
An aluminium-based fluorinated counterion for enhanced encapsulation and emission of dyes in biodegradable polymer nanoparticles†
Bohdan
Andreiuk
ab,
Andreas
Reisch
a,
Vasyl G.
Pivovarenko
 b and
Andrey S.
Klymchenko
*a
b and
Andrey S.
Klymchenko
*a
aLaboratoire de Biophotonique et Pharmacologie, UMR CNRS 7213, University of Strasbourg, 74 route du Rhin, 67401 Illkirch Cedex, France. E-mail: andrey.klymchenko@unistra.fr
bOrganic Chemistry Department, Chemistry Faculty, Taras Shevchenko National University of Kyiv, 01601 Kyiv, Ukraine
First published on 15th August 2017
Abstract
Dye-loaded polymer nanoparticles, due to their high brightness and potential biodegradability, emerge as a powerful alternative to quantum dots in bioimaging applications. To minimize aggregation-caused quenching of the loaded dyes, we have recently proposed the use of cationic dyes with bulky hydrophobic counterions (also known as weakly coordinating anions), which serve as spacers preventing dye pi-stacking inside nanoparticles. However, so far this approach of counterion-enhanced emission inside polymer NPs has been limited to one fluorinated tetraphenylborate (tetrakis(pentafluorophenyl)borate, F5-TPB). Herein, we show that the counterion-enhanced emission approach is not limited to tetraphenylborates and can be extended to other types of anions, such as the aluminium-based anion, Al[OC(CF3)3]4− (F9-Al), which is much easier to scale up, compared to F5-TPB. It is found that F9-Al strongly improves the encapsulation efficiency of the octadecyl rhodamine B dye compared to the perchlorate counterion (97 ± 2 vs. 51 ± 2%), being slightly better than F5-TPB (92 ± 4%). Similarly to F5-TPB, F9-Al can effectively prevent aggregation-caused quenching of rhodamine inside NPs made of the biodegradable polymer, poly(lactide-co-glycolide) (PLGA), even at 50 mM dye loading. According to single-particle microscopy, the obtained NPs are 33-fold brighter than commercial quantum dots QD585 at 532 nm excitation and exhibit complete ON/OFF switching (blinking), as was originally observed for NPs based on F5-TPB. Importantly, NPs loaded with the rhodamine/F9-Al ion pair entered the cells by endocytosis, showing no signs of dye leaching, in contrast to rhodamine perchlorate, which exhibited severe leakage from NPs with characteristic accumulation inside mitochondria. Moreover, F9-Al surpassed the F5-TPB anion in stability of dye-loaded NPs against leaching, which can be attributed to the higher hydrophobicity of the former. Overall, this work shows that counterion-enhanced encapsulation and emission of cationic dyes inside polymer NPs is a general approach for the preparation of stable and highly fluorescent nanomaterials for bioimaging applications.
Introduction
Fluorescent nanoparticles (NPs) attract great attention in biological and biomedical research, as they are much brighter than organic dyes and can serve as multifunctional platforms for theranostics. The interest in fluorescent nanoparticles has grown rapidly.1 The most representative are quantum dots,2 dye-doped silica NPs,3 carbon nanostructures4 and NPs based on organic materials. Fluorescent organic NPs are an emerging class of functional nanomaterials, which includes conjugated polymer NPs,5 dye-loaded polymer6,7 and lipid8 NPs as well as dye-based NPs, notably using aggregation-induced emission (AIE) dyes.7,9 Dye-loaded polymer NPs present several key features, attractive for biological applications.6 Their polymer matrix can be biodegradable and can be employed for encapsulating other contrast agents and drugs. The rich variety of available organic dyes makes it possible to prepare dye-loaded NPs of almost any desired spectroscopic characteristics.10–12 However, two key challenges remain in the field of dye-loaded NPs. First, at high loading dyes tend to self-quench, which results in poor particle brightness.6 Second, dye encapsulation can be inefficient and the dye may simply adsorb onto the surface of NPs. The latter leads to dye leaching (also called leakage) in biological medium, which is detrimental to the signal-to-noise ratio in fluorescence microscopy imaging.13,14 Several approaches were developed to solve these two problems. The use of long hydrocarbon chains in the case of cyanine dyes allowed their efficient encapsulation into polymer NPs and minimal leaching inside cells, although relatively high self-quenching was observed at high dye loading.15 The use of bulky side groups can significantly decrease self-quenching, as exemplified in the case of BODIPY16 and perylenediimide11,17 derivatives. Aggregation-induced emission has become a highly popular approach,9 which has led to the development of a variety of organic dye loaded NPs in recent years.7,18,19 In this case, the dyes are often not dispersed inside the polymer matrix, but form a core, built from pure dyes or their polymerized monomers which is further stabilized by polymeric amphiphiles.7,18 A relatively recent approach to prevent aggregation-caused quenching has been to use bulky hydrophobic counterions.6,10 Many of them belong to the class of weakly coordinating (also called non-coordinating) anions20 because of their large radius and charge delocalization. Typical examples are derivatives of tetraphenylborate (TPB), tetraalkoxyaluminates, bis(alkylsulfonyl)imides, etc. Weakly coordinating anions have been successfully used in catalysis,21 electrochemistry,22 stabilization of reactive cations,23 as components of ionic liquids,24 microporous materials,25 lithium batteries,26 ion-selective electrodes,27 ion sensors,28 solar cells29 and organic light-emitting diodes.30 Recently, they were proposed for assembly of cationic dyes into fluorescent nanoparticles31–33 and ultra-small fluorescent micelles.34However, in biological media these nanomaterials tend to disassemble,33 which is a strong limitation for their bioimaging applications. Encapsulation of ion pairs of cationic dyes with such counterions inside a polymer matrix ensures both efficient fluorescence and high stability against dye leaching.10 Indeed, we recently showed that fluorinated derivatives of tetraphenylborate, due to their high hydrophobicity, ensured efficient encapsulation of the cationic dye, octadecyl rhodamine B (R18), inside polymer NPs.10,35 Remarkably, only a perfluorinated analogue of TPB, tetrakis(pentafluorophenyl)borate (F5-TPB), could efficiently prevent dye self-quenching inside polymer NPs, which allowed preparation of 40-nm NPs that were ∼6-fold brighter than quantum dots (QD605) and showed an excellent signal-to-noise ratio after entering living cells. Moreover, the obtained NPs showed unprecedented ON/OFF switching (blinking) due to the collective behaviour of the dyes, assembled with F5-TPB inside the polymer matrix.10 This collective behaviour enabled controlled photoswitching of the dye-loaded NPs.36 However, at the moment the approach of counterion-enhanced encapsulation and emission of dyes inside polymer NPs is still in its infancy, and it relies exclusively on one counterion, F5-TPB. It appears that fluorination in TPB ions is essential for preventing self-quenching of cationic dyes.31,33 So, the key question is whether it is possible to extend this unique approach to other anions. The key to broad applications is an easy scale up of the counterion through a straightforward synthesis from inexpensive materials, which is not the case for F5-TPB. Finally, it is also important to replace the aromatic pentafluorophenyl groups of F5-TPB with aliphatic perfluorinated groups in order to understand whether the aromatic residues are absolutely required to achieve efficient encapsulation and prevent self-quenching of dyes inside NPs.
Herein, we explored an Al-based anion, tetrakis[perfluoro-tert-butoxy]aluminate (denoted as F9-Al), as a potential replacement for F5-TPB. In this anion, four perfluorinated tert-butylate groups are coordinated to the central aluminium ion (Fig. 1). This anion was described in the literature as a weakly coordinating anion,37 similarly to F5-TPB.20 Importantly, this anion can be synthesized in large quantities in one step from inexpensive materials, namely LiAlH4 and perfluorinated tert-butyl alcohol.38 Our results show that F9-Al perfectly replaces F5-TPB. It ensures more effective encapsulation of rhodamine dyes inside polymer NPs and prevents their self-quenching, especially in comparison with perchlorate, a small inorganic counterion. The obtained NPs are much brighter than reference quantum dots of the same emission colour. Moreover, cellular studies revealed that at high dye loading the Al-based counterion prevents dye leakage better than F5-TPB, indicating that it encapsulates dyes more efficiently. Finally, the obtained fluorescent NPs showed minimal cytotoxicity for both F9-Al and F5-TPB counterions. The obtained results propose an easy to scale-up aluminium-based weakly coordinating anion as a highly efficient agent for preparation of bright dye-loaded polymer NPs exhibiting minimized dye self-quenching and leaching.
 | ||
| Fig. 1 (A) Chemical structures and molecular models of counterions used in this study to encapsulate R18 inside polymer nanoparticles. (B) Thin layer chromatography of R18 dye salts with different counterions: F5-TPB (F5), perchlorate (Cl) and F9-Al (Al). The eluent was dichloromethane/methanol, 95/5. | ||
Results and discussion
Synthesis of dye salts
Basic optimization of the geometry of F9-Al showed that this anion is close in effective diameter to F5-TPB (1.04 vs. 1.13 nm). However, F9-Al contains 36 fluorine atoms vs. 20 in the case of F5-TPB, which leads to their higher density at the surface of F9-Al (Fig. 1A). Li[F9-Al] was synthesized similarly to previously described protocols, which consist of reacting LiAlH4 and perfluorinated tert-butyl alcohol.38,39 The procedure required a gradual increase in temperature to ensure a controlled reaction without rapid evaporation of perfluorinated tert-butyl alcohol. The reaction was accompanied by a shift of the perfluorinated tert-butyl alcohol signal in 19F NMR in DMSO-d6 from −73.88 to −75.10 after the aluminium alcoholate formation. Then, the ion pair of rhodamine B octadecyl ester (R18) with F9-Al was prepared similarly to that of F5-TPB: R18 perchlorate (R18/ClO4) was mixed with the lithium salt of F9-Al in dichloromethane and the product of ion exchange was purified by column chromatography. It should be noted that upon performing thin layer chromatography the ion pair R18/F9-Al showed a much higher Rf compared to the parent R18/ClO4, but nearly the same Rf as R18/F5-TPB (Fig. 1B). According to these results, F9-Al formed a stable and highly apolar ion pair with R18 in organic solvents, similarly to F5-TPB.Preparation and characterization of NPs
The three different R18 salts with ClO4, F5-TPB and F9-Al anions were encapsulated into PLGA NPs. To this end, acetonitrile solutions of PLGA and R18 salts were nanoprecipitated in phosphate buffer (pH 7.4). According to our earlier studies, nanoprecipitation of PLGA at neutral pH should result in small NPs, stabilized by negative charges of deprotonated carboxylate groups of PLGA at the NP's surface.10,35 Indeed, as expected, at low dye loading (5 mM of dye salt with respect to the polymer), small NPs with hydrodynamic diameters of around 40 nm and low polydispersity (∼0.1) were obtained according to dynamic light scattering (DLS) for all three dye salts (Fig. 2A and Fig. S5, Table S1 in the ESI†). Nevertheless, R18/ClO4 NPs were slightly larger. A further increase in R18/ClO4 dye loading significantly increased the size of NPs, reaching ∼300 nm at 50 mM loading. The colloidal stability of these NPs was poor, as they precipitated overnight. This is in line with our earlier studies, which suggested that R18 salts with small inorganic counterions, being of amphiphilic nature, tend to accumulate at the NP surface, where cationic R18 neutralizes negative charges of PLGA carboxylates, thus resulting in NP aggregation. In sharp contrast, NPs loaded with R18/F5-TPB and R18/F9-Al ion pairs were small, independently of dye loading (Fig. 2A), showing no signs of precipitation over weeks. Transmission electron microscopy (TEM) confirmed the DLS data: at 50 mM loading of R18/F5-TPB (Fig. 2H) and R18/F9-Al (Fig. 2I), small particles were observed with average sizes of 38 ± 7 and 35 ± 5 nm, respectively, whereas R18/ClO4 loading resulted in large irregular aggregates (Fig. 2G). Zeta potential also showed relatively stable negative values for all dye loadings with both F9-Al and F5-TPB anions (Table S1 in the ESI†), indicating that in these cases dye encapsulation did not influence the particle surface properties. These results suggest that, similarly to F5-TPB, the aluminium-based anion F9-Al is capable of encapsulating dyes without interference with the nanoprecipitation process. | ||
| Fig. 2 Characterization of PLGA NPs loaded with R18 and different counterions. Hydrodynamic diameter (A), fluorescence quantum yield (B), fluorescence anisotropy (C) and encapsulation efficiency (D) of NPs. (E and F) Absorption and fluorescence spectra of NPs at 5 (E) and 50 mM (F) dye loading. (G–I) TEM images of R18/ClO4 (G), R18/F5-TPB (H) and R18/F9-Al (I) NPs at 50 mM dye loading. | ||
To quantitatively evaluate the efficiency of dye encapsulation into NPs, we developed a protocol to distinguish between encapsulated and adsorbed dyes. In this protocol, the dye-loaded NPs were dialyzed using a solution of β-cyclodextrin as a recipient medium. As R18 is a hydrophobic molecule, poorly soluble in water, β-cyclodextrin, which is known to solubilize hydrophobic molecules,40 was essential to ensure transfer of poorly encapsulated dyes (i.e. those adsorbed onto the NP surface) to the aqueous phase during dialysis. Absorption spectroscopy confirmed that 1 mM β-cyclodextrin can effectively solubilize R18/ClO4 in water at micromolar concentration, as the initially broadened absorption band of aggregates of the free dye in water was converted into a narrow band similar to that in organic solvents (Fig. S6 in the ESI†). The dialysis membrane was chosen to be permeable for small molecules, like R18 and β-cyclodextrin, but impermeable for our NPs (MWCO 14000). Then, using absorption measurements of R18 after 24 h of dialysis (see Materials and methods for details), we found that both R18/F5-TPB and R18/F9-Al were encapsulated almost quantitatively for the highest (50 mM) loading, with slightly higher efficiency for F9-Al, whereas R18/ClO4 was encapsulated at only about 50% (Fig. 2D and Table S2 in the ESI†). This result is in good agreement with observations on the NP size, confirming that a large part of R18/ClO4 is simply adsorbed on the particle surface. In a control experiment, we used R18/ClO4 without the polymer, and only 13% of the remaining dye was observed using the same dialysis protocol (Fig. 2D and Fig. S7 in the ESI†). To understand the role of the counterion hydrophobicity in the encapsulation of dyes, we studied their partition behaviour in a two-phase system formed with a heptane/acetonitrile/water mixture. In this case heptane and water form, respectively, highly apolar and polar phases, whereas acetonitrile was used to improve the solubility of the dye salts in water. The obtained partitioning constant values for R18/Al-F9, R18/F5-TPB and R18/ClO4 in this ternary solvent mixture (log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) PHWA) were 1.43, 0.74 and −2.74, respectively. These differences in hydrophobicity of dye salts correlate well with their encapsulation efficiency, where the most hydrophobic salt R18/Al-F9 shows the most efficient encapsulation. Thus, the encapsulation efficiency of dyes is directly linked to the counterion hydrophobicity.
PHWA) were 1.43, 0.74 and −2.74, respectively. These differences in hydrophobicity of dye salts correlate well with their encapsulation efficiency, where the most hydrophobic salt R18/Al-F9 shows the most efficient encapsulation. Thus, the encapsulation efficiency of dyes is directly linked to the counterion hydrophobicity.
The fluorescence quantum yield (QY), which is the key parameter of NP brightness, was then evaluated for the obtained NPs (Fig. 2B and Table S3 in the ESI†). As expected, NPs loaded with R18/ClO4 displayed a drop in quantum yield from 0.8 to 0.07 with an increase in the dye loading from 5 to 50 mM, demonstrating the classical phenomenon of aggregation-caused quenching of rhodamine dyes.10 This conclusion is also supported by the red-shifted absorption and fluorescence spectra, observed for R18/ClO4 NPs at 50 mM loading compared to NPs at low loading and the dye in methanol (Fig. 2E, F and Fig. S8 in the ESI†). In sharp contrast, the dye salts R18/F5-TPB and R18/F9-Al displayed significantly lower self-quenching behaviour. Indeed, at the highest dye loading (50 mM) the QY value was 0.55 for both salts. Moreover, the absorption and emission spectra of these NPs were almost the same, and the positions of their maxima were close to those of R18/ClO4 in methanol (Fig. 2E). These results suggest that the Al-based counterion F9-Al effectively prevents aggregation-caused quenching of R18, similarly to the boron-based anion F5-TPB. We can conclude that, being close in size to F5-TPB and having even higher fluorination, F9-Al can also serve as a spacer between the cationic rhodamine dyes, thus preventing their π-stacking. It should be noted that so far only aromatic F5-TPB has been reported to prevent dye self-quenching in polymer NPs.10,35 The present result shows that the phenomenon of counterion-enhanced emission in polymer NPs is more general and can be extended to perfluorinated aliphatic Al-based ions. Taking into account the quantum yield of NPs, the extinction coefficient of R18 (125000 M−1 cm−1) and the calculated number of R18 dyes per NP (680–870 for NPs of 35–38 nm diameter), we can estimate the brightness of R18/F5-TPB and R18/F9-Al NPs at 50 mM loading to be 4.7–6.0 × 107 M−1 cm−1.
The other important point is that at high loading dyes may start communicating through excitation energy transfer (EET).10,36 The diffusion of energy within randomly oriented fluorophores leads to a loss of fluorescence anisotropy. Earlier, we showed that R18/F5-TPB inside PLGA NPs displayed very low anisotropy even for relatively low dye loading, which suggested that F5-TPB brings dyes in close proximity within the PLGA matrix and thus ensures fast EET.10 At low dye loading (0.5 mM), NPs based on all three counterions displayed similarly high values of fluorescence anisotropy (0.22–0.23, Table S4 in the ESI†), indicating that dyes were well immobilized inside the polymer matrix of NPs. The anisotropy values for R18/F9-Al NPs decreased with dye loading similarly to those for R18/F5-TPB NPs, whereas for R18/ClO4 NPs these values were systematically much higher (Fig. 2C and Table S4 in the ESI†). Thus, the Al-based anion also brings dyes in close proximity within the polymer matrix, which ensures fast EET.
Single-particle fluorescence
PLGA NPs with the highest tested loading were then deposited on a glass surface and further characterized by wide-field fluorescence microscopy. NPs loaded with R18/F5-TPB and R18/F9-Al appeared as dots and their intensities were similar (Fig. 3A and B). NPs loaded with R18/ClO4 were not tested because of their poor colloidal stability. Remarkably, the mean intensities of R18/F9-Al NPs and R18/F5-TPB NPs were, respectively, 33-fold and 34-fold higher than those of commercial quantum dots QD585 (Fig. 3A and B). Thus, the F9-Al counterion also enables preparation of NPs that are much brighter than QDs of comparable size, which is an attractive property for bioimaging applications. A remarkable feature of polymer NPs, loaded with R18/F5-TPB, is the ON/OFF switching of the whole particle, caused by the collective behaviour of the encapsulated dyes.10 In the present work, we confirmed the previous data for R18/F5-TPB NPs, as can be seen from the intensity traces of individual particles (Fig. 3C and Fig. S9 in the ESI†). Importantly, very similar ON/OFF switching was also observed for F9-Al NPs (Fig. 3D and Fig. S9 in the ESI†). Together with the low anisotropy values, observed for this counterion at high dye loading, the switching behaviour of the NPs suggests that F9-Al also induces collective behaviour of hundreds of dyes coupled by EET. In this case, a single dark species among encapsulated dyes (e.g. their triplet or radical state) in the NP can quench all encapsulated dyes of the particle. | ||
| Fig. 3 Single-particle fluorescence properties of dye-loaded PLGA NPs. (A and B) Wide-field fluorescence images of different NPs in the form of 2D (A) and 3D (B) graphs. In the case of the 2D image of quantum dots, the intensity was multiplied 10-fold to make the signal visible. All imaging conditions were identical for dye-loaded NPs and QDs. The excitation power density at 532 nm was 0.6 W cm−2. (C and D) Representative single-particle traces of NPs loaded with R18/F5-TPB (C) and R18/F9-Al (D), recorded under a wide-field microscope. The excitation power density at 532 nm was 0.6 W cm−2. | ||
Cellular data
The obtained NPs at the lowest and highest dye loading were incubated with HeLa cells for 3 h. NPs loaded with 5 mM R18/ClO4 showed continuous fluorescent structures inside the cells, delimited by the membrane marker (WGA-Alexa488 in green, Fig. 4). Very similar images were observed earlier with HeLa cells, stained with the free R18 dye, where this cationic dye accumulated inside mitochondria.33 Thus, without bulky hydrophobic counterions, the dye is not well-encapsulated inside NPs, and therefore upon coming into contact with the cells it is readily released into the cytoplasm. This result is in agreement with our dialysis data, which shows that only 50% of the dye is efficiently encapsulated. In sharp contrast, PLGA NPs, containing 5 mM of R18/F5-TPB or R18/F9-Al, appeared in the form of bright dots, located inside the cells (Fig. 4). This result is in agreement with our previous work, where PLGA NPs containing 1 wt% R18/F5-TPB (6.3 mM) exhibited minimal dye leakage after entering cells by endocytosis.10 The present data suggest that R18/F9-Al also prevents dyes from leaking into the biological medium. Importantly, high dye loading (50 mM) revealed additional differences among the counterions used. While R18/ClO4 NPs again showed strong dye leaching, observed as continuous intracellular fluorescence, for R18/F5-TPB NPs both particles and continuous extended fluorescent structures were observed (Fig. 4E). This result suggested that at the highest dye loading encapsulation is not complete for the R18/F5-TPB ion pair and even a tiny fraction of dye that is adsorbed onto the NP surface can lead to significant amounts of nonencapsulated dyes entering the cells. The probable reason here is contact-mediated dye transfer from the NP surface to the cell membrane, which occurs more effectively than endocytosis of NPs.14 Remarkably, despite high loading, R18/F9-Al NPs displayed exclusively dotted fluorescence of endocytosed NPs without signs of continuous extended structures. Thus, F9-Al is advantageous over F5-TPB in terms of the stability of dye loading against leaching inside the cells, which corroborates with the encapsulation efficiency studies based on dialysis. This property could be attributed to the higher hydrophobicity of the R18/F9-Al dye salt vs. R18/F5-TPB, which was confirmed in our two-phase partition experiments in the ternary mixture of solvents (see above). It can be explained by higher level of fluorination of F9-Al vs. F5-TPB with the “Teflon-coated”, extremely hydrophobic surface of the former.37,41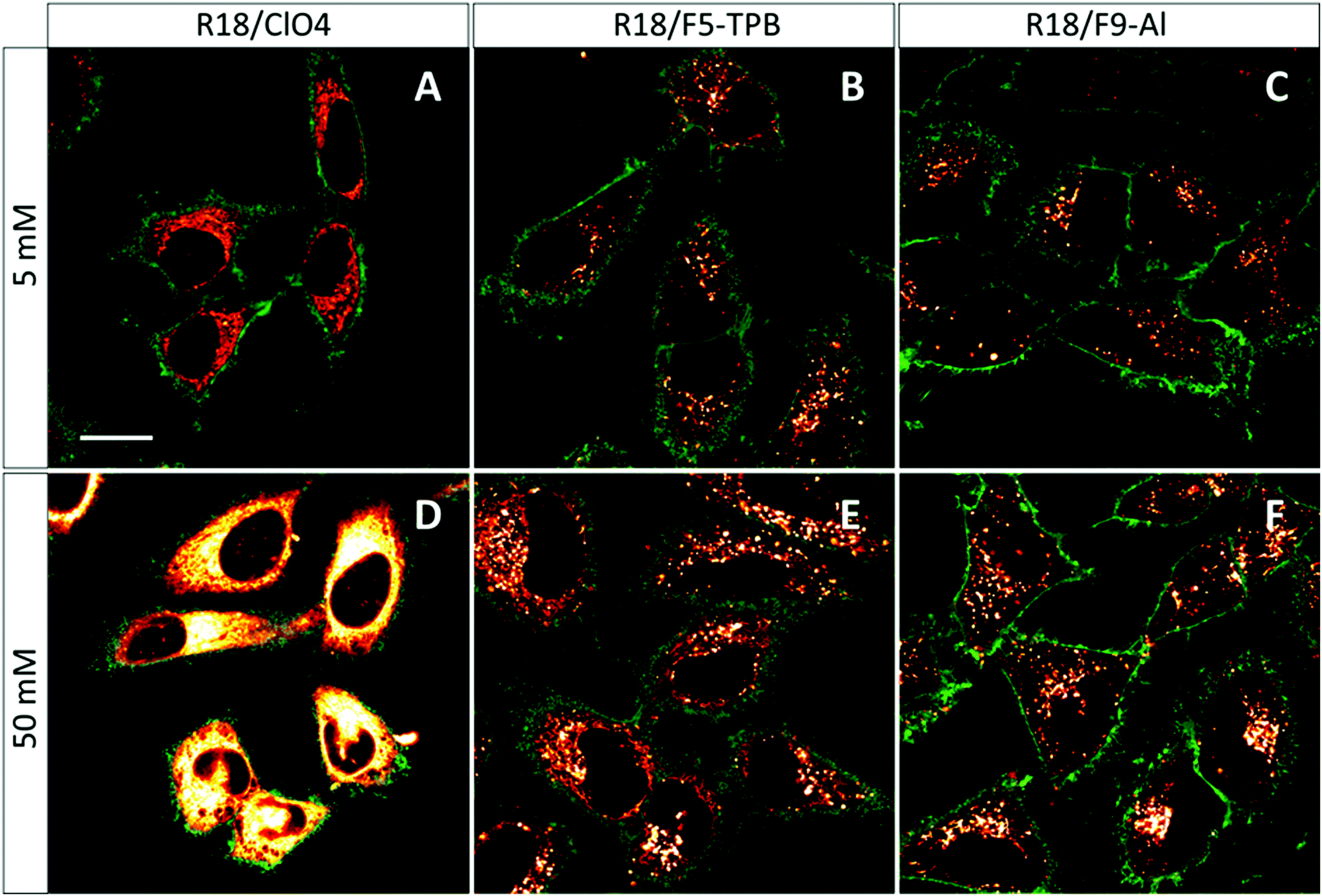 | ||
| Fig. 4 Laser scanning confocal images of HeLa cells incubated for 3 h with dye-loaded PLGA NPs. NPs were loaded with R18/ClO4, R18/F5-TPB and R18/Al-F9 at 5 and 50 mM concentrations. The excitation wavelength was 561 nm, while the emission was collected from 570 to 650 nm. The image size was 116.8 × 116.8 μm. | ||
Cytotoxicity
Finally, we verified the cytotoxicity of our dye-loaded NPs. As R18/ClO4 NPs were not suitable for bioimaging because of the strong leakage, we did not study them further. All dye-loaded NPs at the highest dye loading showed the same low cytotoxicity as the blank NPs without the dye (Fig. 5). Importantly, low values of cytotoxicity were observed even for 0.05 mg ml−1 of polymer, which is 25-fold higher than the concentration used for cell imaging experiments. Increased cytotoxicity of the highest concentration of polymer (0.084 mg ml−1) for both blank and dye-loaded particles could be explained by a significant increase in the concentration of low-salt phosphate buffer (used for NP preparation) in the growth medium, as compared to cases with lower concentration (0.0005–0.05 mg ml−1) (see Materials and methods for the protocol). | ||
| Fig. 5 Viability of HeLa cells incubated with blank and dye-loaded NPs for 48 h according to the MTT assay. The dye/counterion pairs were loaded at 50 mM concentration. | ||
Materials and methods
PLGA (lactide 50 mole%, glycolide 50 mole%, Mn 24000; PDI 1.7), rhodamine B octadecyl ester perchlorate, and lithium aluminium hydride solution (1.0 M in diethyl ether) were purchased from Sigma-Aldrich and used as received. Lithium Tetrakis(pentafluorophenyl)borate-ethyl ether complex (Li/F5-TPB) was purchased from TCI and used as received. Perfluoro-tert-butanol was purchased from Fluorochem and used as received. Toluene was purchased from Alfa Aesar. Sodium phosphate monobasic (99.0%, Sigma-Aldrich) and sodium phosphate dibasic dihydrate (99.0%, Sigma-Aldrich) were used to prepare 20 mM phosphate buffer solutions at pH 7.4. MilliQ-water (Millipore) was used in all experiments.
NMR spectra were recorded at 20 °C on Bruker Avance III 400 MHz and Bruker Avance III 500 MHz spectrometers. Mass spectra were obtained using an Agilent Q-TOF 6520 mass spectrometer.
For details of chemical synthesis, see the ESI.†
Preparation of fluorescent NPs
PLGA was dissolved at a concentration of 2 mg ml−1 in acetonitrile containing different concentrations of dyes. The solutions were rapidly added with a micropipette to a ninefold volume excess of 20 mM phosphate buffer at pH 7.4 and under stirring. The obtained suspension of NPs was then quickly diluted fivefold with the same buffer.Characterization of NPs
DLS measurements were performed on a Zetasizer Nano series DTS 1060 (Malvern Instruments S.A.). Electron microscopy was performed on a Philips CM120 transmission electron microscope. The particle size was obtained as mean value ± standard deviation for >120 NPs. Absorption and emission spectra were recorded on a Cary 400 Scan ultraviolet-visible spectrophotometer (Varian) and a FluoroMax-4 spectrofluorometer (Horiba Jobin Yvon) equipped with a thermostated cell compartment, respectively. QYs were calculated using rhodamine B in methanol (QY = 0.7)42 as a reference.The encapsulation efficiency of dyes inside NPs was measured as the ratio of dye quantity (in moles) in the sample after and before dialysis (MEMBRA-CEL MD34 14 × 100 membrane, MWCO 14000) in 1 mM beta-cyclodextrin solution according to the following protocol. 450 μl of freshly prepared NP solution was added to 550 μl of 1 mM beta-cyclodextrin solution, and the resulting mixture was dialysed for 24 hours with 1 mM beta-cyclodextrin solution as a recipient medium. The mole quantities of the dye inside NPs before and after dialysis were measured using absorption spectroscopy taking into account the volume change during the dialysis.
For control experiment, 20 μl of acetonitrile solution of the R18/ClO4 dye were added to 1 ml of 1 mM solution of beta-cyclodextrin in 20 mM phosphate buffer (pH 7.4), and this sample was treated according to the protocol above, showing that 87% of the dye got washed out after 24 h dialysis, and the rest contained probably too large aggregates of the dye to pass through the membrane (see Fig. S7, ESI†).
Determination of the partitioning coefficient
To compare hydrophobicities of the ion pairs, a characteristic, similar to conventional logP (which is applied for octanol/water partition), was introduced. It is a logarithm of the partition of a substance between two phases, heptane as the non-aqueous phase and water/acetonitrile 50/50 as the aqueous phase. It was called logPHWA. In this method, mutually presaturated phases A (heptane), 1 ml, and B (water/acetonitrile 50/50), 1 ml, were placed in an Eppendorf tube, and 10–25 μl of acetonitrile solution of a R18-counterion ion pair was added to the mixture. The mixture was vigorously vortexed and then centrifuged to ensure complete phase partition. Then 50 μl of each phase was added to 1 ml of ethanol and concentrations of the R18 dye in the resulting solutions were measured. In the case of R18/ClO4, which showed very poor partition into the heptane phase, the absorbance of the heptane phase was measured without dilution. logPHWA was calculated as the logarithm of the ratio of dye concentration in phase A to dye concentration in phase B.
Cellular studies
Conclusions
Recently we introduced a concept of counterion-enhanced emission of dyes inside polymer nanoparticles using perfluorinated tetraphenylborate (F5-TPB), which enabled preparation of ultrabright switchable fluorescent NPs. Here, we show that this concept can be extended to another type of anion, based on aluminate of an aliphatic fluorinated alcohol. In comparison to F5-TPB, the Al-based counterion (F9-Al) is much easier to prepare. Our results show that F9-Al can perfectly replace F5-TPB for dye encapsulation, because it effectively prevents aggregation-caused quenching, ensures high brightness of NPs and favours collective behaviour of encapsulated dyes. Importantly, F9-Al enhances encapsulation of rhodamine dyes into PLGA NPs, as they did not show any sign of leakage after entering living cells, in contrast to NPs loaded with the dye perchlorate salt. Moreover, cellular imaging data suggested that F9-Al encapsulates cationic rhodamine even more efficiently than F5-TPB, which can be assigned to the higher hydrophobicity of F9-Al compared to F5-TPB.This work proposes an efficient methodology for preparation of highly fluorescent nanomaterials and it opens an important direction in the application of Al-based counterions. Moreover, this work shows that counterion-enhanced encapsulation and emission of cationic dyes inside polymer nanoparticles is a universal approach, extendable to other families of counterions beyond fluorinated tetraphenylborates.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported by the European Research Council ERC Consolidator grant BrightSens 648528. BA was supported by LabEx Chimie des Systèmes Complexes. Prof. Yves Mely and Prof. Pascal Didier are acknowledged for providing access to wide-field microscopy. We thank Christine Ruhlmann and Anne Runser for help with electron microscopy.References
- O. S. Wolfbeis, Chem. Soc. Rev., 2015, 44, 4743–4768 RSC.
- X. Michalet, F. F. Pinaud, L. A. Bentolila, J. M. Tsay, S. Doose, J. J. Li, G. Sundaresan, A. M. Wu, S. S. Gambhir and S. Weiss, Science, 2005, 307, 538–544 CrossRef CAS PubMed.
- M. Montalti, L. Prodi, E. Rampazzo and N. Zaccheroni, Chem. Soc. Rev., 2014, 43, 4243–4268 RSC.
- V. N. Mochalin, O. Shenderova, D. Ho and Y. Gogotsi, Nat. Nanotechnol., 2012, 7, 11–23 CrossRef CAS PubMed; P. J. G. Luo, S. Sahu, S. T. Yang, S. K. Sonkar, J. P. Wang, H. F. Wang, G. E. LeCroy, L. Cao and Y. P. Sun, J. Mater. Chem. B, 2013, 1, 2116–2127 RSC.
- C. Wu and D. T. Chiu, Angew. Chem., Int. Ed., 2013, 52, 3086–3109 CrossRef CAS PubMed.
- A. Reisch and A. S. Klymchenko, Small, 2016, 12, 1968–1992 CrossRef CAS PubMed.
- K. Li and B. Liu, Chem. Soc. Rev., 2014, 43, 6570–6597 RSC.
- V. N. Kilin, H. Anton, N. Anton, E. Steed, J. Vermot, T. E. Vandamme, Y. Mely and A. S. Klymchenko, Biomaterials, 2014, 35, 4950–4957 CrossRef CAS PubMed.
- Y. N. Hong, J. W. Y. Lam and B. Z. Tang, Chem. Soc. Rev., 2011, 40, 5361–5388 RSC; J. Mei, N. L. C. Leung, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, Chem. Rev., 2015, 115, 11718–11940 CrossRef CAS PubMed.
- A. Reisch, P. Didier, L. Richert, S. Oncul, Y. Arntz, Y. Mely and A. S. Klymchenko, Nat. Commun., 2014, 5, 4089 CAS.
- K. Trofymchuk, A. Reisch, I. Shulov, Y. Mely and A. S. Klymchenko, Nanoscale, 2014, 6, 12934–12942 RSC.
- A. Wagh, F. Jyoti, S. Mallik, S. Qian, E. Leclerc and B. Law, Small, 2013, 9, 2129–2139 CrossRef CAS PubMed.
- S. Snipstad, S. Hak, H. Baghirov, E. Sulheim, Y. Morch, S. Lelu, E. von Haartman, M. Back, K. P. Nilsson, A. S. Klymchenko, C. de Lange Davies and A. K. Aslund, Cytometry, Part A DOI:10.1002/cyto.a.22853.
- H. T. Chen, S. W. Kim, L. Li, S. Y. Wang, K. Park and J. X. Cheng, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 6596–6601 CrossRef CAS PubMed.
- A. Wagh, S. Y. Qian and B. Law, Bioconjugate Chem., 2012, 23, 981–992 CrossRef CAS PubMed.
- R. Méallet-Renault, A. Hérault, J.-J. Vachon, R. B. Pansu, S. Amigoni-Gerbier and C. Larpent, Photochem. Photobiol. Sci., 2006, 5, 300–310 Search PubMed; C. Grazon, J. Rieger, R. Méallet-Renault, B. Charleux and G. Clavier, Macromolecules, 2013, 46, 5167–5176 CrossRef CAS.
- Z. Tian, A. D. Shaller and A. D. Q. Li, Chem. Commun., 2009, 180–182 RSC.
- X. Y. Zhang, K. Wang, M. Y. Liu, X. Q. Zhang, L. Tao, Y. W. Chen and Y. Wei, Nanoscale, 2015, 7, 11486–11508 RSC.
- X. Zhang, L. Tao, Z. Chi, J. Xu and Y. Wei, J. Mater. Chem. B, 2014, 2, 4398–4414 RSC; W. Qin, D. Ding, J. Z. Liu, W. Z. Yuan, Y. Hu, B. Liu and B. Z. Tang, Adv. Funct. Mater., 2012, 22, 771–779 CrossRef CAS; P. Zhang, X. Nie, M. Gao, F. Zeng, A. Qin, S. Wu and B. Z. Tang, Mater. Chem. Front., 2017, 1, 838–845 RSC.
- I. Krossing and I. Raabe, Angew. Chem., Int. Ed., 2004, 43, 2066–2090 CrossRef CAS PubMed.
- M. Q. Jia and M. Bandini, ACS Catal., 2015, 5, 1638–1652 CrossRef CAS.
- W. E. Geiger and F. Barriere, Acc. Chem. Res., 2010, 43, 1030–1039 CrossRef CAS PubMed.
- T. A. Engesser, M. R. Lichtenthaler, M. Schleep and I. Krossing, Chem. Soc. Rev., 2016, 45, 789–899 RSC.
- A. B. A. Rupp and I. Krossing, Acc. Chem. Res., 2015, 48, 2537–2546 CrossRef CAS PubMed; M. Kaliner and T. Strassner, Tetrahedron Lett., 2016, 57, 3453–3456 CrossRef; I. Raabe, K. Wagner, K. Guttsche, M. K. Wang, M. Gratzel, G. Santiso-Quinones and I. Krossing, Chem. – Eur. J., 2009, 15, 1966–1976 CrossRef PubMed.
- S. Fischer, J. Schmidt, P. Strauch and A. Thomas, Angew. Chem., Int. Ed., 2013, 52, 12174–12178 CrossRef CAS PubMed.
- A. Rupp, N. Roznyatovskaya, H. Scherer, W. Beichel, P. Klose, C. Sturm, A. Hoffmann, J. Tubke, T. Koslowski and I. Krossing, Chem. – Eur. J., 2014, 20, 9794–9804 CrossRef CAS PubMed; W. Xu and C. A. Angell, Electrochem. Solid-State Lett., 2001, 4, E1–E4 CrossRef; M. L. Aubrey and J. R. Long, J. Am. Chem. Soc., 2015, 137, 13594–13602 CrossRef PubMed.
- E. Bakker, P. Buhlmann and E. Pretsch, Chem. Rev., 1997, 97, 3083–3132 CrossRef CAS PubMed.
- X. J. Xie, A. Gutierrez, V. Trofimov, I. Szilagyi, T. Soldati and E. Bakker, Anal. Chem., 2015, 87, 9954–9959 CrossRef CAS PubMed.
- L. E. Chen, Y. Tan, X. F. Liu and Y. W. Chen, Nano Energy, 2016, 27, 492–498 CrossRef CAS; A. C. Veron, H. Zhang, A. Linden, F. Nuesch, J. Heier, R. Hany and T. Geiger, Org. Lett., 2014, 16, 1044–1047 CrossRef PubMed.
- D. X. Ma, C. Zhang, Y. Qiu and L. Duan, J. Mater. Chem. C, 2016, 4, 5731–5738 RSC; D. X. Ma, L. Duan and Y. Qiu, J. Mater. Chem. C, 2016, 4, 5051–5058 RSC.
- H. Yao and K. Ashiba, RSC Adv., 2011, 1, 834–838 RSC.
- D. K. Bwambok, B. El-Zahab, S. K. Challa, M. Li, L. Chandler, G. A. Baker and I. M. Warner, ACS Nano, 2009, 3, 3854–3860 CrossRef CAS PubMed; A. N. Jordan, S. Das, N. Siraj, S. L. de Rooy, M. Li, B. El-Zahab, L. Chandler, G. A. Baker and I. M. Warner, Nanoscale, 2012, 4, 5031–5038 RSC; M. Soulie, C. Frongia, V. Lobjois and S. Fery-Forgues, RSC Adv., 2015, 5, 1181–1190 RSC; W. I. S. Galpothdeniya, F. R. Fronczek, M. Y. Cong, N. Bhattarai, N. Siraj and I. M. Warner, J. Mater. Chem. B, 2016, 4, 1414–1422 RSC.
- I. Shulov, S. Oncul, A. Reisch, Y. Arntz, M. Collot, Y. Mely and A. S. Klymchenko, Nanoscale, 2015, 7, 18198–18210 RSC.
- I. Shulov, Y. Arntz, Y. Mely, V. G. Pivovarenko and A. S. Klymchenko, Chem. Commun., 2016, 52, 7962–7965 RSC.
- A. Reisch, A. Runser, Y. Arntz, Y. Mély and A. S. Klymchenko, ACS Nano, 2015, 9, 5104–5116 CrossRef CAS PubMed.
- K. Trofymchuk, L. Prodi, A. Reisch, Y. Mély, K. Altenhöner, J. Mattay and A. S. Klymchenko, J. Phys. Chem. Lett., 2015, 6, 2259–2264 CrossRef CAS PubMed.
- I. Krossing and A. Reisinger, Coord. Chem. Rev., 2006, 250, 2721–2744 CrossRef CAS.
- S. M. Ivanova, B. G. Nolan, Y. Kobayashi, S. M. Miller, O. P. Anderson and S. H. Strauss, Chem. – Eur. J., 2001, 7, 503–510 CrossRef CAS.
- I. Krossing, Chem. – Eur. J., 2001, 7, 490–502 CrossRef CAS.
- E. M. M. Del Valle, Process Biochem., 2004, 39, 1033–1046 CrossRef CAS.
- M. Cametti, B. Crousse, P. Metrangolo, R. Milani and G. Resnati, Chem. Soc. Rev., 2012, 41, 31–42 RSC.
- R. A. Velapoldi and H. H. Tonnesen, J. Fluoresc., 2004, 14, 465–472 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7qm00248c |
| This journal is © the Partner Organisations 2017 |