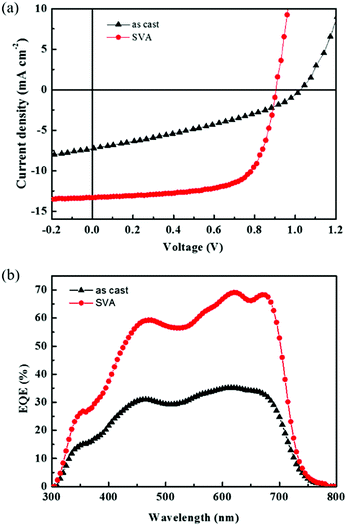 |
| | Fig. 3 (a) External quantum efficiency spectra of STB-4:PC71BM (1:0.8) before and after SVA. (b) The corresponding J–V characteristics. | |
Table 2 Photovoltaic performance of STB-4-based solar cells
| Cpd. |
Treatment |
V
oc [V] |
J
sc [mA cm−2] |
J
cal [mA cm−2] |
Fill factor (FF) |
PCEa [%] |
μ
e [×10−4 cm2 V−1 s−1] |
μ
h [×10−4 cm2 V−1 s−1] |
μ
e/μh |
|
The values in parentheses indicate the average values of PCEs obtained from more than 10 devices.
Device structure: ITO/PEDOT:PSS/STB-4:PC71BM (1:0.8 weight ratio)/Ca/Al.
|
|
STB-4
|
None |
1.02 |
7.27 |
7.11 |
0.34 |
2.57 (2.48)b |
2.17 |
1.20 |
1.81 |
| SVA |
0.904 |
13.33 |
12.98 |
0.67 |
8.17 (8.02)b |
3.58 |
3.02 |
1.19 |
|
STB-2
|
None |
0.961 |
12.12 |
12.01 |
0.47 |
5.50 (5.34)b |
2.71 |
1.92 |
1.41 |
| SVA |
0.901 |
13.12 |
12.91 |
0.66 |
7.84 (7.61)b |
4.69 |
3.16 |
1.49 |
The effect of SVA on the charge transport in these blends was investigated by space-charge-limited current (SCLC) measurement. By fitting the J–V curves from both hole- and electron-only devices, the carrier mobilities of STB-4:PC71BM blends were determined; the results are summarized in Table 2. For the blends before SVA, the hole and electron mobilities were 1.20 × 10−4 and 2.17 × 10−4 cm2 V−1 s−1, respectively, with an imbalanced μe/μh ratio of 1.81. For the active layer processed by SVA, the hole and the electron mobilities increased to 3.02 × 10−4 and 3.58 × 10−4 cm2 V−1 s−1, respectively, with a higher and more balanced μe/μh of 1.19, which is beneficial for enhanced charge transport and the efficiency of charge carrier collection.
Thin-film morphology
Morphology optimization is an important factor to achieve a finely nanoscaled phase separation of the active blend for improving device performance in OPVs. We first characterized the nanostructure of STB-4:PC71BM thin films by atomic force microscopy (AFM). From AFM measurements, the root-mean-square (RMS) roughness values are 0.32 nm and 0.65 nm for blend films before and after SVA, respectively, which reveals that both of the blend films are smooth with high quality (Fig. 4a and b). In order to get a deeper insight into the active layer morphology, we also used transmission electron microscopy (TEM) to study the blend films (Fig. 4c and d). Before SVA treatment, the blend film shows no obvious phase separation of the donor and the acceptor, which could be harmful for charge transport, thus leading to low Jsc and FF. After SVA, the blend film shows a uniform interpenetrating network structure and good nanoscaled phase separation, which is beneficial for charge separation and transport, helping to improve the performance of solar cell devices.
 |
| | Fig. 4 Tapping-mode AFM height and TEM images of optimal blend films cast from chloroform solution: (a) and (c) without SVA treatment, (b) and (d) with SVA treatment. | |
2D GIXD scattering images and the corresponding out-of-plane (OOP) and in-plane (IP) line cut profiles are shown in Fig. S4 (ESI†). The as-cast STB-4:PC71BM blended thin films showed crystalline features, with the lamellae packing distance of 24.1 Å and a PC71BM scattering ring at 1.35 Å−1. The π–π stacking peak was quite weak, and no obvious crystal orientation was evident. After SVA, a sharp scattering of the (100) peak showed up at 0.31 Å−1, which corresponds to a decreased lamella packing distance of 20.3 Å with stronger molecular interactions. The obvious (200) peak at the OOP direction further demonstrated the enhanced crystallinity, which was well matched with the increased hole and electron mobilities. Moreover, a weak peak was found in the IP direction at approximately 1.65 Å−1 after SVA and resulted in the stacking distance of 3.80 Å which may be attributed to the π–π interactions, indicating the edge-on orientations to the substrate.
3. Conclusions
A new solution-processable small-molecule donor material STB-4, in which alkyl-substituted TbT served as an electron-rich bridge to link the weak electron-donating dithienobenzodithiophene and the strongly electron-withdrawing rhodanine was designed and synthesized. BHJ OPVs based on STB-4 exhibited a low PCE of 2.57% without SVA. After SVA treatment, the PCE significantly improved to 8.17%, with higher Jsc and FF. The relationship between thin-film morphology, and device performance was investigated. The blend film formed continuous interpenetrating networks after SVA treatment, which benefited the exciton separation and charge transport, leading to higher Jsc and FF. By comparing the STB-2 performance, we revealed that extending the π-conjugation of BDT to DTBDT is effective to further promote the photovoltaic performance of TbT-based small-molecule donor materials.
4. Experimental
Materials and synthesis methods
All reactions involving air- or moisture-sensitive compounds were carried out in a dry reaction vessel under a positive pressure of nitrogen. Unless stated otherwise, starting materials were obtained from Adamas, Aldrich, or J&K and were used without further purification. Anhydrous THF, toluene, and DMF were distilled over Na/benzophenone prior to use. Anhydrous DMF was distilled over CaH2 prior to use. 1H and 13C NMR spectra were measured with Bruker Advance 400 spectrometers. Chemical shifts for hydrogens are reported in parts per million (ppm, δ scale) downfield from tetramethylsilane and are referenced to the residual protons in the NMR solvent (CDCl3: δ 7.26). 13C NMR spectra were recorded at 100 MHz. Chemical shifts for carbons are reported in parts per million (ppm, δ scale) downfield from tetramethylsilane and are referenced to the carbon resonance of the solvent (CDCl3: δ 77.2). The data are presented as follows: chemical shifts, multiplicity (s singlet, d doublet, t triplet, m multiplet and/or multiple resonances, br broad), coupling constant in hertz (Hz), and integration. MALDI measurements were performed with a MALDI-FT 9.4 T, Bruker solariX, or MALDI-TOF MS Bruker Autoflex III. Elemental analyses were performed with a Flash EA 1112 Series from ThermoQuest. UV-vis and fluorescence spectra were recorded with Jasco V-570 and Jasco FP-6600 spectrometers, respectively. Cyclic voltammetry (CV) was performed with a CHI620D potentiostat. All measurements were carried out in a one-compartment cell under a nitrogen atmosphere, equipped with a glassy-carbon electrode, a platinum counter-electrode, and an Ag/Ag+ reference electrode with a scan rate of 100 mV s−1. The supporting electrolyte was a 0.1 mol L−1 dichloromethane solution of tetrabutylammonium perchlorate (TBAP). All potentials were corrected against Fc/Fc+. CV was measured with a scan rate of 100 mV s−1. Thermogravimetric analysis (TGA) was performed using a Shimadzu DTG 60 instrument at a heating rate of 10 °C min−1 under a nitrogen atmosphere with runs recorded from room temperature to 550 °C.
5,10-Bis(5-(2-hexyldecyl)thiophen-2-yl)dithieno[2,3-d′:2′,3′-d′]benzo[1,2-b:4′,5′-b′]dithiophene (2)
2-(2-Hexyldecyl)thiophene (277 mg, 0.9 mmol) was dissolved in anhydrous THF (10 mL) and cooled to 0 °C under a nitrogen atmosphere. n-BuLi (0.9 mmol, 0.56 mL, 1.60 M in hexane) was added via a syringe. After stirring at 0 °C for 90 min, the mixture was heated to 50 °C for 90 min before being cooled down to room temperature. Compound 1 (100 mg, 0.3 mmol) was added in one portion and the mixture was stirred at 50 °C for 90 min again. Tin(II) chloride (349 mg, 1 mL) in 10% aqueous hydrochloric acid was added after the reaction solution was cooled to room temperature. After stirring overnight, the reaction solution was poured into water and extracted n-hexane for three times. The organic layers were combined and washed with water, dried over MgSO4, and concentrated under reduced pressure. The residue was purified by silica-gel column chromatography to give compound 2 (0.244 g, 89%) as a yellow oil. 1H NMR (400 MHz, CDCl3): δ 7.37 (d, 3J = 5.2 Hz, 2H), 7.23 (d, 3J = 5.1 Hz, 2H), 7.16 (d, 3J = 3.2 Hz, 2H), 6.97 (d, 3J = 3.4 Hz, 2H), 2.91 (d, 3J = 6.7 Hz, 4H), 1.76 (m, 2H), 1.50–1.27 (m, 48H), 0.88 (m, 12H).
2,7-Diiodo-5,10-bis(5-(2-hexyldecyl)thiophen-2-yl)-dithieno[2,3-d′:2′,3′-d′]benzo[1,2-b:4′,5′-b′]dithiophene (3)
Compound 2 (0.244 g, 0.267 mmol) was dissolved in anhydrous THF (10 mL) under a nitrogen atmosphere and cooled to −78 °C, to which n-BuLi (0.35 mL, 0.55 mmol, 1.60 M in hexane) was added. After stirring at −78 °C for 60 min, 1,2-diiodoethane (0.18 g, 0.64 mmol) was added and the mixture was stirred for 30 min. The clear reaction solution was warmed to room temperature for 30 min, poured into water and extracted three times with dichloromethane. The organic layer was separated, dried over MgSO4, and concentrated under reduced pressure. The residue was purified by silica-gel column chromatography to give compound 3 (0.22 mg, 70%) as a yellow solid. 1H NMR (400 MHz, CDCl3): δ 7.38 (m, 2H), 7.13 (d, 3J = 3.4 Hz, 2H), 6.97 (d, 3J = 3.6 Hz, 2H), 2.92 (d, 3J = 6.7 Hz, 4H), 1.77 (m, 2H), 1.50–1.27 (m, 48H), 0.87 (m, 12H).
2,7-Bis(2-(2-ethylhexyl)thieno[3,4-b]thiophen-6-yl)-5,10-bis(5-(2-hexyldecyl)thiophen-2-yl)dithieno[2,3-d′:2′,3′-d′]benzo[1,2-b:4′,5′-b′]dithiophene (4)
To an oven-dried round-bottom flask loaded with compound 3 (0.22 g, 0.19 mmol) in anhydrous DMF (2.5 mL) and toluene (2.5 mL) under a nitrogen atmosphere was added tributyl(2-(2-ethylhexyl)thieno[3,4-b]thiophen-6-yl)stannane (0.271 g, 0.50 mmol) and Pd(PPh3)4 (40 mg, 0.035 mmol). The reaction was stirred and heated to 90 °C for 2 days in the dark. The reaction mixture was poured into water and extracted three times with dichloromethane. The organic layers were separated, dried over MgSO4, and concentrated under reduced pressure. The residue was quickly passed through a silica-gel column and used for the next step without further purification.
4,4′-(5,10-Bis(5-(2-hexyldecyl)thiophen-2-yl)dithieno[2,3-d′:2′,3′-d′]benzo[1,2-b:4′,5′-b′]dithiophene-2,7-diyl)bis(2-ethylhexyl)thieno[3,4-b]thiophene-6-carbaldehyde (5)
Compound 4 (0.208 g, 0.147 mmol) was dissolved in anhydrous THF (10 mL) under a nitrogen atmosphere and cooled to −78 °C, to which n-BuLi (0.24 mL, 0.38 mmol, 1.60 M in hexane) was added. After stirring at −78 °C for 30 min, DMF (0.2 mL) was added and the mixture was stirred at −78 °C for another 30 min. The clear reaction solution was warmed to room temperature for 30 min, then poured into water and extracted three times with CH2Cl2. The organic layers were separated, dried over MgSO4, and concentrated under reduced pressure. The residue was purified by silica-gel column chromatography to give compound 5 (0.11 g, 30%, two step from compound 3) as a red solid. 1H NMR (400 MHz, CDCl3): δ 9.96 (s, 2H), 7.57 (s, 2H), 7.24 (d, 3J = 3.4 Hz, 2H), 7.16 (s, 2H), 7.05 (d, 3J = 3.4 Hz, 2H), 2.97 (d, 3J = 6.6 Hz, 4H), 2.82 (d, 3J = 7.0 Hz, 4H), 1.81 (m, 2H), 1.73 (m, 2H), 1.47–1.22 (m, 64H), 0.93 (m, 24H), 0.84 (m, 24H); 13C NMR (100 MHz, CDCl3) δ 179.8, 159.8, 152.3, 147.8, 143.6, 140.6, 138.9, 136.6, 134.9, 133.7, 133.6, 130.4, 129.0, 126.2, 124.8, 124.3, 118.9, 115.0, 41.0, 40.3, 36.6, 34.8, 33.6, 33.5, 32.7, 32.1, 32.1, 30.2, 29.9, 29.8, 29.5, 29.0, 26.9, 25.8, 23.1, 22.9, 22.8, 14.28, 14.26, 10.9; HRMS (MALDI-TOF) calcd for C84H110O2S10 [M]+: 1470.5707, found 1470.5709.
(5Z,5′Z)-5,5′-((4,4′-(5,10-Bis(5-(2-hexyldecyl)thiophen-2-yl))dithieno[2,3-d′:2′,3′-d′]benzo[1,2-b:4′,5′-b′]dithiophene-2,7-diyl)bis(2-(2-ethylhexyl)thieno[3,4-b]thiophene-4,6-diyl))bis(methanylylidene)bis(3-ethyl-2-thioxothiazolidin-4-one) (STB-4)
A solution of compound 5 (110 mg, 0.074 mmol), β-alanine (2 mg, 0.021 mmol) and 3-ethyl-rhodanine (0.171 g, 1.06 mmol) in ethanol (8 mL) and 1,2-dichloroethane (10 mL) was stirred at 75 °C for 3 days. The solvent was evaporated and the residue was purified by silica-gel column chromatography using dichloromethane as the eluent. The crude product was recrystallized from ethanol and 1,2-dichloroethane three times to afford STB-4 (0.109 g, 84%) as a black solid. 1H NMR (400 MHz, CDCl3): δ 7.79 (s, 2H), 7.34 (d, 3J = 3.2 Hz, 2H), 7.12 (s, 2H), 7.11 (d, 3J = 3.2 Hz, 2H), 6.85 (s, 2H), 4.05 (m, 4H), 3.03 (d, 3J = 6.4 Hz, 4H), 2.80 (m, 4H), 1.86 (m, 2H), 1.74 (m, 2H), 1.55–1.15 (m, 64H), 1.03–0.93 (m, 12H), 0.87–0.78 (m, 12H); 13C NMR (100 MHz, CDCl3): δ 191.1, 166.8, 157.5, 152.2, 147.3, 143.1, 140.3, 138.4, 135.8, 134.3, 133.5, 131.5, 129.6, 129.2, 125.7, 123.3, 122.5, 122.0, 117.5, 117.0, 114.5, 40.8, 40.0, 39.7, 36.4, 34.5, 33.3, 32.6, 31.9, 31.9, 30.1, 29.7, 29.7, 29.3, 28.9, 26.7, 25.6, 23.0, 22.6, 22.6, 14.1, 14.0, 12.2, 10.8; MS (MALDI-TOF): m/z = 1758.7 [M]+; anal. calcd for C94H120N2O2S14: C, 64.19; H, 6.88; N, 1.59; found: C, 64.15; H, 6.96; N, 1.69.
Solar cell fabrication and characterization
Indium tin oxide (ITO)-coated glass substrates were cleaned by ultrasonic treatment with detergent, deionized water, acetone, and ethanol for 20 min each, and were then UV/ozone treated. A thin layer of PEDOT:PSS (Clevios P VP Al 4083) was spin-coated at 3000 r.p.m. for 30 s and then baked at 150 °C for 10 min. The substrates were then transferred to a nitrogen-filled glove box. A solution of STB-4 and PC71BM (25 mg mL−1) in chloroform was spin-coated at different spin rates on the top of the substrate. The films were solvent vapor annealed by placing the substrates in a glass Petri dish containing 100 μL of chloroform. An 80 nm thin layer of Al was then deposited under high vacuum. The active area of the solar cells was 3.08 mm2 as defined by shadow masks. The J–V curves were measured with a Keithley 2400 source meter. The AM 1.5G solar irradiation was simulated by using a 300 W xenon arc solar simulator (Oriel) under 100 mW cm−2. The illumination intensity was calibrated with a standard Si solar cell. The external quantum efficiency (EQE) was obtained with IPCE equipment (XES-70S1).
SCLC mobility measurements were tested in electron-only devices with the device structures of glass/Al/active layer/Al and hole-only device structure of ITO/PEDOT:PSS/active layer/Au. The mobilities were calculated by fitting the SCLC model: 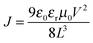 , where J is the current, ε0 is the zero-field mobility, εr is the permittivity of free space, μ0 is the relative permittivity of the material, L is the thickness of the active layer, and V is the effective voltage in the devices.
, where J is the current, ε0 is the zero-field mobility, εr is the permittivity of free space, μ0 is the relative permittivity of the material, L is the thickness of the active layer, and V is the effective voltage in the devices.
AFM and TEM characterization
Atomic force microscopy investigation was performed using a Nanoscopy IIIa in “tapping mode”. The transmission electron microscope investigation was performed on a JEOL JEM2010FIF operated at 200 kV. The specimen for TEM measurement was prepared by spin casting the blend solution on ITO/PEDOT:PSS substrates, then floating the film on a water surface, and transferring to TEM grids.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We thank the National Basic Research Program of China (973 Program) (No. 2014CB643502) for financial support, the Strategic Priority Research Program of the Chinese Academy of Sciences (XDB12010200), and the National Natural Science Foundation of China (91333113 and 21572234).
Notes and references
- L. Lu, T. Zheng, Q. Wu, A. M. Schneider, D. Zhao and L. Yu, Chem. Rev., 2015, 115, 12666–12731 CrossRef CAS PubMed.
- L. Dou, J. You, Z. Hong, Z. Xu, G. Li, R. A. Street and Y. Yang, Adv. Mater., 2013, 25, 6642–6671 CrossRef CAS PubMed.
-
C. Brabec and U. S. A. V. Dyakonov, Organic Photovoltaics: Materials, Device Physics, and Manufacturing Technologies, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2nd edn, 2014 Search PubMed.
- J. Zhao, Y. Li, G. Yang, K. Jiang, H. Lin, H. Ade, W. Ma and H. Yan, Nat. Energy, 2016, 1, 15027 CrossRef CAS.
- W. Zhao, S. Li, S. Zhang, X. Liu and J. Hou, Adv. Mater., 2017, 29, 1604059 CrossRef PubMed.
- F. Zhao, S. Dai, Y. Wu, Q. Zhang, J. Wang, L. Jiang, Q. Ling, Z. Wei, W. Ma, W. You, C. Wang and X. Zhan, Adv. Mater., 2017, 29, 1700144 CrossRef PubMed.
- L. Ye, W. Zhao, S. Li, S. Mukherjee, J. H. Carpenter, O. Awartani, X. Jiao, J. Hou and H. Ade, Adv. Energy Mater., 2017, 7, 1602000 CrossRef.
- S. Li, L. Ye, W. Zhao, S. Zhang, S. Mukherjee, H. Ade and J. Hou, Adv. Mater., 2016, 28, 9423–9429 CrossRef CAS PubMed.
- J. Wan, X. Xu, G. Zhang, Y. Li, K. Feng and Q. Peng, Energy Environ. Sci., 2017, 10, 1739–1745 CAS.
- D. Deng, Y. Zhang, J. Zhang, Z. Wang, L. Zhu, J. Fang, B. Xia, Z. Wang, K. Lu, W. Ma and Z. Wei, Nat. Commun., 2016, 7, 13740 CrossRef CAS PubMed.
- Y. Lin, Y. Li and X. Zhan, Chem. Soc. Rev., 2012, 41, 4245 RSC.
- Y. Lin and X. Zhan, Acc. Chem. Res., 2016, 49, 175 CrossRef CAS PubMed.
- H. Shang, H. Fan, Y. Liu, W. Hu, Y. Li and X. Zhan, Adv. Mater., 2011, 23, 1554 CrossRef CAS PubMed.
- Q. Zhang, B. Kan, F. Liu, G. Long, X. Wan, X. Chen, Y. Zuo, W. Ni, H. Zhang, M. Li, Z. Hu, F. Huang, Y. Cao, Z. Liang, M. Zhang, T. P. Russell and Y. Chen, Nat. Photonics, 2014, 9, 35–41 CrossRef.
- L. Yuan, K. Lu, B. Xia, J. Zhang, Z. Wang, Z. Wang, D. Deng, J. Fang, L. Zhu and Z. Wei, Adv. Mater., 2016, 28, 5980–5985 CrossRef CAS PubMed.
- H. Yao, L. Ye, J. Hou, B. Jang, G. Han, Y. Cui, G. M. Su, C. Wang, B. Gao, R. Yu, H. Zhang, Y. Yi, H. Y. Woo, H. Ade and J. Hou, Adv. Mater., 2017, 29, 1700254 CrossRef PubMed.
- H. Yao, Y. Cui, R. Yu, B. Gao, H. Zhang and J. Hou, Angew. Chem., Int. Ed., 2017, 56, 3045–3049 CrossRef CAS PubMed.
- J. L. Wang, K. K. Liu, J. Yan, Z. Wu, F. Liu, F. Xiao, Z. F. Chang, H. B. Wu, Y. Cao and T. P. Russell, J. Am. Chem. Soc., 2016, 138, 7687–7697 CrossRef CAS PubMed.
- Y. Lin, F. Zhao, Q. He, L. Huo, Y. Wu, T. C. Parker, W. Ma, Y. Sun, C. Wang, D. Zhu, A. J. Heeger, S. R. Marder and X. Zhan, J. Am. Chem. Soc., 2016, 138, 4955–4961 CrossRef CAS PubMed.
- B. Kan, H. Feng, X. Wan, F. Liu, X. Ke, Y. Wang, Y. Wang, H. Zhang, C. Li, J. Hou and Y. Chen, J. Am. Chem. Soc., 2017, 139, 4929–4934 CrossRef CAS PubMed.
- K. Gao, J. Miao, L. Xiao, W. Deng, Y. Kan, T. Liang, C. Wang, F. Huang, J. Peng, Y. Cao, F. Liu, T. P. Russell, H. Wu and X. Peng, Adv. Mater., 2016, 28, 4727–4733 CrossRef CAS PubMed.
- S. Xu, Z. Zhou, H. Fan, L. Ren, F. Liu, X. Zhu and T. P. Russell, J. Mater. Chem. A, 2016, 4, 17354–17362 CAS.
- Y. Sun, G. C. Welch, W. L. Leong, C. J. Takacs, G. C. Bazan and A. J. Heeger, Nat. Mater., 2011, 11, 44–48 CrossRef PubMed.
- B. Kan, Q. Zhang, M. Li, X. Wan, W. Ni, G. Long, Y. Wang, X. Yang, H. Feng and Y. Chen, J. Am. Chem. Soc., 2014, 136, 15529–15532 CrossRef CAS PubMed.
- H. Yao, L. Ye, H. Zhang, S. Li, S. Zhang and J. Hou, Chem. Rev., 2016, 116, 7397–7457 CrossRef CAS PubMed.
- M. Li, F. Liu, X. Wan, W. Ni, B. Kan, H. Feng, Q. Zhang, X. Yang, Y. Wang, Y. Zhang, Y. Shen, T. P. Russell and Y. Chen, Adv. Mater., 2015, 27, 6296–6302 CrossRef CAS PubMed.
- Z. Zhou, S. Xu, W. Liu, C. Zhang, Q. Hu, F. Liu, T. P. Russell and X. Zhu, J. Mater. Chem. A, 2017, 5, 3425–3433 CAS.
- C. J. Takacs, Y. Sun, G. C. Welch, L. A. Perez, X. Liu, W. Wen, G. C. Bazan and A. J. Heeger, J. Am. Chem. Soc., 2012, 134, 16597–16606 CrossRef CAS PubMed.
- Y. Wu, Z. Li, X. Guo, H. Fan, L. Huo and J. Hou, J. Mater. Chem., 2012, 22, 21362 RSC.
- H. J. Son, L. Lu, W. Chen, T. Xu, T. Zheng, B. Carsten, J. Strzalka, S. B. Darling, L. X. Chen and L. Yu, Adv. Mater., 2013, 25, 838–843 CrossRef CAS PubMed.
- Y. Wu, Z. Li, W. Ma, Y. Huang, L. Huo, X. Guo, M. Zhang, H. Ade and J. Hou, Adv. Mater., 2013, 25, 3449–3455 CrossRef CAS PubMed.
- H. J. Yun, Y. J. Lee, S. J. Yoo, D. S. Chung, Y. H. Kim and S. K. Kwon, Chem. – Eur. J., 2013, 19, 13242–13248 CrossRef CAS PubMed.
- Y. R. Cheon, Y. J. Kim, J. Y. Back, T. k. An, C. E. Park and Y.-H. Kim, J. Mater. Chem. A, 2014, 2, 16443–16451 CAS.
- T. I. Ryu, Y. Yoon, J.-H. Kim, D.-H. Hwang, M. J. Ko, D.-K. Lee, J. Y. Kim, H. Kim, N.-G. Park, B. Kim and H. J. Son, Macromolecules, 2014, 47, 6270–6280 CrossRef CAS.
- S. Sun, P. Zhang, J. Li, Y. Li, J. Wang, S. Zhang, Y. Xia, X. Meng, D. Fan and J. Chu, J. Mater. Chem. A, 2014, 2, 15316 CAS.
- T. Zheng, L. Lu, N. E. Jackson, S. J. Lou, L. X. Chen and L. Yu, Macromolecules, 2014, 47, 6252–6259 CrossRef CAS.
- L. Huo, T. Liu, X. Sun, Y. Cai, A. J. Heeger and Y. Sun, Adv. Mater., 2015, 27, 2938–2944 CrossRef CAS PubMed.
- M. Jung, D. Seo, K. Kwak, A. Kim, W. Cha, H. Kim, Y. Yoon, M. J. Ko, D.-K. Lee, J. Y. Kim, H. J. Son and B. Kim, Dyes Pigm., 2015, 115, 23–34 CrossRef CAS.
- Y. J. Kim, M. J. Kim, T. K. An, Y. H. Kim and C. E. Park, Chem. Commun., 2015, 51, 11572–11575 RSC.
- H. S. Lee, H. G. Song, H. Jung, M. H. Kim, C. Cho, J.-Y. Lee, S. Park, H. J. Son, H.-J. Yun, S.-K. Kwon, Y.-H. Kim and B. Kim, Macromolecules, 2016, 49, 7844–7856 CrossRef CAS.
- Y. S. Lee, S. Song, Y. J. Yoon, Y.-J. Lee, S.-K. Kwon, J. Y. Kim and Y.-H. Kim, J. Polym. Sci., Part A: Polym. Chem., 2016, 54, 3182–3192 CrossRef CAS.
- W. Zhong, J. Xiao, S. Sun, X.-F. Jiang, L. Lan, L. Ying, W. Yang, H.-L. Yip, F. Huang and Y. Cao, J. Mater. Chem. C, 2016, 4, 4719–4727 RSC.
- H. Feng, M. Li, W. Ni, B. Kan, Y. Wang, Y. Zhang, H. Zhang, X. Wan and Y. Chen, Sci. China: Chem., 2017, 60, 552–560 CrossRef CAS.
- H. Bai, Y. Wang, P. Cheng, Y. Li, D. Zhu and X. Zhan, ACS Appl. Mater. Interfaces, 2014, 6, 8426 CAS.
- C. Zhang and X. Zhu, Acc. Chem. Res., 2017, 50, 1342–1350 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Details on the physical properties of STB-4, SCLC mobility plot and NMR spectra of all new compounds. See DOI: 10.1039/c7qm00291b |
|
| This journal is © the Partner Organisations 2017 |

 *ab
*ab





 , where J is the current, ε0 is the zero-field mobility, εr is the permittivity of free space, μ0 is the relative permittivity of the material, L is the thickness of the active layer, and V is the effective voltage in the devices.
, where J is the current, ε0 is the zero-field mobility, εr is the permittivity of free space, μ0 is the relative permittivity of the material, L is the thickness of the active layer, and V is the effective voltage in the devices.