
Synergetic regulation of CO2 and light for controllable inversion of Pickering emulsions
Yuanyuan
Cao
,
Zhen
Wang
,
Shiming
Zhang
and
Yapei
Wang
 *
*
Department of Chemistry, Renmin University of China, Beijing, 100872, China. E-mail: yapeiwang@ruc.edu.cn
First published on 31st July 2017
Abstract
Manipulating inversion of Pickering emulsions via “green” and “erasable” external stimuli is at the forefront of developing emulsion-templated materials on account of no chemical addition and convenience. Herein, a type of synergetic CO2- and light-triggered Pickering emulsion was constructed using a special emulsifier formulated by poly[2-(diethylamino)ethyl methacrylate] (PDEAEMA)-grafted-polydopamine (PDA) nanoparticles. Taking advantage of the remarkable photothermal-conversion ability of PDA and tunable amphiphilicity of PDEAEMA, water-in-oil (W/O) emulsions stabilized by PDA–PDEAEMA particles could be converted into oil-in-water (O/W) types upon CO2 treatment, and then switched between the W/O type and O/W type reversibly by turning light irradiation on and off. Such controllable emulsion inversion through synergetic CO2 and light stimuli could be applied in temperature-dependent heterogeneous catalysis and separation.
1. Introduction
As an alternative to conventional surfactant-stabilized emulsion systems, Pickering emulsions that are stabilized by solid particles as interfacial emulsifiers have attracted enormous attention due to the surfactant-free character and enhanced emulsion stability.1,2 Therefore, the areas in which Pickering emulsions can be applied have been expanded to certain cosmetic and pharmaceutical industries in which conventional surfactants might cause inevitable adverse impacts, such as tissue irritation and cell damage.3,4 The long-term stability against coalescence of Pickering emulsions ensures the preparation of multitudinous micrometer-sized or even millimeter-sized emulsions which are difficult to construct using conventional surfactants. However, the stabilized emulsion systems need to be demulsified in some cases of oil recovery,5,6 heterogeneous catalysis,7–9 and emulsion polymerization.10,11 How to manipulate emulsion stability as well as phase inversion on demand is a critical bottleneck that restricts the practical applications of Pickering emulsions.Regulating the amphiphilicity of nanoparticle-based emulsifiers by changing their surface wettability has become the most convenient and effective method to construct controllable Pickering emulsions. Molecular adsorption and chemical grafting of stimuli-responsive small molecules or polymers on a solid particle surface could provide it with tunable hydrophilic/hydrophobic properties.12,13 Chemical addition, such as acids, bases or salts, have been applied to alter particle amphiphilicity through changing the environmental pH or ionic strength.14–17 However, these regulation conditions are difficult to remove and may lead to contamination by chemical residues. In this regard, developing “green” and “erasable” external stimuli is highly desirable. CO2 is an attractive stimulus owing to its excellent advantages of low cost, good biocompatibility and complete erasability.18–20 Near infrared light (NIR) is another type of neat and non-invasive external stimulus. More importantly, the light energy could be converted to thermal energy with the assistance of photothermal conversion materials.21–28 This highly efficient pathway of energy conversion might be the perfect way to increase the temperature of the system by increasing regional selectivity and negligible functional damage.29,30 This light-actuated microheater could serve as a supplier of thermal power to tune the amphiphilicity of thermoresponsive emulsifiers in a “remote control” strategy. Integration of these two triggers is believed to regulate the Pickering emulsion in a more convenient way. Nevertheless, construction of CO2 and NIR dual-responsive emulsifiers using this simple (yet extremely versatile) approach is challenging.
Herein, a synergetic responsive Pickering emulsifier was designed through grafting poly[2-(diethylamino)ethyl methacrylate] (PDEAEMA) onto a polydopamine (PDA) particle surface. The internal PDA core served as a thermal reservoir that could supply thermal energy because of its remarkable ability for photothermal conversion. The grafted PDEAEMA chain acted as the CO2 and temperature responder due to its CO2- and thermo-responsive features. Consequently, the stability and types of Pickering emulsion were successfully regulated by tuning the amphiphilicity of PDA–PDEAEMA under the synergetic stimuli of CO2 and NIR light.
2. Experimental
2.1 Chemicals and materials
Dopamine hydrochloride (DA, >98% purity) was purchased from Sangon Biothech Company. Trometamol (Tris, 99.5%) was obtained from LanYi Reagent Company. 2-Bromoisobutyryl bromide (BIBB, 98%), N,N-diethylaminoethyl methacrylate (DEAEMA, 99.5%) and copper(I) bromide (CuBr, 98%) were supplied by J&K Scientific. N,N,N′,N′′,N′′-Pentamethylene-diethylenetriamine (PMDETA, 99%), triethylamine (TEA, 99%), N,N-dimethylformamide (DMF), ethanol (EtOH, 95%), ethyl acetate (EA) and Nile red were provided by Beijing Chemical Reagent Company.2.2 Characterization methods
Fourier transform infrared (FTIR) spectra were measured using a Bruker Tensor 27 spectrometer with a resolution of 4 cm−1 and a scanning number of 64 from 400 to 4000 cm−1. The thermal stability of PDA, PDEAEMA and PDA–PDEAEMA were tested on Thermogravimetric Analyzer Q50 (TA Instrument Company). Temperature-dependent 1H NMR and 13C NMR were done on a Bruker 500 MHz spectrometer. The diameter and zeta potential of PDA–PDEAEMA particles were measured by a Zetasizer Nano ZS instrument (Malvern Instruments). The temperature range of temperature-dependent dynamic light scattering (DLS) measurements was varied from 25 °C to 70 °C at an interval of 5 °C. An 808 nm laser (HTOE) was used as the NIR light source with spot diameter of ≈8 mm. A thermometer was employed to monitor the temperature change in the solution under irradiation with a 808 nm laser with different power settings. The macroscopic images of PDA–PDEAEMA solubility in ethyl acetate and water were recorded by a commercial digital SLR camera (D7100; Nikon) and the emulsion droplets were investigated by a fluorescence microscope (Axio Scope A1; Zeiss).2.3 Synthesis of PDA–Br initiator and PDA–PDEAEMA particles
DA (1.0 g, 5.27 mmol), TEA (375.5 μL, 2.71 mmol) and redistilled DMF (20 mL) were mixed in a dried 250 mL three-necked flask. Under N2 protection, 2-bromoisobutyryl bromide (BIBB; 325 μL, 2.63 mmol) was added dropwise into the flask. After stirring for 2 h at room temperature, Tris (3.5 g) and deionized water (50 mL) were added, and the mixture was stirred at room temperature for polymerization. The yielded PDA–Br particles were separated and purified by centrifugation to remove the unreacted DA monomer.The PDA–Br initiator (40 mg), DEAEMA monomer (17 mL) and DMF (3 mL) were added into a 50 mL Schlenk flask. After three freeze–pump–thaw cycles, CuBr (30.24 mg, dissolved in DMF solution) and PMDETA (44 μL) were added through a gas-tight syringe. The flask was then placed in an oil bath at 65 °C for 15 h. Then, the polymerization reaction was quenched by exposing the reaction flask to air. The reaction mixture was washed with ethanol several times to remove the CuBr catalyst and unreacted DEAEMA monomer. Finally, the PDA–PDEAEMA particles were dissolved in ethyl acetate solution.
2.4 Preparation and regulation of Pickering emulsion
The ethyl acetate phase with dispersed PDA–PDEAEMA emulsifier (1.5 mg mL−1, 200 μL) was mixed with 100 μL of water. After vortex vibration at 1500 rpm for 30 s, the emulsion was formed and observed by fluorescence microscopy with Nile red as the indicator. The obtained emulsion was then bubbled with CO2 for 1 min to induce inversion of the emulsion. Light controlled-emulsion regulation was conducted by illuminating the emulsion with NIR light at 2.0 W for 1 min and the solution temperature was recorded by a thermometer.2.5 Calculation of the grafting weight ratio of PDEAEMA on PDA
As indicated in Fig. 2b, pure PDA and PDEAEMA exhibited 51.6% and 97.5% weight loss, respectively, from room temperature to 800 °C. For PDA–PDEAEMA particles, the weight loss was 77.9%. Then, the weight fraction of PDEAEMA grafted onto the PDA surface could be calculated using the following equation:| 97.5%mPDEAEMA + 51.6%mPDA = 77.9%mPDA–PDEAEMA | (1) |
| mPDEAEMA + mPDA = mPDA–PDEAEMA | (2) |
3. Results and discussion
The dual responsive PDEAEMA-grafted-PDA nanoparticles as a type of smart Pickering emulsifier were synthesized through atom transfer radical polymerization (ATRP) (Fig. 1). In particular, the ATRP initiator (PDA–Br) was synthesized through bio-inspired PDA chemistry.31 Due to the presence of hydroxyl and amine groups in the dopamine (DA) monomer, DA can be readily modified with other functional groups. Herein, BIBB was first reacted with the DA monomer in TEA before polymerization into PDA. The molar ratio of BIBB to DA monomer was fixed at 0.5 to introduce a certain amount of DA–BIBB while leaving some unreacted DA monomer to ensure polymerization. After this reaction had proceeded with N2 protection for 2 h, Tris buffer solution was added to activate the polymerization reaction with the pH fixed at 8.5. The one-pot synthesis of PDA–Br initiator was environmentally friendly because the synthesis was carried out in aqueous solution at room temperature. The obtained PDA–Br initiator was then dispersed in DEAEMA monomer with DMF as the solvent and CuBr/PMDETA as the catalyst. After ATRP polymerization at 65 °C for 15 h, the PDA–PDEAEMA products were acquired and purified through a centrifugal washing procedure.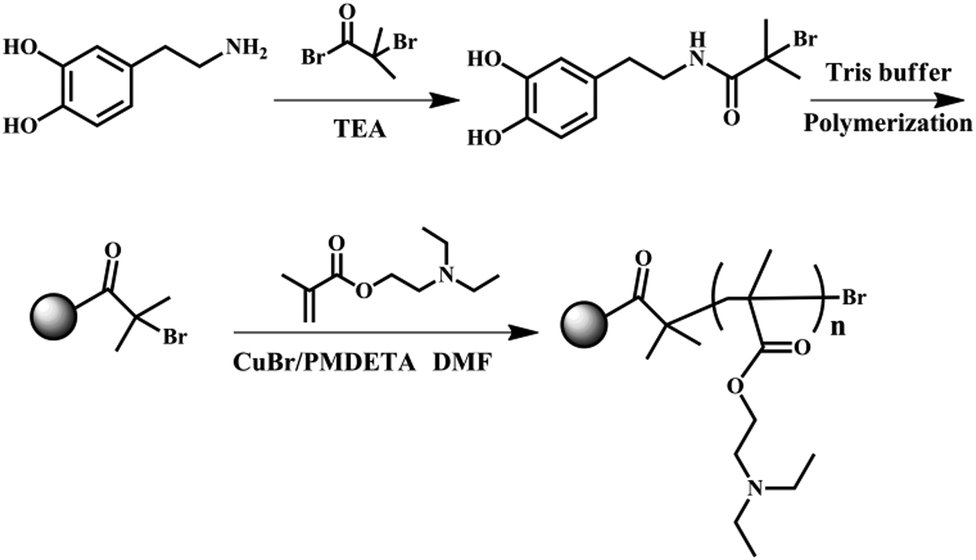 | ||
| Fig. 1 Synthetic procedure of PDA–PDEAEMA nanoparticles by atom transfer radical polymerization (ATRP) (schematic). | ||
FTIR spectroscopy was adopted to confirm the existence of PDEAEMA on the PDA surface (Fig. 2a). Compared with PDA, PDA–Br and PDA–PDEAEMA appeared a new peak at ≈2900 cm−1, which corresponded to the stretching vibration of methyl groups. New peaks at 1660 cm−1 and 1408 cm−1 assigned to the stretching vibration of the amide I band demonstrated the formation of the PDA–Br initiator. The stretching vibration of the ester carbonyl peak at 1732 cm−1 was also observable in PDA–PDEAEMA.32,33 These representative characteristic peaks indicated that PDEAEMA was grafted on the PDA surface. The grafting weight ratio of PDEAEMA was confirmed further by thermogravimetric analysis (TGA). As shown in Fig. 2b, the weight of PDA decreased gradually with an increase in temperature, whereas the TGA curve of PDEAEMA homopolymer exhibited a maximum thermal decomposition temperature at about 364.8 °C and 445.5 °C during the degradation process. The degradation curve of PDA–PDEAEMA showed the same trend as that of the PDEAEMA homopolymer but the maximum thermal decomposition temperature was slightly different (about 283.5 °C and 415.2 °C). According to the weight loss from room temperature to 800 °C, the weight fraction of PDEAEMA grafted onto the PDA surface was calculated to be 57.3% and the detailed calculation process is provided in the experimental section.
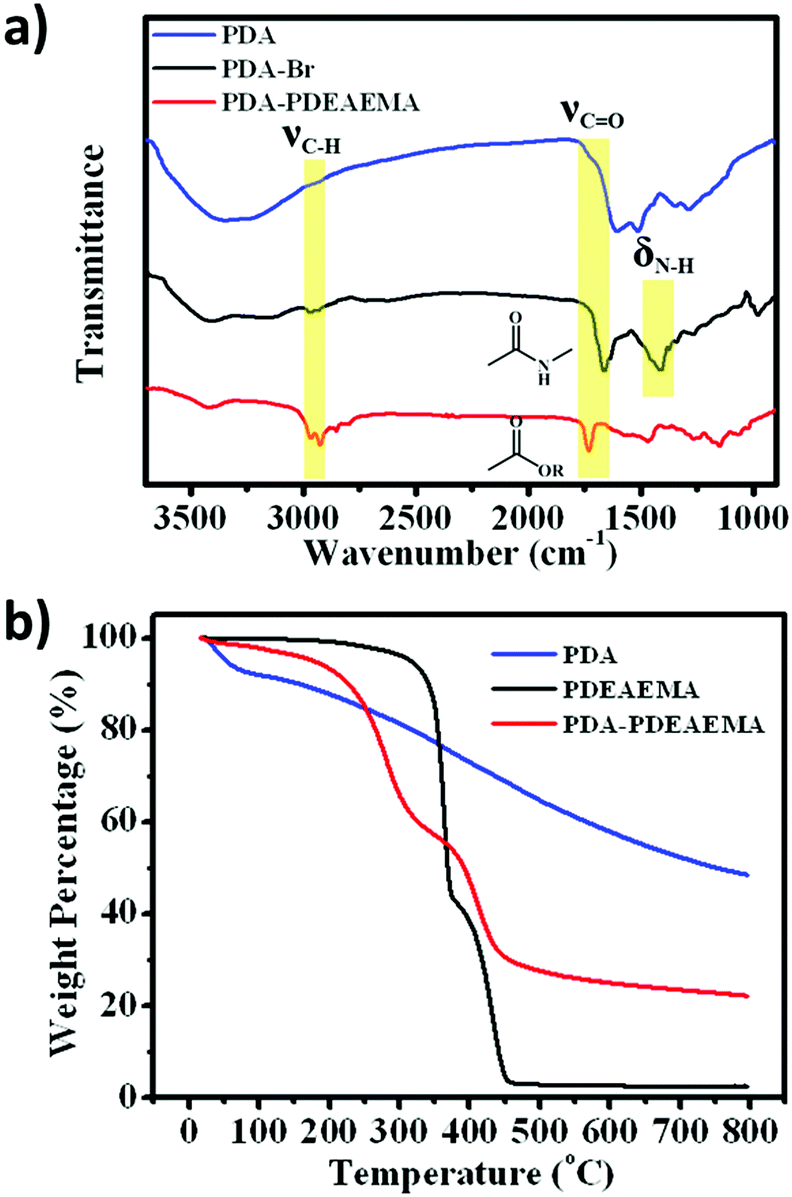 | ||
| Fig. 2 (a) FTIR spectra of PDA, PDA–Br and PDA–PDEAEMA. (b) TGA curves of PDA, PDEAEMA and PDA–PDEAEMA from room temperature to 800 °C. | ||
Owing to the insolubility of the resultant PDA–PDEAEMA particles in most deuterated solvents, the 1H NMR of PDA–PDEAEMA was recorded in a D2O system through bubbling with CO2 because of the hydrophilic nature of PDA–PDEAEMA after CO2 treatment (the detailed expression is noted in the following section). The characteristic chemical shifts of PDEAEMA chain were very discernible and the proton signals at 3.32, 3.03, 2.11, 1.43 and 0.97 ppm were assigned to –NCH2CH3, –OCH2CH2–, –CH2C–, –NCH2CH3 and –CH3C–, which are labelled as f, e, b, g and c, respectively, in Fig. 3. The integration area of individual peaks was in accordance with their stoichiometric ratio. Simultaneously, the proton signal in the 2-bromoisobutyryl ester moiety was also detectable (labelled as a). Taken together, these results confirmed the attachment of PDEAEMA onto the PDA backbone.
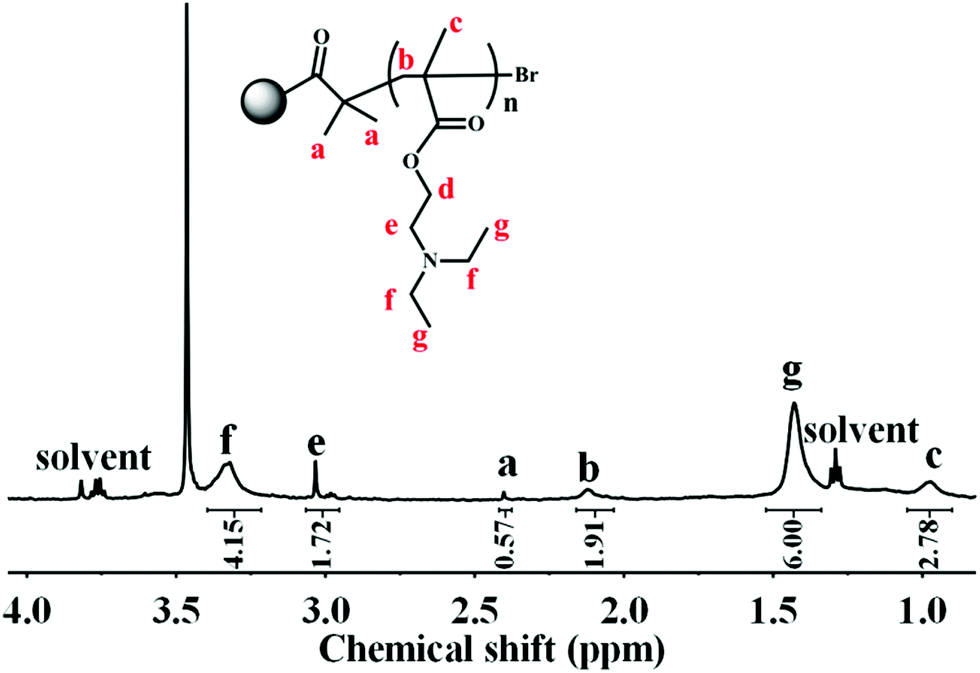 | ||
| Fig. 3 1H NMR of PDA–PDEAEMA particles dissolved in D2O after bubbling with CO2. | ||
CO2 response is a unique feature of PDEAEMA due to the presence of the diethylamino group. Under CO2 treatment, the diethylamino groups are protonated and, thus, PDEAEMA is converted to a hydrophilic cationic polymer. Moreover, such a hydrophilic polymer quickly switches to a hydrophobic polymer upon removal of the CO2 stimulus by bubbling with N2.34–36 By analogy, the synthesized PDA–PDEAEMA particles might also be responsive to CO2 on account of attachment of the PDEAEMA chain. Therefore, the variation in the solubility and zeta potential of PDA–PDEAEMA particles was investigated systematically through alternating the CO2 and N2 triggers. As expected, the original PDA–PDEAEMA particles were hydrophobic and insoluble in water, but they could disperse in an organic solvent such as ethyl acetate (Fig. 4). Mixing the ethyl acetate phase containing PDA–PDEAEMA particles and Nile red indicator with an equal volume of water, the PDA–PDEAEMA particles remained dissolved in ethyl acetate and the aqueous phase was colourless. However, upon treatment with CO2 for 30 s, the PDA–PDEAEMA particles became hydrophilic and were transferred completely from the ethyl acetate phase to the water phase. As a result, the aqueous phase became brown and the ethyl acetate phase preserved the colour of Nile red.
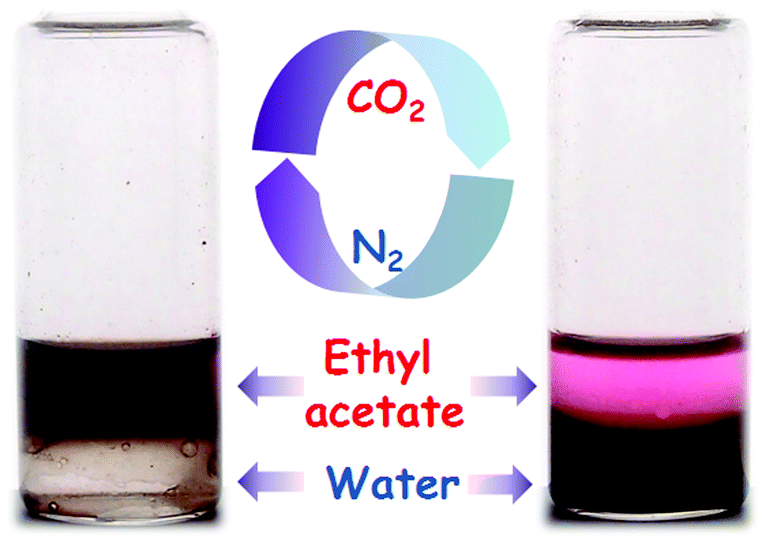 | ||
| Fig. 4 Solubility variation of PDA–PDEAEMA particles in ethyl acetate and water through CO2 or N2 triggers. | ||
Apart from visual observations, measurement of the zeta potential also provided powerful evidence to confirm the change in amphiphilicity under CO2 or N2 triggers. The original PDA–PDEAEMA particles were insoluble in water and the zeta potential was ≈13.5 mV, indicating that the PDA–PDEAEMA particles were unstable and prone to aggregation in water. The zeta potential increased dramatically to 51.5 mV upon purging with CO2 due to the protonation of the PDEAEMA chain. The protonated particles quickly became deprotonated upon the removal of bound CO2 by purging with N2, and correspondingly the zeta potential decreased to 14.0 mV once again (Fig. 5). The repeatable cycles of zeta potential indicated the reversible protonation and deprotonation behaviour of PDEAEMA chain through alternative bubbling of CO2 or N2.
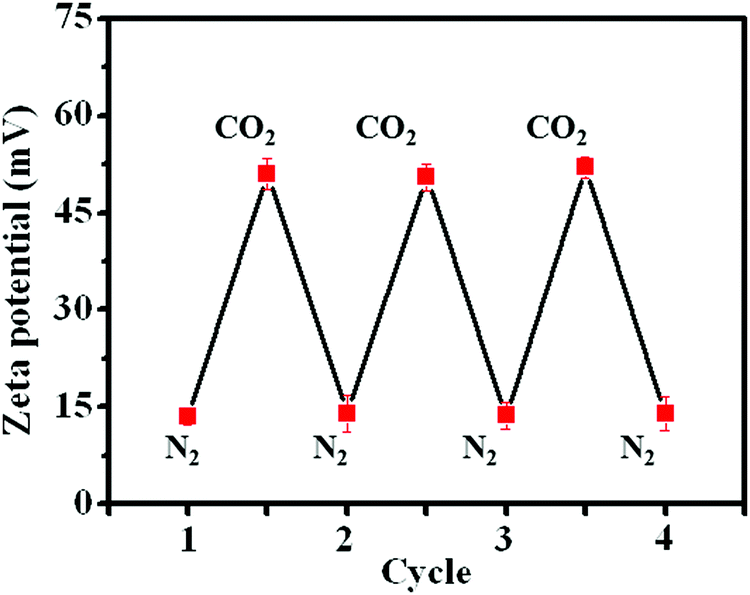 | ||
| Fig. 5 Measurement of the zeta potential of PDA–PDEAEMA particles dissolved in water through alternative bubbling of CO2 or N2. | ||
In addition to the CO2 response, PDEAEMA was sensitive to temperature. The transformation from hydrophilicity to hydrophobicity occurs if the temperature reaches above the lower critical solution temperature (LCST).37,38 To elucidate the thermoresponsive behaviour of PDA–PDEAEMA particles, temperature-dependent DLS was done to characterize the hydrodynamic diameter of PDA–PDEAEMA particles under different temperatures. CO2 was preferentially bubbled due to the poor solubility of PDA–PDEAEMA particles in water. As summarized in Fig. 6, the hydrodynamic diameter displayed a slight change from room temperature to 55 °C, whereas it decreased sharply to 310 nm upon heating to 70 °C, demonstrating that the LCST of PDA–PDEAEMA particles was near 55 °C and that the PDEAEMA chain started to shrink once the temperature reached 55 °C. To analyze the shrinkage behavior at the molecular level, temperature-dependent 1H NMR was employed, and Fig. 7 exhibits the changes in peak intensities at different temperatures. The peak intensities corresponding to e, f and g in the PDEAEMA moiety were attenuated significantly along with the temperature increase, indicating the poor solubility of the PDEAEMA chain in D2O at high temperature. In accordance with temperature-dependent DLS measurements, all these results revealed the thermoresponsive property of PDA–PDEAEMA particles.
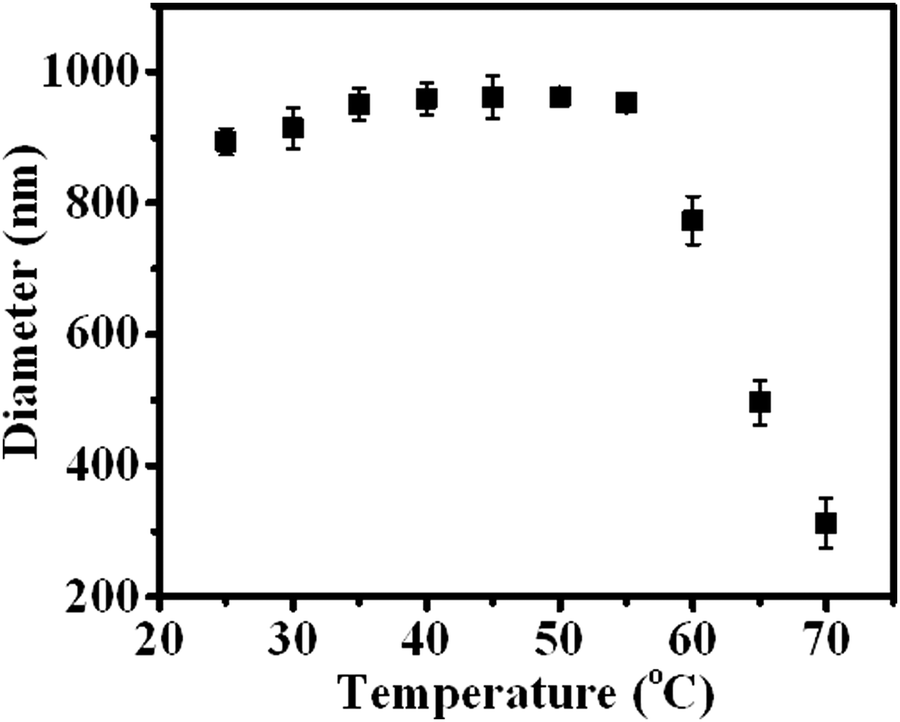 | ||
| Fig. 6 Temperature-dependent DLS measurement of PDA–PDEAEMA particles dissolved in water with CO2 bubbling. | ||
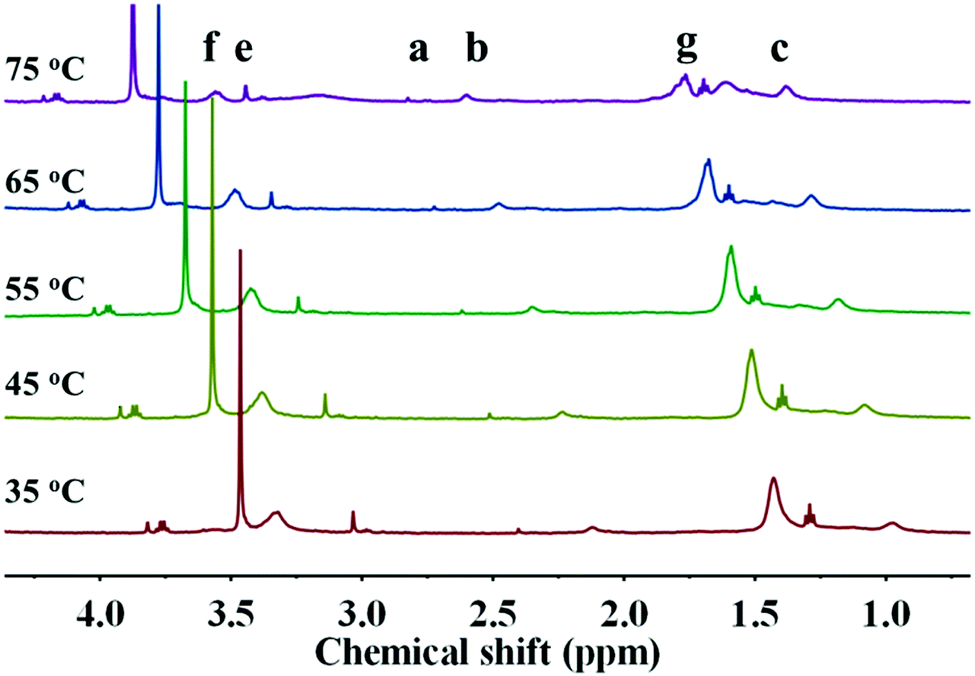 | ||
| Fig. 7 Temperature-dependent 1H NMR measurement of PDA–PDEAEMA particles dissolved in D2O with CO2 bubbling. | ||
After demonstrating the excellent CO2 and temperature dual-responsive performance of PDEAEMA segments, the outstanding ability of photothermal conversion of PDA segments was investigated. PDA displayed a broad absorption band ranging from ultraviolet to the NIR region, which enabled it to convert NIR light energy to thermal energy effectively.39 To ascertain if PDA–PDEAEMA particles are responsive to NIR light, the temperature change of oil-in-water (O/W) emulsions stabilized by PDA–PDEAEMA particles (bubbling with CO2) under 808 nm irradiation was evaluated at different light power. As shown in Fig. 8, the emulsion temperature increased dramatically under exposure to an 808 nm laser and the equilibrium temperature increased from 51.9 °C to 71.6 °C, which was nearly proportional to the laser power. In contrast, there was a slight temperature increase when irradiating the solution without PDA–PDEAEMA particles under 1.2 W power. The excellent ability for photothermal conversion of PDA–PDEAEMA particles enables it to be a perfect thermal reservoir to realize light-controlled inversion of Pickering emulsions.
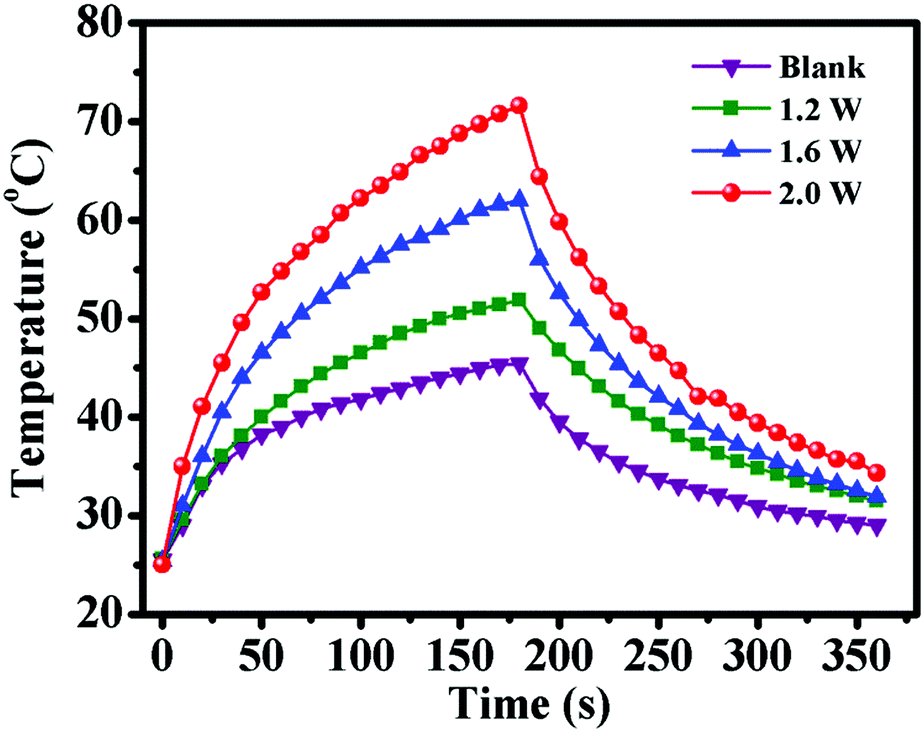 | ||
| Fig. 8 Temperature increase and decrease of O/W emulsions stabilized by PDA–PDEAEMA particles (bubbling with CO2) through alternative turning on and off of an 808 nm laser (3 min) under different power. | ||
By integrating the dual-responsive feature of PDEAEMA and remarkable ability for photothermal conversion of PDA, a controllable Pickering emulsion was constructed via synergetic regulation of CO2 and light. The ethyl acetate phase consisting of PDA–PDEAEMA particles was selected as the oil phase and the oil![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :water ratio was fixed at 2:1. A stable W/O emulsion was formed through vortex vibration at 1500 rpm for 30 s. Fluorescence microscopy revealed the formation of a W/O emulsion by probing the oil phase with Nile red (Fig. 9a). Interestingly, the W/O emulsion was converted to an O/W emulsion after bubbling CO2 for only 1 min. This rapid phase inversion was attributed to the amphiphilicity transformation of PDA–PDEAEMA emulsifier under a CO2 stimulus. As discussed above, the original PDA–PDEAEMA particles were hydrophobic and oil-wetted, which is beneficial for the stabilization of W/O emulsions according to the Bancroft rule. Importing CO2 to the system switched the PDA–PDEAEMA emulsifier to become protonated, hydrophilic and water-wetted, thereby resulting in the phase inversion from a W/O type to an O/W type.
:water ratio was fixed at 2:1. A stable W/O emulsion was formed through vortex vibration at 1500 rpm for 30 s. Fluorescence microscopy revealed the formation of a W/O emulsion by probing the oil phase with Nile red (Fig. 9a). Interestingly, the W/O emulsion was converted to an O/W emulsion after bubbling CO2 for only 1 min. This rapid phase inversion was attributed to the amphiphilicity transformation of PDA–PDEAEMA emulsifier under a CO2 stimulus. As discussed above, the original PDA–PDEAEMA particles were hydrophobic and oil-wetted, which is beneficial for the stabilization of W/O emulsions according to the Bancroft rule. Importing CO2 to the system switched the PDA–PDEAEMA emulsifier to become protonated, hydrophilic and water-wetted, thereby resulting in the phase inversion from a W/O type to an O/W type.
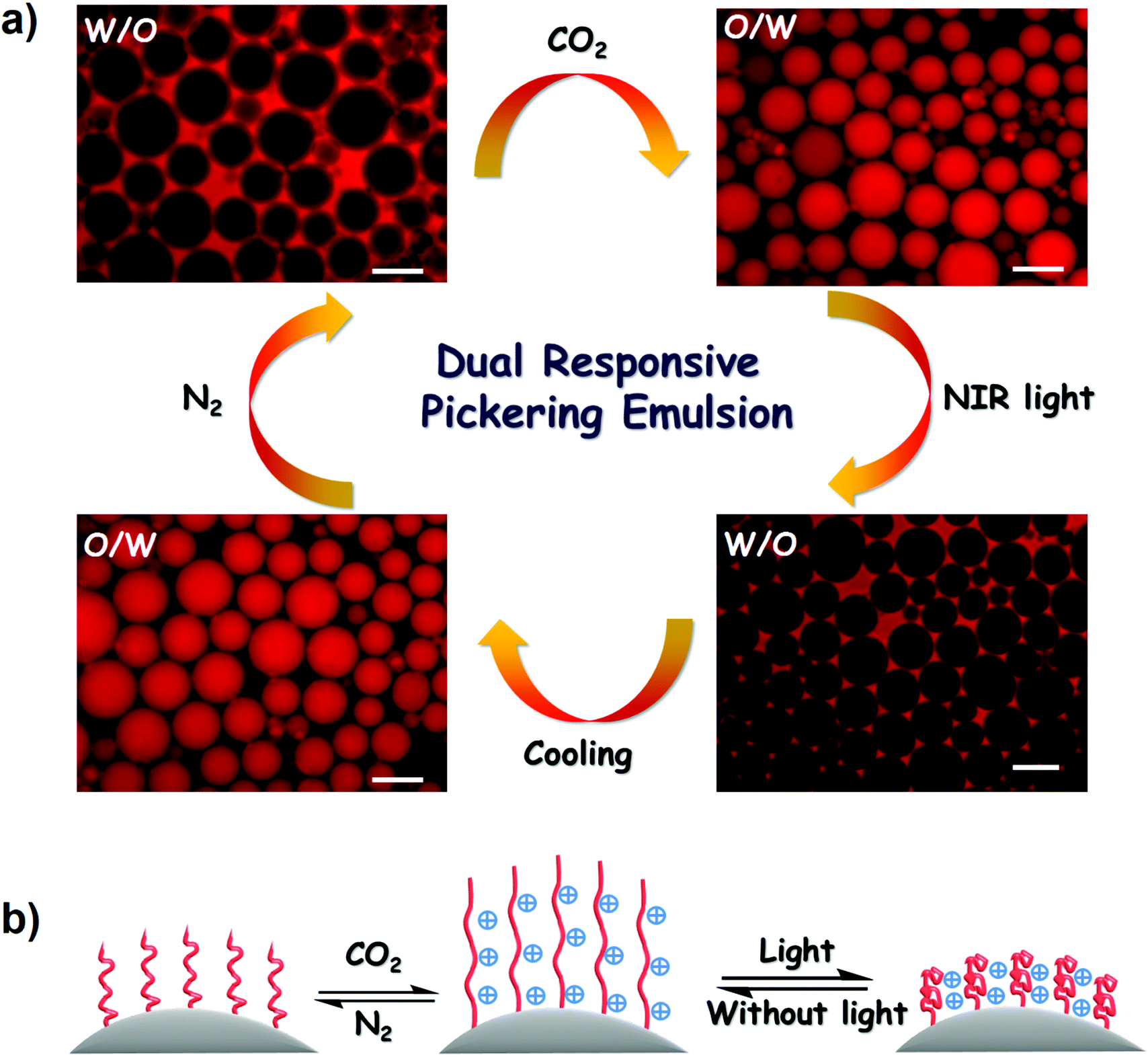 | ||
| Fig. 9 (a) Fluorescence microscopic images of inversion of Pickering emulsions stabilized by PDA–PDEAEMA particles under CO2 and NIR light triggers. Scale bar: 100 μm. (b) Charge and conformation transformation of a PDEAEMA chain under CO2 and NIR light triggers (schematic). | ||
In addition to CO2-triggered phase inversion, light-triggered phase inversion was also investigated. As shown in Fig. 9a, the obtained O/W emulsion under a CO2 stimulus could apparently convert to a W/O emulsion upon illuminating the emulsion system under 2.0 W NIR light for 1 min. This phase inversion originated directly from the increase in solution temperature due to the photothermal conversion of PDA–PDEAEMA particles. The emulsion temperature reached ≈58 °C under illumination with NIR light, relatively higher than the LCST of PDA–PDEAEMA. Therefore, the conformation of PDA–PDEAEMA particles was transformed from an expansion to a contraction state, and correspondingly the particles became hydrophobic and tended to stabilize the W/O emulsion. The high temperature may have slightly decreased the degree of protonation of PDA–PDEAEMA particles due to the reduced solubility of CO2 at high temperature. Hence, the synergetic effect of contraction and deprotonation could account for the phase inversion from the O/W type to the W/O type. To ascertain if the CO2 stimulus was removed under the light trigger, temperature-dependent 13C NMR of DEAEMA monomer was analyzed from room temperature to 65 °C. The HCO3− signal (160 ppm) emerged immediately upon CO2 bubbling and this signal did not disappear with an increase in temperature (Fig. 10). This NMR study showed that CO2 was present even at high temperature. Therefore, the W/O emulsion became an O/W emulsion again upon hand shaking if light illumination was switched off and the emulsion was cooled to room temperature. A schematic illustration of CO2 and light synergetic-regulated inversion of a Pickering emulsion is displayed in Fig. 9b. Hence, the reversible protonation, contraction, extension and deprotonation processes under CO2 and light stimuli have crucial roles in the controllable inversion of emulsions between W/O and O/W alternatively.
 | ||
| Fig. 10 Temperature-dependent 13C NMR of the DEAEMA monomer dissolved in D2O with CO2 bubbling. | ||
4. Conclusions
We synthesized a CO2 and light dual-responsive Pickering emulsifier based on grafting a CO2-responsive PDEAEMA chain on light-sensitive PDA nanoparticles. By combining the outstanding photothermal-conversion ability of PDA and CO2/temperature dual-responsive features of PDEAEMA chain, the controllable inversion of Pickering emulsions between W/O and O/W types was constructed via synergetic regulation of CO2 and NIR light. These two external stimuli have the advantages of complete erasability, non-pollution and easy manipulation. Based on the integration of these two stimuli, on-demand inversion of Pickering emulsions could be realized in a more convenient and efficient strategy. This approach is expected to show great potential in temperature-dependent heterogeneous catalysis and separation.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported financially by the National Natural Science Foundation of China (21674127, 51373197, and 21422407). We thank Dr Yu Chen for his help on NMR measurements.Notes and references
- J. Tang, P. J. Quinlan and K. C. Tam, Soft Matter, 2015, 11, 3512–3529 RSC.
- Y. Chevalier and M.-A. Bolzinger, Colloids Surf., A, 2013, 439, 23–34 CrossRef CAS.
- M. Pera-Titus, L. Leclercq, J.-M. Clacens, F. De Campo and V. Nardello-Rataj, Angew. Chem., Int. Ed., 2015, 54, 2006–2021 CrossRef CAS PubMed.
- A. Schrade, K. Landfester and U. Ziener, Chem. Soc. Rev., 2013, 42, 6823–6839 RSC.
- X. Wang, Y. Shi, R. W. Graff, D. Lee and H. Gao, Polymer, 2015, 72, 361–367 CrossRef CAS.
- Y. Chen, Y. Bai, S. Chen, J. Jup, Y. Li, T. Wang and Q. Wang, ACS Appl. Mater. Interfaces, 2014, 6, 13334–13338 CAS.
- M. Zhang, L. Wei, H. Chen, Z. Du, B. P. Binks and H. Yang, J. Am. Chem. Soc., 2016, 138, 10173–10183 CrossRef CAS PubMed.
- S. Wiese, A. C. Spiess and W. Richtering, Angew. Chem., Int. Ed., 2013, 52, 576–579 CrossRef CAS PubMed.
- Z. Chen, C. Zhao, E. Ju, H. Ji, J. Ren, B. P. Binks and X. Qu, Adv. Mater., 2016, 28, 1682–1688 CrossRef CAS PubMed.
- A. Walther, M. Hoffmann and A. H. E. Müller, Angew. Chem., Int. Ed., 2008, 47, 711–714 CrossRef CAS PubMed.
- D. J. Voorn, W. Ming and A. M. van Herk, Macromolecules, 2006, 39, 2137–2143 CrossRef CAS.
- Z. Wang and Y. Wang, Materials, 2016, 9, 903 CrossRef PubMed.
- C. Yi, N. Liu, J. Zheng, J. Jiang and X. Liu, J. Colloid Interface Sci., 2012, 380, 90–98 CrossRef CAS PubMed.
- J. Huang, F. Cheng, B. P. Binks and H. Yang, J. Am. Chem. Soc., 2015, 137, 15015–15025 CrossRef CAS PubMed.
- H. Yang, T. Zhou and W. Zhang, Angew. Chem., Int. Ed., 2013, 52, 7455–7459 CrossRef CAS PubMed.
- C. Ma, X. Bi, T. Ngai and G. Zhang, J. Mater. Chem. A, 2013, 1, 5353–5360 CAS.
- Z. Li and T. Ngai, Colloid Polym. Sci., 2011, 289, 489–496 CAS.
- A. Feng, C. Zhan, Q. Yan, B. Liu and J. Yuan, Chem. Commun., 2014, 50, 8958–8961 RSC.
- Y. Chen, Z. Wang, D. Wang, N. Ma, C. Li and Y. Wang, Langmuir, 2016, 32, 11039–11042 CrossRef CAS PubMed.
- S. Lin and P. Theato, Macromol. Rapid Commun., 2013, 34, 1118–1133 CrossRef CAS PubMed.
- Q. Qian, X. Huang, X. Zhang, Z. Xie and Y. Wang, Angew. Chem., Int. Ed., 2013, 52, 10625–10629 CrossRef CAS PubMed.
- Q. Qian, J. Wang, F. Yan and Y. Wang, Angew. Chem., Int. Ed., 2014, 53, 4465–4468 CrossRef CAS PubMed.
- J. Wang, J. Zhao, Y. Li, M. Yang, Y.-Q. Chang, J.-P. Zhang, Z. Sun and Y. Wang, ACS Macro Lett., 2015, 392–397 CrossRef.
- Y. He, S. Liao, H. Jia, Y. Cao, Z. Wang and Y. Wang, Adv. Mater., 2015, 27, 4622–4627 CrossRef CAS PubMed.
- Y. Cao, J.-H. Dou, N.-J. Zhao, S. Zhang, Y.-Q. Zheng, J.-P. Zhang, J.-Y. Wang, J. Pei and Y. Wang, Chem. Mater., 2017, 29, 718–725 CrossRef CAS.
- Y. Liu, K. Ai, J. Liu, M. Deng, Y. He and L. Lu, Adv. Mater., 2013, 25, 1353–1359 CrossRef CAS PubMed.
- Z. Li, X. Zhang, S. Wang, Y. Yang, B. Qin, K. Wang, T. Xie, Y. Wei and Y. Ji, Chem. Sci., 2016, 7, 4741–4747 RSC.
- R. Zheng, S. Wang, Y. Tian, X. Jiang, D. Fu, S. Shen and W. Yang, ACS Appl. Mater. Interfaces, 2015, 7, 15876 CAS.
- L. Tang, F.-X. Liang, Q. Wang, X.-Z. Qu, B.-Y. Jiang and Z.-Z. Yang, Chin. J. Polym. Sci., 2017, 35, 799–808 CrossRef CAS.
- M. D. J. Quinn, V. Khu, S. Madden and S. M. Notley, ACS Appl. Mater. Interfaces, 2016, 8, 10609–10616 CAS.
- B. Zhu and S. Edmondson, Polymer, 2011, 52, 2141–2149 CrossRef CAS.
- H. Yang, F. Liang, X. Wang, Y. Chen, C. Zhang, Q. Wang, X. Qu, J. Li, D. Wu and Z. Yang, Macromolecules, 2013, 46, 2754–2759 CrossRef CAS.
- Z. Zhao, F. Liang, G. Zhang, X. Ji, Q. Wang, X. Qu, X. Song and Z. Yang, Macromolecules, 2015, 48, 3598–3603 CrossRef CAS.
- J. Tang, R. M. Berry and K. C. Tam, Biomacromolecules, 2016, 17, 1748–1756 CrossRef CAS PubMed.
- Y. Chen, T. Zhao, B. Wang, D. Qiu and N. Ma, Langmuir, 2015, 31, 8138–8145 CrossRef CAS PubMed.
- D. Han, X. Tong, O. Boissière and Y. Zhao, ACS Macro Lett., 2012, 1, 57–61 CrossRef CAS.
- A. Bousquet, E. Ibarboure, E. Papon, C. Labrugère and J. Rodríguez-Hernández, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 1952–1961 CrossRef CAS.
- T. Thavanesan, C. Herbert and F. A. Plamper, Langmuir, 2014, 30, 5609–5619 CrossRef CAS PubMed.
- Y. Cao, Z. Wang, S. Liao, J. Wang and Y. Wang, Chem. – Eur. J., 2016, 22, 1152–1158 CrossRef CAS PubMed.
| This journal is © the Partner Organisations 2017 |