
Pt-embedded-CeO2 hollow spheres for enhancing CO oxidation performance†
Ke
Wu
a,
Liang
Zhou
a,
Chun-Jiang
Jia
 b,
Ling-Dong
Sun
*a and
Chun-Hua
Yan
*a
b,
Ling-Dong
Sun
*a and
Chun-Hua
Yan
*a
aBeijing National Laboratory for Molecular Sciences, State Key Laboratory of Rare Earth Materials Chemistry and Applications, PKU-HKU Joint Laboratory in Rare Earth Materials and Bioinorganic Chemistry, and College of Chemistry and Molecular Engineering, Peking University, Beijing 100871, China. E-mail: sun@pku.edu.cn; yan@pku.edu.cn
bKey Laboratory for Colloid and Interface Chemistry, Key Laboratory of Special Aggregated Materials, School of Chemistry and Chemical Engineering, Shandong University, Jinan 250100, China
First published on 29th June 2017
Abstract
Exploring high-performance catalysts has always been a general concern for promoting extended heterocatalysis reactions. Here, Pt embedded highly-porous CeO2 hollow sphere (Pt/CeO2 HS) composites are developed by a one-pot template-free solvothermal method. Evolution mechanism studies unravel that the Pt/CeO2 HS composites are derived from a self-assembly–reduction–Ostwald ripening process, where Pt nanoparticles (Pt NPs) are embedded into ceria mesoporous hollow spheres. The as-developed embedment strategy is more facile, sustainable and cost-effective than conventional deposition approaches, ensuring the precise control of the location, distribution, and uniformity of Pt NPs throughout the outer shell of hollow spherical CeO2. Importantly, the Pt NP embedding process could be determined to play a key role in creating oxygen vacancies and activating surface chemisorbed oxygen. Besides that, the aggregation of Pt NPs can be efficiently inhibited in the Pt/CeO2 HS composites, and more active sites should be involved in catalytic reactions. All these advantages contribute to the strikingly improved performance as well as excellent stability of the Pt/CeO2 HS composites toward CO oxidation reaction compared with mesoporous CeO2 nanospheres (CeO2 NS) and Pt/CeO2 NS reference catalysts. These findings present a decent protocol for desgining noble metal/oxide hollow structural composites in heterogeneous catalysis.
Introduction
Pt-based nanocatalysts have been widely studied in recent years because of their extensive applications in various catalysis reactions, such as volatile organic compound (VOC) elimination, auto-mobile exhaust purification, selective hydrogenations, and organocatalytic reactions, etc.1,2 In particular, Pt-based nanocomposite catalysts have attracted much attention for catalyzing the oxidation of carbon monoxide (CO), which is highly toxic and can cause serious health damage as well as environmental pollution.3,4–7 Pt-based nanocatalysts prepared by conventional methods, including coprecipitation, impregnation, deposition–precipitation, etc.,8 feature small Pt NPs dispersed on support nanomaterials with high surface area, in which the Pt NPs tend to migrate and grow into larger particles to minimize their surface energy when used at high temperatures.9 As a result, the inevitable sintering of supported Pt NPs results in serious catalytic degradation and even complete inactivation of heterogeneous catalysts.10–12 Hence, the synthesis of Pt related catalysts with excellent anti-sintering capabilities has become an important area. In order to satisfy the urgent demand for thermally stable catalysts, it is desirable to inhibit the active centers of Pt NPs in inexpensive and robust supports with isolated cavities to prevent particle migration and the corresponding decay of the catalytic activity.13–16 Therefore, on the one hand, hollow micro-/nanostructures are considered as potential candidates for catalyst supports, since the hollow geometry provides high surface area and internal void space, open porous channels, favoring a more homogeneous dispersion of Pt NPs.17–23 On the other hand, embedding Pt NPs into the outer shell of hollow materials to produce Pt-embedded composites can probably maximize the interaction between the Pt NPs and the support, which can efficiently suppress the migration, aggregation and deactivation of Pt NPs during catalytic reactions, thus eventually contributing to activity improvement.24,25Compared to inert supports such as alumina, cerium dioxide (CeO2), one of the most abundant rare earth oxides on earth, is an active substrate that can greatly increase the reaction rates of redox steps owing to its numerous oxygen vacancy defects, high oxygen storage capacity, and ability to shift facilely between Ce(IV) and Ce(III).26–35 Therefore, the unique electronic configuration and structural properties of ceria make Pt-embedded CeO2 hollow materials effective promoters in three-way-catalysts for CO oxidation.36–39 To date, the template method has been commonly used to fabricate Pt-embedded CeO2 hollow structures, while the template selection and the subsequent removal of the template are critical and complicated.40,41 As a result, the designed approach to embedding small Pt NPs into the outer shell of porous CeO2 hollow structures with a well-controlled rigid structure is of particular interest and has been explored to be one of the most efficient strategies to design high-performance catalysts since it allows efficient control of the location, distribution and uniformity of supported Pt NPs with short processing time, thus leading to efficient enhancement of the catalytic activities.
Herein, we report a facile and one-pot acetic acid assisted ethylene glycol solvothermal method for the well-controlled synthesis of Pt-embedded-CeO2 porous hollow sphere (Pt/CeO2 HS) composites. The as-prepared Pt/CeO2 HS composites with porous hollow interiors, uniform dispersion of Pt NPs, and self-supported micro-/nanostructures can meet the requirements of high catalytic activity and superior durability for CO oxidation reaction as a result of their novel hollow Pt-embedded structure, which not only provides Pt/CeO2 HS composites with a synergistic effect between the active sites that consist of self-assembled CeO2 NPs and the embedded Pt NPs, but also effectively enables Pt NPs to separate from each other to prevent their aggregation during repetitive CO oxidation reactions.
Experimental section
Chemicals
Cerium nitrate hexahydrate (Ce(NO3)3·6H2O, 99%), ethylene glycol (EG, 99%), potassium tetrachloroplatinate(II) (K2PtCl4), rhodium chloride trihydrate(III) (RhCl3·3H2O), chloroauric acid trihydrate(III) (HAuCl4·3H2O), sodium tetrachloropalladate(II) (H2PdCl4), formic acid (HCOOH), acetic acid (CH3COOH), and propionic acid (CH3CH2COOH) were purchased from Sinopharm Chemical Reagent Co., Ltd (Shanghai, China). N,N-Dimethylformamide (DMF, A.R.,) was purchased from Xilong Chemical Co. Ltd, China. All reagents were of analytical reagent grade and used without further purification. All of the aqueous solutions were prepared with ultrapure water (Millipore, 18.2 MΩ cm).Synthesis of Pt/CeO2 HS composite catalysts
In a typical synthesis, 1.0 g of Ce(NO3)3·6H2O was dissolved in 1.0 mL of deionized water. Then, 1.0 mL of acetic acid was added with vigorous stirring to form a uniform solution. The mixed solution and 30 mL ethylene glycol were added into a 50 mL Teflon-lined stainless-steel autoclave under stirring at room temperature. After stirring for several minutes, a certain amount of K2PtCl4 was added to the above homogeneous solution under intensive stirring for 30 min. After that, the autoclave was sealed and heated at 180 °C for 150 min to get the products. After the solvothermal treatment, the obtained suspensions were collected by centrifugation at 8000 rpm for 10 min and then washed with water/ethanol several times. Finally, the products were dried at 60 °C overnight and calcined at 300 °C for 4 hours, and denoted as Pt/CeO2 HS composites. For comparison, single component CeO2 nanospheres (CeO2 NS) were also prepared under similar experimental conditions without addition of K2PtCl4.Synthesis of Pt/CeO2 NS reference catalysts
Pt NPs were synthesized using a facile strategy similar to that of the Pt/CeO2 HS composites except with the absence of Ce(NO3)3, and the Pt/CeO2 NS reference catalysts were further prepared by physically mixing the Pt NPs with CeO2 NS, while carefully controlling the loading of Pt to be approximately equal to that of the Pt/CeO2 HS composite catalysts. The as-prepared mixtures were further stirred at room temperature for 2 hours and then centrifuged at 8000 rpm for 10 min. Finally, the products were washed with water/ethanol several times, dried in air at 60 °C overnight and then calcined in still air at 300 °C for 4 hours.Synthesis of noble metal (M = Pd, Au, Rh)/CeO2 (M/CeO2) composites
H2PdCl4, HAuCl4, and RhCl3 were separately used to replace K2PtCl4 to produce M/CeO2 composites (M = Pd, Au, Rh). The synthesis process was similar to that of the Pt/CeO2 HS composites.Catalytic tests
The catalytic activities of the samples for CO oxidation were evaluated in a plug flow reactor using 50 mg of sieved (20–40 mesh) catalysts in a reactant gas mixture of 1% CO/20% O2/79% N2 (from Deyang Gas Co., Jinan, 99.997% purity), at an hourly space velocity of 80![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 mL h−1 gcat−1, corresponding to 67 mL min−1. Before the catalytic tests, the catalysts were pretreated in synthetic air (21 vol% O2/79 vol% N2) for 30 min at 300 °C for activation. Then, the stainless-steel reactor was cooled to room temperature under a flow of pure N2 gas. Upon ramping the catalysts’ temperature (3 °C min−1) from room temperature to 300 °C, the catalytic tests of CO oxidation were carried out in the reactant gas atmosphere. Nondispersive IR spectroscopy (Gasboard 3500, Wuhan Sifang Company, China) was used to monitor the outlet gas compositions of CO and CO2 online. The CO conversion ratio was calculated conforming to the following equation: CO conversion = (COinput–Coout)/COinput. The stability tests were carried out under a cycling experiment. The cycle was repeated five times. Long-term stability tests were performed under the reactant gas mixture at 152 °C for 12 hours.
000 mL h−1 gcat−1, corresponding to 67 mL min−1. Before the catalytic tests, the catalysts were pretreated in synthetic air (21 vol% O2/79 vol% N2) for 30 min at 300 °C for activation. Then, the stainless-steel reactor was cooled to room temperature under a flow of pure N2 gas. Upon ramping the catalysts’ temperature (3 °C min−1) from room temperature to 300 °C, the catalytic tests of CO oxidation were carried out in the reactant gas atmosphere. Nondispersive IR spectroscopy (Gasboard 3500, Wuhan Sifang Company, China) was used to monitor the outlet gas compositions of CO and CO2 online. The CO conversion ratio was calculated conforming to the following equation: CO conversion = (COinput–Coout)/COinput. The stability tests were carried out under a cycling experiment. The cycle was repeated five times. Long-term stability tests were performed under the reactant gas mixture at 152 °C for 12 hours.
Apparatus
Transmission electron microscopy (TEM), high-resolution transmission electron microscopy (HRTEM), high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM), linear EDX scanning and energy dispersive spectroscopy mapping (EDX-Mapping) measurements were taken using a JEOL JEM-2100F transmission electron microscope operated at 200 kV. X-ray diffraction (XRD) patterns were obtained with a Rigaku D/MAX-2000 diffractometer (Japan) with a slit of 1/2° at a scanning rate of 3° min−1 using a Cu Kα radiation source (λ = 1.5406 Å) and operating at 40 kV and 40 mA. The surface compositions of the catalysts were determined using X-ray photoelectron spectroscopy (XPS) carried out on an Axi Ultra X-ray imaging photoelectron spectrometer with an Al Kα radiator, and the vacuum in the analysis chamber was maintained at about 10−9 mbar. The binding energy was calibrated by means of the C 1s peak energy of 284.8 eV. The surface Ce(III) concentrations (Ce(III) atom%) were determined by integrating the areas of the Ce(III) 3d peaks with the CasaXPS software. Nitrogen adsorption–desorption isotherm (SADI) experiments were performed at 77 K on an Accelerated Surface Area & Porosimetry system (Micromeritics, ASAP 2010) to determine the Brunauer–Emmett–Teller (BET) surface area and pore size distribution. Fourier transform infrared spectroscopy (FT-IR) was carried out using a Bruker Tensor27 spectrophotometer. Raman spectra were recorded on a Jobin-Yvon HR800 Laser Raman Microscope with 488 nm laser excitation. Thermogravimetric analysis (TGA) was performed on a Q600SDT under N2 flow (100 mL min−1). Inductively coupled plasma optical emission spectroscopy (ICP-OES) results were obtained with a Profile Spec spectrometer (Leeman, USA). Gas Chromatography-Mass Spectrometer (GC-MS) spectra were measured on an Agilent 7890A/5975C GC/MSD system equipped with a DB-17MS (30 m, 0.25 mm, 0.25 µm) column.H2 temperature-programmed reduction (H2-TPR) was carried on a Builder PCSA-1000 apparatus with a thermal conductivity detector (TCD). The sieved catalysts (20–40 mesh, 30 mg) were pretreated in pure O2 at 300 °C for 30 min prior to the measurement. H2-TPR was run from room temperature to 900 °C at a ramping rate of 10 °C min−1, with the reduction gas consisting of a 5% H2/Ar gas mixture at a flow rate of 30 mL min−1. CO temperature-programmed reduction (CO-TPR) results were obtained using a TILON LC-D200M quadrupole mass spectrometer, which can online detect the reaction products. The sample (50 mg) was mixed with 250 mg quartz sand and then placed in a quartz tube. Prior to the CO-TPR experiments, the sieved catalysts were pretreated in pure O2 at 300 °C for 30 min. Then, CO-TPR was performed in a gas mixture of 5% CO/He (30 mL min−1) with heating from room temperature to 800 °C at a ramping rate of 10 °C min−1.
Results and discussion
Structural characterizations of the Pt/CeO2 HS composite catalysts
Fig. 1 shows the morphological, structural and compositional characterizations of the as-prepared Pt/CeO2 HS composite catalysts. The large area SEM image in Fig. 1a directly reveals spherical particles with an average particle diameter of ∼200 nm. The magnified SEM image in Fig. 1b confirms the rough surface of the Pt/CeO2 HS composites, indicating a three-dimensionally interconnected porous structure with self-assembled CeO2 nanograins as building blocks. Furthermore, a single cracked Pt/CeO2 HS in Fig. 1c clearly shows that the thickness of the hollow sphere is ca. 35 nm. Besides that, it can be seen from the TEM images in Fig. 1d and e that the Pt NPs (red arrows) surrounded by self-supported CeO2 NPs can be distinguished by their deeper contrast to the inner brighter part. The corresponding selected-area electron diffraction (SAED) image in Fig. S1 (ESI†) demonstrates that the as-prepared Pt/CeO2 HS composites should have a polycrystalline structure. The high-resolution TEM (HRTEM) image in Fig. 1f further indicates that each hollow sphere is composed of many single-crystalline grains with random crystallographic orientation. The interplanar spacing of 0.32 nm is observed at most regions on the surface of the Pt/CeO2 HS composite catalysts, which is related to the (111) lattice spacing of cerium dioxide (0.31 nm). Additionally, high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) also verifies the hollow structure through image contrast (Fig. 1g). EDS mapping and the EDS line scanning profile are performed to clearly distinguish the distribution of Pt and Ce elements in the Pt/CeO2 HS composites. The EDS line scanning in Fig. 1g across a single hollow sphere further confirms the dispersity of CeO2 and Pt NPs. Moreover, the resulting EDS mapping patterns in Fig. 1h show that both Ce and O are distributed throughout the whole Pt/CeO2 HS while the more dense points in the mapping image indicate that Pt is much more rich in the outer part of the Pt/CeO2 HS composites, which is strong evidence for the formation of a hollow embedded nanostructure. The metal loading of Pt in the Pt/CeO2 HS composites is ca. 1.4 wt% according to the result of EDS data in Fig. 1i, which is in accordance with ICP-OES results (1.6 wt%, Table S1 in ESI†) and the theoretical stoichiometric proportion (1.5 wt%).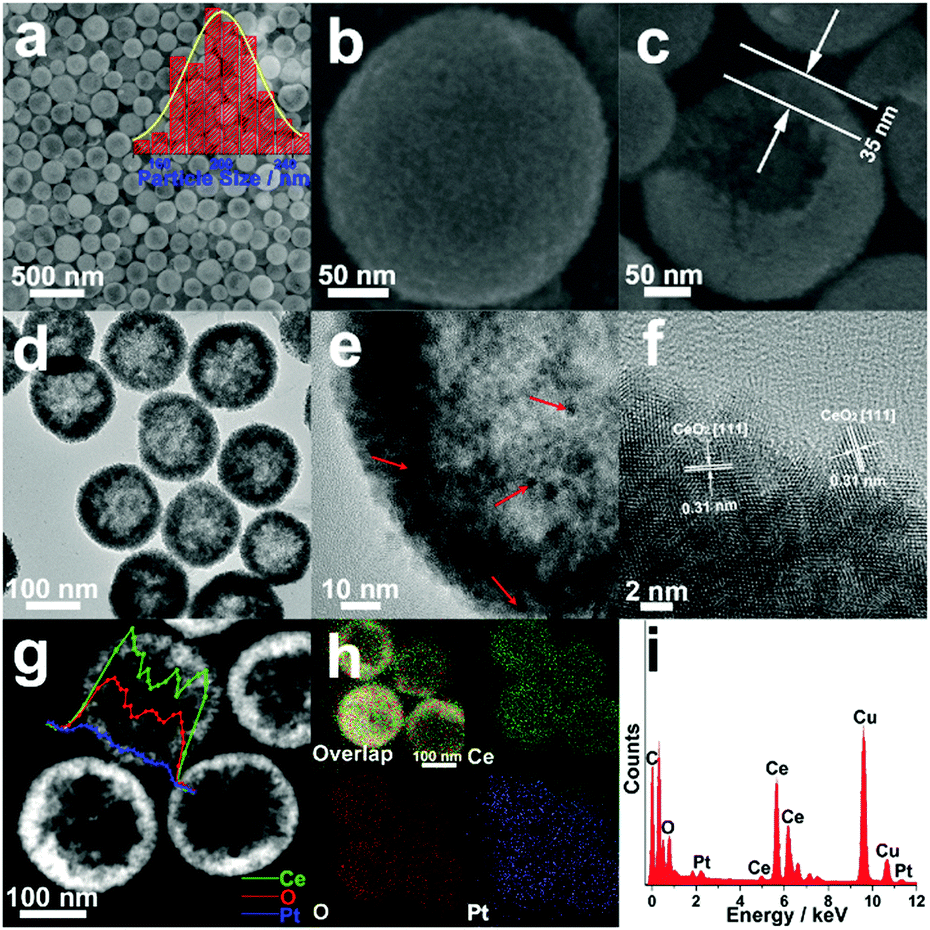 | ||
| Fig. 1 Morphological, structural and compositional characterizations of Pt/CeO2 HS composites: (a–c) SEM images, the illustration in (a) is the corresponding size distribution diagram; (d and e) low resolution TEM images; (f) HRTEM image; (g) HAADF-STEM image, the inset in (g) shows the linear EDS line scanning image. (h) EDS mapping images. (i) EDX spectrum. | ||
Fig. 2 shows the XRD patterns of the Pt/CeO2 HS composites and CeO2 NS. A weak and broad peak of the Pt/CeO2 HS composites at about 40° is clearly observed, which could be attributed to the (111) crystallographic facet of the face-centered cubic (fcc) structure of Pt NPs (JCPDS 04-0802), confirming that Pt is successfully reduced. Except for the peak of Pt NPs, the Pt/CeO2 HS composite catalysts show similar diffraction peaks to those for CeO2 NS, which match well with the fluorite structured CeO2 (JCPDS no. 34-0394). According to the XRD pattern, the width of the XRD peaks can provide detailed analysis for the average crystallite size. Generally, the broader the XRD peak, the smaller the size of the corresponding NPs.42 According to the Scherrer equation, the average particle size (dXRD) of the small Pt NPs and CeO2 grains of the Pt/CeO2 HS composites is calculated to be 5.7 nm and 6.5 nm, respectively. Meanwhile, the dXRD value of CeO2 NS (5.5 nm) is the same as that of the Pt/CeO2 HS composites. The similar dXRD value further confirms that the Pt/CeO2 HS composites and CeO2 NS share the same crystalline structure.
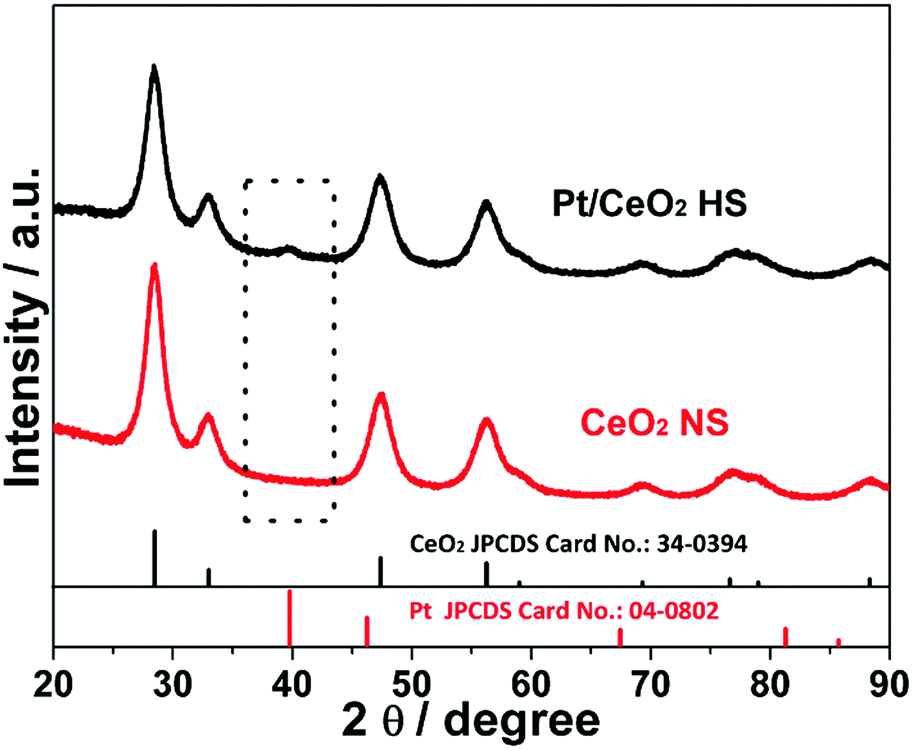 | ||
| Fig. 2 XRD patterns of the Pt/CeO2 HS composites and CeO2 NS. | ||
The porous structure of the Pt/CeO2 HS composites was further confirmed by means of nitrogen adsorption–desorption isotherm (SADI) experiments (Fig. 3a). The steep increase in the curve at a higher relative pressure exhibits a typical type IV nitrogen adsorption branch with two hysteresis loops in the relative pressure range of 0.4–1.0, which is assigned to the typical adsorption–desorption characteristics of porous materials.43 Correspondingly, the pore size distribution of Pt/CeO2 HS composites and CeO2 NS calculated by the Barrett–Joyner–Halenda (BJH) formula derived from the desorption branch are similar, and the pore diameters are mainly distributed at about 4 nm (Fig. 3b), indicating that the nearly narrow pore size distributions and the building blocks of the CeO2 grains are not tightly adhered to each other. The BET surface area of the Pt/CeO2 HS composites is measured to be 190.1 m2 g−1, which is a little bit smaller than the value of 210 m2 g−1 calculated for the CeO2 NS. For the Pt/CeO2 HS composites, the Pt NPs are surrounded by CeO2 nanograins, which somewhat blocks the pores of the CeO2 hollow spheres and leads to a decreased surface area.44 Obviously, the porous Pt/CeO2 HS composites can be expected to promote the CO oxidation reaction and achieve higher catalytic performance due to the unique hollow embedded micro-/nanostructure and the high surface area.45
 | ||
| Fig. 3 (a) Typical nitrogen adsorption/desorption isotherm of the Pt/CeO2 HS composites and CeO2 NS; (b) the corresponding pore-size distribution curve obtained from the Barrett–Joyner–Halenda method. | ||
FT-IR spectra were used to characterize the adsorption species on the surface of the Pt/CeO2 HS composites. As Fig. S2 (ESI†) shows, the uncalcinated CeO2 NS and Pt/CeO2 HS composites both display strong characteristic bands at around 1650 cm−1, which are ascribed to the stretching vibrations of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O (carboxyl or carbonyl).46 After being calcinated at 300 °C for 4 hours, the band at about 1650 cm−1 for the CO stretch dramatically decreases, which means that the acetate ions on the surface of the Pt/CeO2 HS composites have been entirely removed. Thermal gravity analysis (TGA) is further employed to study the amounts of acetate ions adsorbed on the Pt/CeO2 HS composites. As shown in Fig. S3 (ESI†), the Pt/CeO2 HS composites experience a total weight loss of 31.6% of the original mass when calcined up to 800 °C.
O (carboxyl or carbonyl).46 After being calcinated at 300 °C for 4 hours, the band at about 1650 cm−1 for the CO stretch dramatically decreases, which means that the acetate ions on the surface of the Pt/CeO2 HS composites have been entirely removed. Thermal gravity analysis (TGA) is further employed to study the amounts of acetate ions adsorbed on the Pt/CeO2 HS composites. As shown in Fig. S3 (ESI†), the Pt/CeO2 HS composites experience a total weight loss of 31.6% of the original mass when calcined up to 800 °C.
XPS analysis was performed to investigate the chemical and oxidation states for the elements of Ce, O and Pt. As shown in Fig. 4a, the Ce 3d high-resolution XPS spectra of the Pt/CeO2 HS composites is fitted to a mixture of both Ce(III) and Ce(IV) oxidation states. The Ce(III)/[Ce(III) + Ce(IV)] ratio of the Pt/CeO2 HS composites can be quasi-quantitatively determined as 41.7% (Table S1, ESI†), which is higher than that of the Pt/CeO2 NS reference catalysts (34.3%) and CeO2 NS (34.7%), respectively.47 Generally, the Ce(III) content may be positively proportional to the oxygen vacancies and defects of ceria, thus the higher proportion of Ce(III) in the Pt/CeO2 HS composite catalysts indicates the existence of more oxygen vacancies.36,48
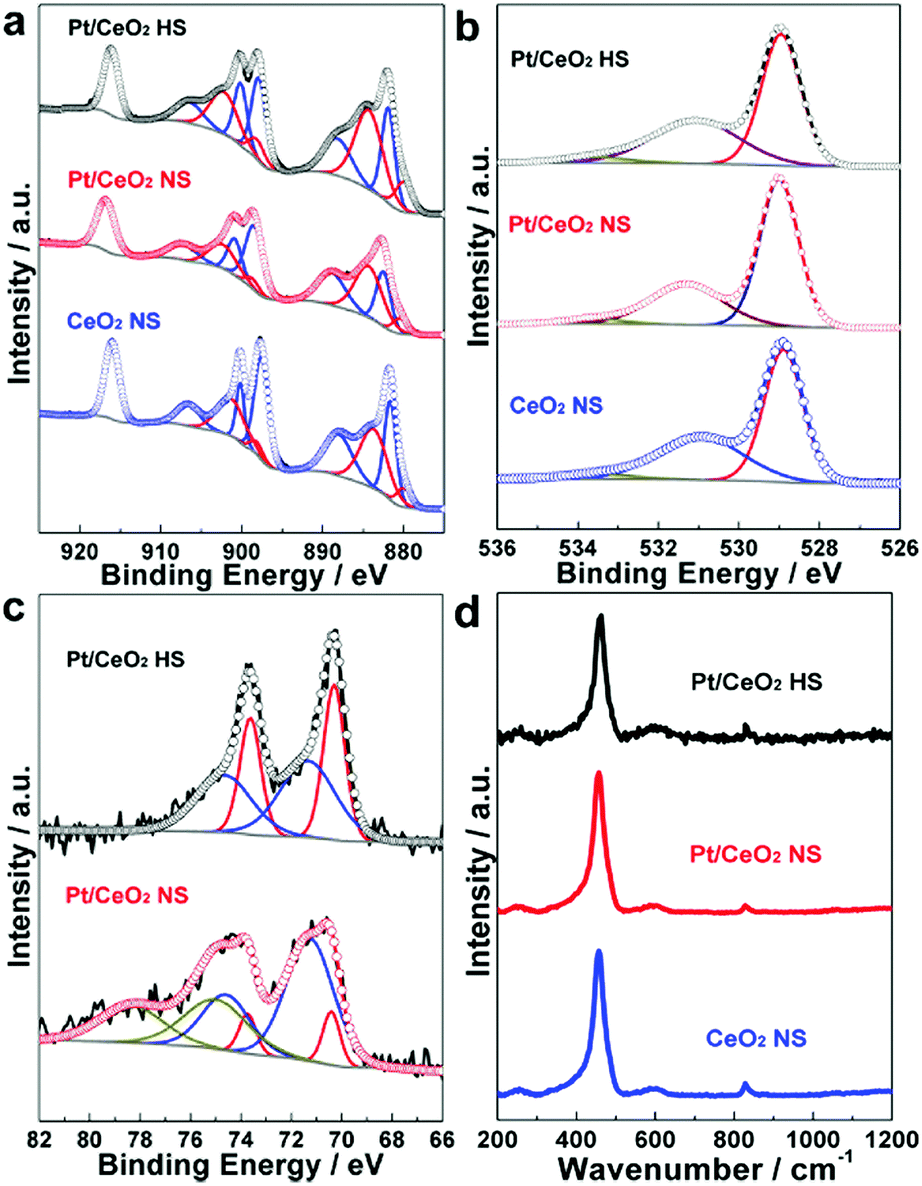 | ||
| Fig. 4 (a) Ce 3d and (b) O 2p XPS spectra of the Pt/CeO2 HS composites, Pt/CeO2 NS reference catalysts and CeO2 NS, respectively; (c) Pt 4f XPS spectra of the Pt/CeO2 HS composites and Pt/CeO2 NS reference catalysts; (d) Raman spectra of the Pt/CeO2 HS composites and CeO2 NS. | ||
The above results are further confirmed by the O 1s XPS spectra depicted in Fig. 4b. The O 1s peak can be deconvolved into three peaks. The lower binding energy at 529.0 eV is attributed to the lattice oxygen of ceria, while the shoulder peak centered at 531.1 eV is assigned to the absorbed oxygen, weakly bonded hydroxyl species, and defective oxygen. Furthermore, the higher binding energy located at 533.5 eV is ascribed to the oxygen in hydroxyl groups and surface bonded water.49 It can be noticed that the relative concentration of adsorbed oxygen (Oad) for the Pt/CeO2 HS composites (41.2%) is higher than those of the Pt/CeO2 NS reference catalysts (32.7%) and CeO2 NS (39.6%), respectively. Combined with the results obtained from the Ce 3d XPS data, the embedment of Pt NPs in the Pt/CeO2 HS composites is likely favorable for the formation of oxygen vacancies.50
The Pt4f signal of the Pt/CeO2 HS composites in Fig. 4c is deconvoluted into two components: 70.3 eV (Pt 4f7/2), 73.6 eV (Pt 4f5/2) and 71.4 eV (Pt 4f7/2), 74.7 eV (Pt 4f5/2), which are assigned to the Pt(0) and Pt–O–Ce species, respectively.51 In addition, Pt(IV) species are also observed in the Pt 4f XPS spectra of the Pt/CeO2 NS reference catalysts at higher binding energy: 74.5 eV (Pt 4f7/2), 77.8 eV (Pt 4f5/2). By measuring the relative deconvoluted peak areas, the proportion of Pt(0) species for the Pt/CeO2 HS composite catalysts and Pt/CeO2 NS reference catalysts are calculated to be 50.2% and 10.3% (Table S1, ESI†), respectively, indicating that Pt(0) species in the Pt/CeO2 HS composites are successfully prevented from being oxidized during the calcination process at 300 °C, which is due to the effective protection by the surrounding CeO2 NPs.52 Overoxidation of the Pt NPs can lead to the deactivation of the Pt sites, thus the Pt/CeO2 HS composites, which exhibit a moderate oxidation state of Pt species, could be suitable for heterocatalysis reactions.53,54
Fig. 4d shows the Raman spectra of the Pt/CeO2 HS composites, Pt/CeO2 NS, and CeO2 NS measured under 488 nm laser excitation. All of the three samples show one prominent vibration at ca. 455 cm−1 and one weak peak at ca. 600 cm−1, arising from the strong Raman-active F2g vibrational mode of the CeO2 fluorite phase and defect-induced (D) mode, respectively.55 The relative intensity I(D)/I(F2g) ratio of the Pt/CeO2 HS composites (0.20) is higher than those of the Pt/CeO2 NS reference catalysts (0.09) and CeO2 NS (0.10), indicating that much more intrinsic defect sites in the CeO2 fluorite structure of Pt/CeO2 HS composites are created.50 Compared to the Pt/CeO2 NS reference catalysts and CeO2 NS, the generation of more oxygen vacancies on the Pt/CeO2 HS composites might be ascribed to the incorporation of Pt into the lattice of cerium dioxide, thus facilitating the activation of lattice oxygen and the formation of oxygen vacancies.56,57
The growth mechanism of the Pt/CeO2 HS composites
The formation mechanism of the Pt/CeO2 HS composites can be discussed in detail based on condition-controlled experiments by altering the reaction parameters. The evolution process of the Pt/CeO2 HS composites is examined at different solvothermal reaction times. When the reaction proceeds to 30 min, CeO2 nanospheres with a rough surface and small amounts of scattered CeO2 NPs can be observed in Fig. 5a as the original transparent solution slowly become blurred. During this period, the hydrolysis of Ce(III) ion could be gradually promoted as the synthesis time increases, followed by the subsequent oxidation of Ce(III) species by NO3− ions to form the nanosized CeO2 precusors.58 After that, the as-prepared nucleated CeO2 nanoparticles self-assemble into the primary CeO2 solid nanospheres with larger diameter step by step. When the reaction time is further extended to 60 min, homogeneous solid CeO2 nanospheres with an apparent size expansion can be found (Fig. 5b), which is probably due to the outward migration of CeO2 nanograins in the Ostwald ripening process.59 In this circumstance, the color of the primary solution changed to grey black, which implies the possible formation of Pt clusters.60 The Pt clusters and CeO2 nanospheres coexist simultaneously in the mixture solution and further co-assemble to produce Pt/CeO2 NS hybrids, where Pt clusters are mainly distributed on the outer surface of the self-supported CeO2 nanospheres. When the solvothermal time increases to 90 min, the tendency for the formation of the hollow structures becomes more and more obvious (Fig. 5c). At the same time, the pristine dense Pt/CeO2 NS hybrids start to become loose and expose an obviously rough surface, which is composed of tiny CeO2 NPs. After 120 min of the solvothermal reaction, the inner nanoparticles in the Pt/CeO2 hollow spheres gradually disappear and attach to the outer shell to form the cavity (Fig. 5d). Finally, when the outward expansion process is accomplished, Pt/CeO2 HS composites will be synthesized successfully. The morphology and structure changes of different intermediate compounds formed in the synthesis process of the Pt/CeO2 HS composites can also be reflected well through the XRD patterns in Fig. S4 (ESI†). The XRD pattern of the as-prepared sample obtained after 40 min of the solvothermal reaction time can be indexed to the cubic fluorite-type structure of cerium dioxide (JCPDS card no. 34-0394). Besides, there are no additional peaks in the XRD pattern, which indicates the phase purity of the as-obtained CeO2 NS. Nevertheless, when the reaction time reaches 90 min, a weak and broad diffraction peak for Pt NPs (2θ = ca. 40°) can be found carefully as for the prepared Pt/CeO2 HS composites, suggesting the existence of crystalline phases of Pt NPs with a low Pt content. Moreover, there is no evidence of Pt in the EDS spectrum of the as-synthesized product after 40 min of solvothermal reaction (Fig. S5a, ESI†), while the specific Pt signal can be observed clearly from the EDS spectrum for the products at the reaction time of 120 min in Fig. S5b (ESI†).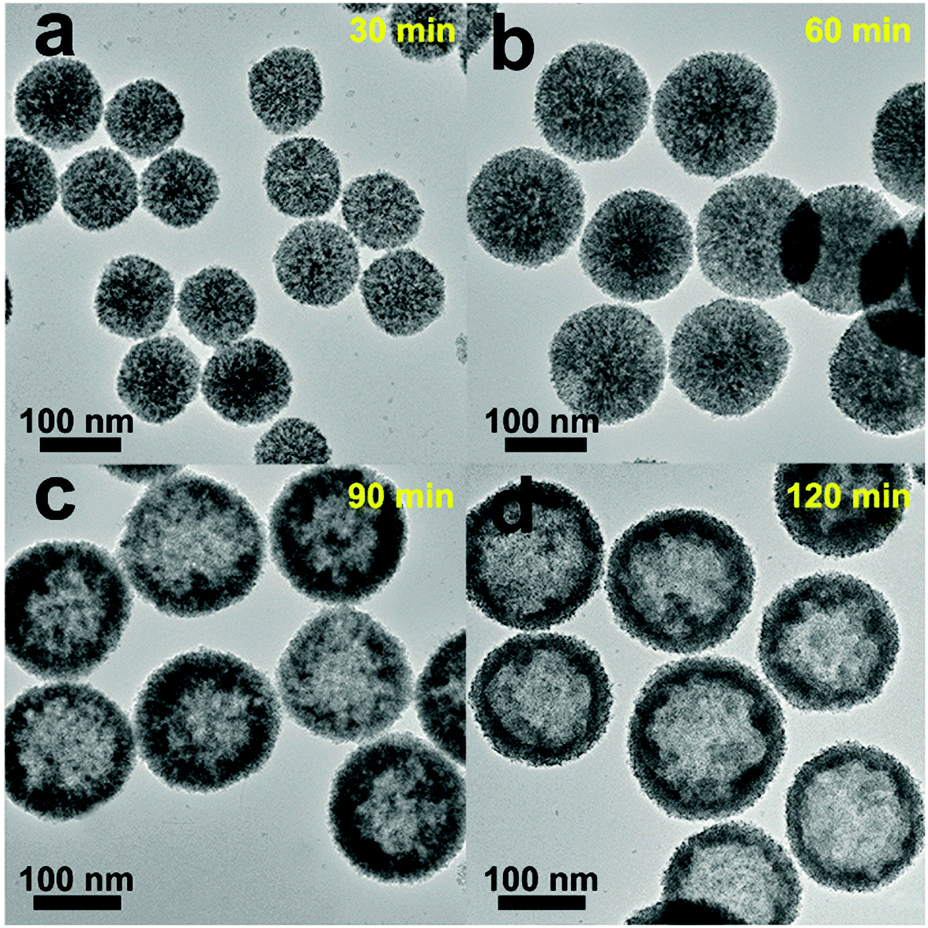 | ||
| Fig. 5 TEM images of the Pt/CeO2 HS composites collected at different growth stages: (a) 30 min, (b) 60 min, (c) 90 min, and (d) 120 min. | ||
To clarify the role of acetic acid, a series of comparative experiments were conducted with different amounts of acetic acid, while the other reaction conditions were kept unchanged. When the reaction was conducted without acetic acid, only some kind of heterogeneous dense nanospheres with diameters around 80 nm could be observed from the TEM image of the as-prepared products (Fig. S6a, ESI†). With an increasing amount of acetic acid, Pt/CeO2 hollow nanostructures start to appear (Fig. S6b, ESI†), while homogeneous mesoporous Pt/CeO2 HS composites are successfully synthesized when the involved amount of acetic acid reaches 1.5 mL (Fig. S6c, ESI†). However, if an excessive amount of acid (3.0 mL) is added, the tiny CeO2 nanocrystals could not be well self-assembled into micro-/nanospheres (Fig. S6d, ESI†). The above results clearly suggest that an appropriate amount of acetic acid is necessary for the formation of the Pt/CeO2 HS composites. These interesting phenomena may be explained in terms of the effect of surface modification arising from the esterification reaction between acetic acid and glycol, which in consequence affects the formation and sell-assembly of primary CeO2 nanocrystals, as well as the co-assembly process to produce Pt/CeO2 HS composites.58,61 Gas-chromatography-mass spectrometry (GC-MS) analysis of the reaction media further confirmed the formation of the ester of 1,2-ethanediol monoacetate through the esterification reaction (Fig. S7a, ESI†).58 Moreover, during the solvothermal reaction process, the increasing pH value of the mixture solution in Fig. S8 (ESI†) further validates the consumption of acetic acid caused by the occurrence of esterification reaction. When acetic acid is replaced with formic acid or propionic acid, unfortunately, only nonuniform Pt/CeO2 hybrid materials can be obtained (Fig. S9, ESI†). The GC-MS results (Fig. S7b and c, ESI†) show that the corresponding esters have been formed through the esterification reaction. However, different carboxylic acids have diverse ionization constants, which remarkably make a difference to the rates and products of the esterification reaction, thus leading to the modulation of the surface modification process to produce samples with distinct morphologies and structures.61
On the one hand, the reaction medium can also play a vital role in determining the structure and morphology of Pt/CeO2 HS composites according to the esterification reaction. When glycol is replaced with polyethylene glycol or glycerol, which both share similar functional groups with glycol, no Pt/CeO2 HS composites can be obtained.61 Moreover, when the solvent with stronger polarity including water or DMF is involved, Pt/CeO2 HS composites could not be produced either (Fig. S10, ESI†). Thus, glycol can also have a great effect on the formation of Pt/CeO2 HS composites. In a controlled experiment without Pt(II) ions, nearly monodisperse CeO2 mesoporous spheres with a porous framework, high surface area and well-defined morphology are obtained in large quantities (Fig. S11, ESI†), consistent with previous literature reported by Li et al.58 In addition, as the load of Pt increases from 0.5 wt% to 2.0 wt% in the starting materials, the as-prepared Pt/CeO2 HS composites become more and more loose, which is likely due to the promoted Ostwald ripening process resulting from the increase of Pt clusters (Fig. S12a–c, ESI†).47 However, if more Pt(II) ions are added, no assembled hollow spheres are formed (Fig. S12d, ESI†). On the other hand, when Pt is replaced by other noble metals, such as Rh, Pd, and Au, it can be seen from the TEM images shown in Fig. S13 (ESI†) that M-CeO2 spheres can be made available. Therefore, the present approach can be a facile and universal method to rationally design and prepare well-controlled micro-/nanoarchitectures of noble metal embedded CeO2 sphere hybrids.
Based on the above experimental results, a possible mechanism can be postulated for the formation of Pt/CeO2 HS composites (Scheme 1). In the first step, small CeO2 nanoparticles are formed firstly through the hydrolysis and oxidation of Ce(III) under solvothermal conditions. As the primary CeO2 nanocrystals are formed in large quantities, they tend to aggregate into self-assembled spheres driven by the minimization of the total surface energy. Subsequently, Pt(II) ions are reduced to produce Pt clusters, and combine with CeO2 NS to co-assemble into Pt/CeO2 NS hybrids with homogeneous Pt cluster dispersion. We infer that the acetic acid and glycol both play a pivotal role as structure-directing agents to dominate the morphology evolution process to produce Pt-embedded CeO2 hollow sphere composites through the appropriate esterification reaction. Importantly, numerous Pt clusters in the reaction system are critical for the formation of a hollow morphology for the Pt/CeO2 HS composites. Considering the absence of Pt(II), no hollow structure will be obtained when the other parameters are constant (Fig. S11, ESI†). Thus, the presence of Pt clusters accelerating the hollowing process is caused by the Ostwald ripening effect and eventually leads to the formation of Pt/CeO2 HS composites.62–64
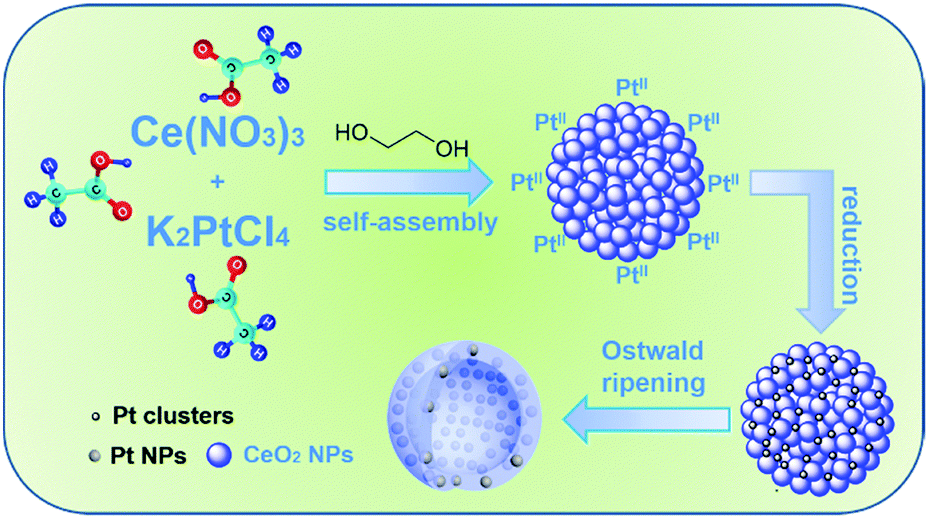 | ||
| Scheme 1 Schematic illustration of the growth process of the Pt/CeO2 HS composites. | ||
Catalytic activity of the Pt/CeO2 HS composites for CO oxidation
The CO oxidation reaction is employed as the probe reaction to investigate the catalytic performance of the as-prepared Pt/CeO2 HS composites. Fig. 6 shows the typical conversion ratio of CO as a function of reaction temperature from 135 °C to 300 °C over the as-prepared catalysts. Compared to the Pt/CeO2 NS reference catalysts, it is found that the Pt/CeO2 HS composites show excellent catalytic reactivity for CO oxidation. The 100% conversion temperature (T100) of Pt/CeO2 HS composites is about 155 °C, which is much lower than that of the Pt/CeO2 NS reference catalysts (186 °C) and CeO2 NS catalysts (288 °C). In addition, as shown in Table S2 (ESI†), the obtained catalytic performance of Pt/CeO2 HS composites for CO oxidation is better than some previously reported data as well, illustrating again the exceptional catalytic activity of the Pt/CeO2 HS composites in this work.7,15,65–67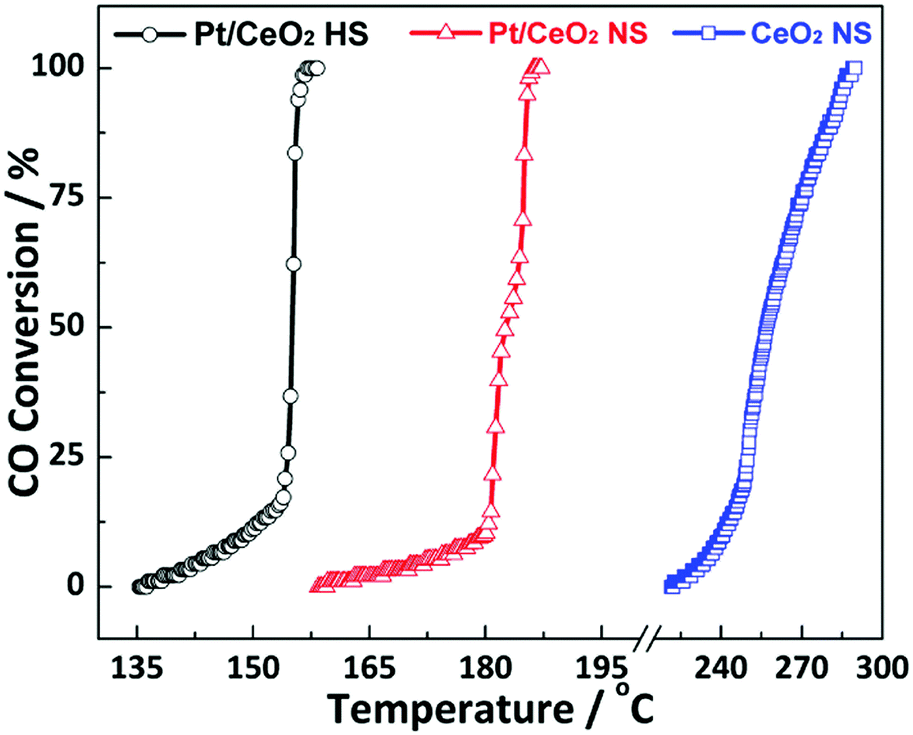 | ||
| Fig. 6 Catalytic performance of the Pt/CeO2 HS composites, Pt/CeO2 NS reference catalysts and CeO2 NS for CO oxidation. | ||
Thermal stability is an important factor to evaluate the catalytic performance of the ceria-based catalysts in real applications. Fig. 7 shows the long-term stability of Pt/CeO2 HS composite catalysts that was investigated by continuous CO oxidation at a constant temperature of 152 °C. As seen in Fig. 7, the CO conversion during 12 h on stream is still kept around 78% on the Pt/CeO2 HS composites. Thus, the slight loss of activity indicates that Pt/CeO2 HS composites have excellent durability and stability for CO oxidation reaction. Besides, as the inset in Fig. 7 shows, even after five successive cycles, the Pt/CeO2 HS composites can maintain a 100% CO conversion rate below 160 °C, which directly confirms their good recyclability. In addition, the TEM image in Fig. S14a and b (ESI†) shows that the morphology of the Pt/CeO2 HS composites is well preserved without the agglomeration or collapse of the hollow sphere structure after the long-term stability test. According to the Scherrer equation, the corresponding average particle size (dXRD) of the embedded Pt NPs in the Pt/CeO2 HS composites that underwent the long-term stability test is calculated to be ca. 6.3 nm from the corresponding labelled diffraction peak of the XRD patterns (Fig. S15a, ESI†). In contrast, it is apparent that the Pt/CeO2 NS reference catalysts experience a dramatic aggregation of small CeO2 units as well as the embedded Pt NPs even after the first cycle of CO oxidation reaction (Fig. S14c and d, ESI†). Larger Pt particles (>20 nm) are observed in the TEM image, indicating a typical coarsening behavior. Besides, the diffraction peak of the XRD pattern in Fig S15b (ESI†) starts to become sharper for the Pt/CeO2 NS reference catalysts after CO oxidation. The relevant average particle size (dXRD) of Pt NPs (ca. 26.9 nm) in Pt/CeO2 NS used after CO oxidation is much larger than that of the primary unused counterparts (ca. 9.8 nm). The different durability behaviours of the Pt/CeO2 HS composite catalysts and Pt/CeO2 NS reference catalysts should be good evidence that the Pt/CeO2 HS composites exhibit superior long-term stability. The enhanced thermal stability of the Pt/CeO2 HS composites could be attributed to the successful embedment of the Pt NPs into the self-supported CeO2 shell and the strong metal–support interaction (SMSI) between the embedded Pt NPs and CeO2 support.68,69
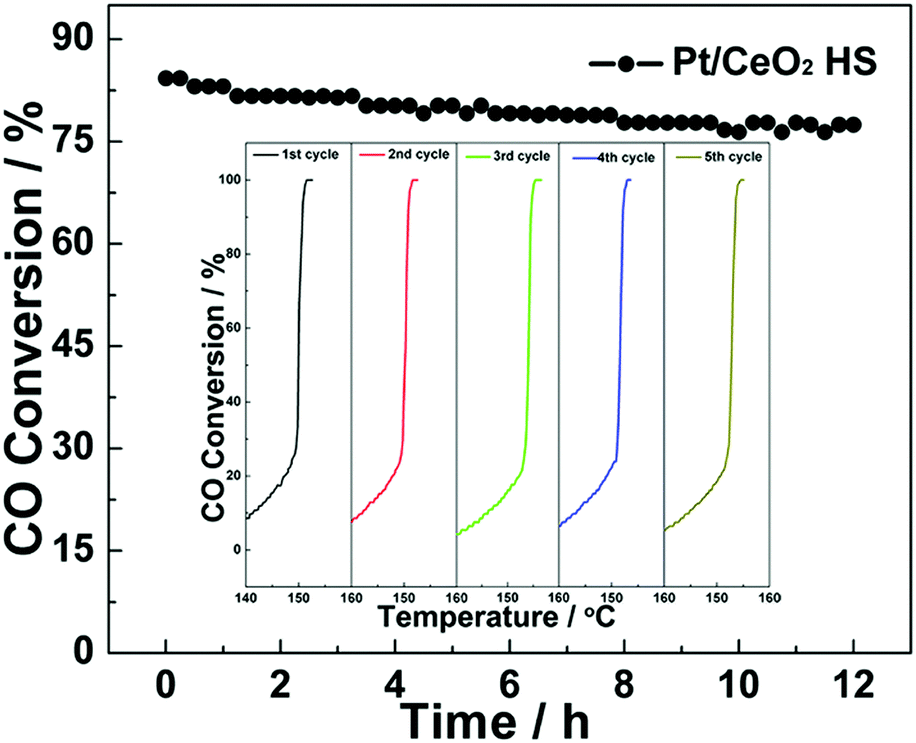 | ||
| Fig. 7 Long-term stability test of the Pt/CeO2 HS composite catalysts for CO oxidation reaction at 152 °C for 12 hours; the inset is the cycling performance of the Pt/CeO2 HS composite catalysts for the CO oxidation reaction. | ||
H2-TPR experiments were used to investigate the reducibility and identify the different surface oxygen species as well as the interaction between Pt species and the CeO2 support of the as-prepared catalysts. The H2-TPR profiles of the Pt/CeO2 HS composites (Fig. 8) apparently show two distinct reduction peaks in the temperature region from 50 °C to 900 °C. The first H2 consumption below 200 °C can be attributed to the reduction of PtOx and the surface active oxygen close to the Pt–ceria interface resulting from the consequent hydrogen spillover from the reduced Pt sites to ceria.54 Meanwhile, the second H2 consumption in the high-temperature region should be ascribed to the reduction of bulk oxygen of ceria. Similarly, two hydrogen reduction peaks of the Pt/CeO2 NS reference catalysts can be observed. The hydrogen reduction peak of the Pt/CeO2 NS reference catalysts in the low temperature region remarkably positively shifts about 45 °C when compared to the Pt/CeO2 HS composites, while the other broad peak is nearly stable at 775 °C. In the sample of Pt/CeO2 NS reference catalysts, Pt NPs are irregularly agglomerated on the CeO2 NS support, resulting in relatively low oxygen at PtOx sites. Thus, the decreasing amount of active sites for CO oxidation leads to notably decreased catalytic performance. However, on the one hand, the embedded Pt NPs in the Pt/CeO2 HS composites are densely located and highly dispersed within the shells of the self-supported porous hollow CeO2 spheres to prevent their severe sintering when used at high temperature. On the other hand, Pt(II) ions could incorporate into the lattice of ceria in the Pt/CeO2 HS composites to create more oxygen vacancies, which has been proved by the results of XPS and Raman spectra, and thus should be beneficial for the surface oxygen activation during CO oxidation.44 In contrast, the reduction peak of CeO2 NS centered at about 260 °C should be ascribed to the reduction of surface lattice oxygen, which cannot be clearly observed for the samples of Pt/CeO2 HS composites and Pt/CeO2 NS reference catalysts.60 This result indicates that Pt can promote the reduction of ceria by the H2 spillover from the Pt to the oxide support. Therefore, CO-TPR tests are also performed to reveal the CO absorption sites and adsorbed states while avoiding the H2 spillover. Fig. S16 (ESI†) presents the CO-TPR profiles of the Pt/CeO2 HS composites and Pt/CeO2 NS reference catalysts obtained by simultaneously monitoring the signals of CO consumption and CO2 formation on mass spectroscopy. For the Pt/CeO2 HS composites, the peak centered at 150 °C can be attributed to the reduction of PtOx. For the reduction of the Pt/CeO2 NS reference catalysts, the corresponding peak shifts to 173 °C, indicating again that the Pt/CeO2 HS composites show a higher reducibility than their Pt/CeO2 NS counterparts.70,71 In addition, the reduction peak of the Pt/CeO2 HS composites seen at about 400 °C is probably due to reduction of some of the CeO2 surface oxygen, which is less negative than that of the Pt/CeO2 NS reference catalysts (428 °C) as well.72 Hence, according to the above H2-TPR and CO-TPR results, the Pt/CeO2 HS composites could provide more active sites than the Pt/CeO2 NS reference catalysts to activate the interfacial oxygen to facilitate the CO oxidation reaction.73
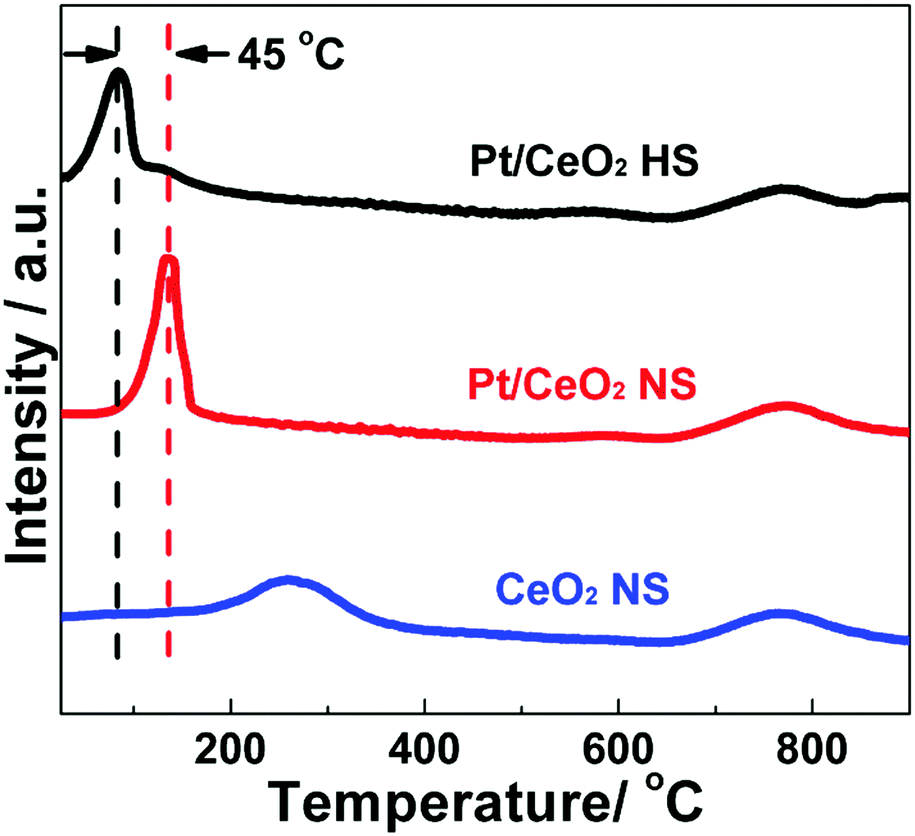 | ||
| Fig. 8 H2-TPR profiles of the Pt/CeO2 HS composite catalysts, Pt/CeO2 NS reference catalysts and CeO2 NS. | ||
Conclusions
In summary, porous micro-/nanostructural Pt-embedded-CeO2 hollow sphere composites are successfully synthesized via a one-pot acetic acid assisted ethylene glycol solvothermal method. The formation of the Pt/CeO2 HS composites can be reasonably attributed to the existence of Pt clusters that co-assemble with CeO2 NS, thus facilitating the Ostwald ripening process. In addition, it is suggested that acetic acid and glycol also play an important role in the assembly process as well as the morphology control of the final Pt-embedded CeO2 hollow micro-/nanostructure. The CO oxidation measurements indicate that Pt/CeO2 HS composites show excellent activity and satisfactory thermal stability when compared to CeO2 NS and Pt/CeO2 NS reference catalysts. The improved activity and structural stability of the Pt/CeO2 HS composites are attributed to the following reasons: (i) the synergistic effect between the highly-dispersed embedded Pt NPs and the surrounding self-assembled CeO2 NPs should be considered for the enhanced catalytic activity;64,74 (ii) the relatively homogeneous distribution of embedded Pt NPs in the self-assembled CeO2 NPs can form porous nanostructures, and thus help to produce large amounts of active sites to make the space-confined CO molecules easily get access to the active sites, and promote the CO oxidation process;75,76 (iii) 3D micro-/nanostructures have high specific surface area together with the rigidly interconnected framework, which not only might be favorable for the heterogeneous reaction due to the ease of diffusion of reactant molecules and fast product transfer, but also effectively prevent the destruction of the structure and overoxidation of active Pt species during the successive CO oxidation reactions.45,77Acknowledgements
The authors are grateful for the financial support of the National Natural Science Foundation of China (no. 21371011, 21590791, 21371011, 21331001) and Ministry of Science and Technology of China (2014CB643800). We also thank Dr Hao Dong and Dr Rui Liu for their kind help in revising this paper.Notes and references
- M. Mao, Y. Li, H. Lv, J. Hou, M. Zeng, L. Ren, H. Huang and X. Zhao, Environ. Sci.: Nano, 2017, 4, 373–384 RSC.
- H. Xu, A.-L. Wang, Y.-X. Tong and G.-R. Li, ACS Catal., 2016, 6, 5198–5206 CrossRef CAS.
- L. D. Prockop and R. I. Chichkova, J. Neurol. Sci., 2007, 262, 122–130 CrossRef CAS PubMed.
- M. V. Twigg, Appl. Catal., B, 2007, 70, 2–15 CrossRef CAS.
- L. Xu, Y. Ma, Y. Zhang, Z. Jiang and W. Huang, J. Am. Chem. Soc., 2009, 131, 16366–16367 CrossRef CAS PubMed.
- S. Zhou, B. Varughese, B. Eichhorn, G. Jackson and K. McIlwrath, Angew. Chem., Int. Ed., 2005, 117, 4615–4619 CrossRef.
- S.-B. Wang, W. Zhu, J. Ke, J. Gu, A.-X. Yin, Y.-W. Zhang and C.-H. Yan, Chem. Commun., 2013, 49, 7168–7170 RSC.
- X. Wang, D. Liu, S. Song and H. Zhang, Chem. Commun., 2012, 48, 10207–10209 RSC.
- J. S. Du, T. Bian, J. Yu, Y. Jiang, X. Wang, Y. Yan, Y. Jiang, C. Jin, H. Zhang and D. Yang, Adv. Sci., 2017, 1700056 CrossRef.
- L. Adijanto, D. A. Bennett, C. Chen, A. S. Yu, M. Cargnello, P. Fornasiero, R. J. Gorte and J. M. Vohs, Nano Lett., 2013, 13, 2252–2257 CrossRef CAS PubMed.
- N. Zhang and Y.-J. Xu, Chem. Mater., 2013, 25, 1979–1988 CrossRef CAS.
- P. Lu, C. T. Campbell and Y. Xia, Nano Lett., 2013, 13, 4957–4962 CrossRef CAS PubMed.
- K. Wu, Q. Zhang, D. Sun, X. Zhu, Y. Chen, T. Lu and Y. Tang, Int. J. Hydrogen Energy, 2015, 40, 6530–6537 CrossRef CAS.
- J. Chen, J. Qi, G. Li, F. Zheng, S. Li and Z. Tang, Chem. – Asian J., 2013, 8, 694–699 CrossRef CAS PubMed.
- J. Qi, J. Chen, G. Li, S. Li, Y. Gao and Z. Tang, Energy Environ. Sci., 2012, 5, 8937–8941 CAS.
- J. Chen, D. Wang, J. Qi, G. Li, F. Zheng, S. Li, H. Zhao and Z. Tang, Small, 2015, 11, 420–425 CrossRef CAS PubMed.
- J. Qi, K. Zhao, G. Li, Y. Gao, H. Zhao, R. Yu and Z. Tang, Nanoscale, 2014, 6, 4072–4077 RSC.
- J. Wang, H. Tang, H. Wang, R. Yu and D. Wang, Mater. Chem. Front., 2017, 1, 414–430 RSC.
- J. Zhang, H. Ren, J. Wang, J. Qi, R. Yu, D. Wang and Y. Liu, J. Mater. Chem. A, 2016, 4, 17673–17677 CAS.
- J. Wang, H. Tang, L. Zhang, H. Ren, R. Yu, Q. Jin, J. Qi, D. Mao, M. Yang and Y. Wang, Nat. Energy, 2016, 1, 16050 CrossRef CAS.
- H. Ren, R. Yu, J. Wang, Q. Jin, M. Yang, D. Mao, D. Kisailus, H. Zhao and D. Wang, Nano Lett., 2014, 14, 6679–6684 CrossRef CAS PubMed.
- J. Wang, H. Tang, H. Ren, R. Yu, J. Qi, D. Mao, H. Zhao and D. Wang, Adv. Sci., 2014, 1, 1400011 CrossRef PubMed.
- X. Lai, J. E. Halpert and D. Wang, Energy Environ. Sci., 2012, 5, 5604–5618 CAS.
- S. Lee, J. Seo and W. Jung, Nanoscale, 2016, 8, 10219–10228 RSC.
- K. Yoon, Y. Yang, P. Lu, D. Wan, H.-C. Peng, K. Stamm Masias, P. T. Fanson, C. T. Campbell and Y. Xia, Angew. Chem., Int. Ed., 2012, 51, 9543–9546 CrossRef CAS PubMed.
- M. Melchionna and P. Fornasiero, Mater. Today, 2014, 17, 349–357 CrossRef CAS.
- M. Cargnello, V. V. T. Doan-Nguyen, T. R. Gordon, R. E. Diaz, E. A. Stach, R. J. Gorte, P. Fornasiero and C. B. Murray, Science, 2013, 341, 771–773 CrossRef CAS PubMed.
- C. Sun, H. Li and L. Chen, Energy Environ. Sci., 2012, 5, 8475–8505 CAS.
- Z.-X. Li, L.-L. Li, Q. Yuan, W. Feng, J. Xu, L.-D. Sun, W.-G. Song and C.-H. Yan, J. Phys. Chem. C, 2008, 112, 18405–18411 CAS.
- K. Wu, L.-D. Sun and C.-H. Yan, Adv. Energy Mater., 2016, 6, 1600501 CrossRef.
- Y. Sun, Q. Liu, S. Gao, H. Cheng, F. Lei, Z. Sun, Y. Jiang, H. Su, S. Wei and Y. Xie, Nat. Commun., 2013, 4, 2899 Search PubMed.
- T. Montini, M. Melchionna, M. Monai and P. Fornasiero, Chem. Rev., 2016, 116, 5987–6041 CrossRef CAS PubMed.
- X. Gong, Y.-Q. Gu, N. Li, H. Zhao, C.-J. Jia and Y. Du, Inorg. Chem., 2016, 55, 3992–3999 CrossRef CAS PubMed.
- P. Xu, R. Yu, H. Ren, L. Zong, J. Chen and X. Xing, Chem. Sci., 2014, 5, 4221–4226 RSC.
- Z. Wang, S. Jiang, Y. Li, P. Xu, K. Zhao, L. Zong, H. Wang and R. Yu, Sci. Chin. Mater., 2017, 1–8, DOI:10.1007/s40843-017-9042-0.
- F. Lin, D. Hoang, C.-K. Tsung, W. Huang, S.-Y. Lo, J. Wood, H. Wang, J. Tang and P. Yang, Nano Res., 2011, 4, 61–71 CrossRef CAS.
- H.-P. Zhou, H.-S. Wu, J. Shen, A.-X. Yin, L.-D. Sun and C.-H. Yan, J. Am. Chem. Soc., 2010, 132, 4998–4999 CrossRef CAS PubMed.
- P. Lu, B. Qiao, N. Lu, D. C. Hyun, J. Wang, M. J. Kim, J. Liu and Y. Xia, Adv. Funct. Mater., 2015, 25, 4153–4162 CrossRef CAS.
- C. Wang, X.-K. Gu, H. Yan, Y. Lin, J. Li, D. Liu, W.-X. Li and J. Lu, ACS Catal., 2017, 7, 887–891 CrossRef CAS.
- S. Fang, Y. Xin, L. Ge, C. Han, P. Qiu and L. Wu, Appl. Catal., B, 2015, 179, 458–467 CrossRef CAS.
- G. Chen, F. Rosei and D. Ma, Nanoscale, 2015, 7, 5578–5591 RSC.
- G. Fu, K. Wu, J. Lin, Y. Tang, Y. Chen, Y. Zhou and T. Lu, J. Phys. Chem. C, 2013, 117, 9826–9834 CAS.
- C. Chen, X. Fang, B. Wu, L. Huang and N. Zheng, ChemCatChem, 2012, 4, 1578–1586 CrossRef CAS.
- L. Ma, D. Wang, J. Li, B. Bai, L. Fu and Y. Li, Appl. Catal., B, 2014, 148–149, 36–43 CAS.
- X. Wang, J. Feng, Y. Bai, Q. Zhang and Y. Yin, Chem. Rev., 2016, 116, 10983–11060 CrossRef CAS PubMed.
- G.-M. Lyu, Y.-J. Wang, X. Huang, H.-Y. Zhang, L.-D. Sun, Y.-J. Liu and C.-H. Yan, Nanoscale, 2016, 8, 7923–7932 RSC.
- W. Liu, T. Deng, L. Feng, A. Xie, J. Zhang, S. Wang, X. Liu, Y. Yang and J. Guo, CrystEngComm, 2015, 17, 4850–4858 RSC.
- L. Artiglia, F. Orlando, K. Roy, R. Kopelent, O. Safonova, M. Nachtegaal, T. Huthwelker and J. A. van Bokhoven, J. Phys. Chem. Lett., 2017, 8, 102–108 CrossRef CAS PubMed.
- S. Gu, Y. Chen, X. Yuan, H. Wang, X. Chen, Y. Liu, Q. Jiang, Z. Wu and G. Zeng, RSC Adv., 2015, 5, 79556–79564 RSC.
- W. Liu, W. Wang, K. Tang, J. Guo, Y. Ren, S. Wang, L. Feng and Y. Yang, Catal. Sci. Technol., 2016, 6, 2427–2434 CAS.
- C. L. Menendez, Y. Zhou, C. M. Marin, N. J. Lawrence, E. B. Coughlin, C. L. Cheung and C. R. Cabrera, RSC Adv., 2014, 4, 33489–33496 RSC.
- X. Wang, D. Liu, S. Song and H. Zhang, J. Am. Chem. Soc., 2013, 135, 15864–15872 CrossRef CAS PubMed.
- S. Reddy, S. Kim, H. Young Jeong, S. Jin, K. Qadir, K. Jung, C. Ho Jung, J. Yeul Yun, J. Yeong Cheon, J.-M. Yang, S. Hoon Joo, O. Terasaki and J. Young Park, Chem. Commun., 2011, 47, 8412–8414 RSC.
- J. Ke, W. Zhu, Y. Jiang, R. Si, Y.-J. Wang, S.-C. Li, C. Jin, H. Liu, W.-G. Song, C.-H. Yan and Y.-W. Zhang, ACS Catal., 2015, 5, 5164–5173 CrossRef CAS.
- Y.-J. Wang, H. Dong, G.-M. Lyu, H.-Y. Zhang, J. Ke, L.-Q. Kang, J.-L. Teng, L.-D. Sun, R. Si, J. Zhang, Y.-J. Liu, Y.-W. Zhang, Y.-H. Huang and C.-H. Yan, Nanoscale, 2015, 7, 13981–13990 RSC.
- Y. Kang, M. Sun and A. Li, Catal. Lett., 2012, 142, 1498–1504 CrossRef CAS.
- J. Lee, Y. Ryou, X. Chan, T. J. Kim and D. H. Kim, J. Phys. Chem. C, 2016, 120, 25870–25879 CAS.
- X. Liang, J. Xiao, B. Chen and Y. Li, Inorg. Chem., 2010, 49, 8188–8190 CrossRef CAS PubMed.
- Z. Wang, J. Qi, K. Zhao, L. Zong, Z. Tang, L. Wang and R. Yu, Mater. Chem. Front., 2017 10.1039/C7QM00134G.
- C. Du, G. Lu, Y. Guo, Y. Guo and X.-q. Gong, ACS Omega, 2016, 1, 118–126 CrossRef CAS.
- Y. Xu, Y. Zhang, Y. Zhou, S. Xiang, Q. Wang, C. Zhang and X. Sheng, RSC Adv., 2015, 5, 58237–58245 RSC.
- C. C. Yec and H. C. Zeng, J. Mater. Chem. A, 2014, 2, 4843–4851 CAS.
- W. Liu, X. Liu, L. Feng, J. Guo, A. Xie, S. Wang, J. Zhang and Y. Yang, Nanoscale, 2014, 6, 10693–10700 RSC.
- N. Zhang, X. Fu and Y.-J. Xu, J. Mater. Chem., 2011, 21, 8152–8158 RSC.
- T. Wu, X. Pan, Y. Zhang, Z. Miao, B. Zhang, J. Li and X. Yang, J. Phys. Chem. Lett., 2014, 5, 2479–2483 CrossRef CAS PubMed.
- J. Jones, H. Xiong, A. T. DeLaRiva, E. J. Peterson, H. Pham, S. R. Challa, G. Qi, S. Oh, M. H. Wiebenga, X. I. Pereira Hernández, Y. Wang and A. K. Datye, Science, 2016, 353, 150–154 CrossRef CAS PubMed.
- C. Du, Y. Guo, Y. Guo, X.-q. Gong and G. Lu, J. Mater. Chem. A, 2015, 3, 23230–23239 CAS.
- N. Ta, J. Liu, S. Chenna, P. A. Crozier, Y. Li, A. Chen and W. Shen, J. Am. Chem. Soc., 2012, 134, 20585–20588 CrossRef CAS PubMed.
- C. Zhang, S. Li, T. Wang, G. Wu, X. Ma and J. Gong, Chem. Commun., 2013, 49, 10647–10649 RSC.
- Q. Fu, H. Saltsburg and M. Flytzani-Stephanopoulos, Science, 2003, 301, 935–938 CrossRef CAS PubMed.
- L. Hu, K. Sun, Q. Peng, B. Xu and Y. Li, Nano Res., 2010, 3, 363–368 CrossRef CAS.
- M.-F. Luo, J.-M. Ma, J.-Q. Lu, Y.-P. Song and Y.-J. Wang, J. Catal., 2007, 246, 52–59 CrossRef CAS.
- X. Hong and Y. Sun, Catal. Lett., 2016, 146, 2001–2008 CrossRef CAS.
- Q. Wang, Y. Li, B. Liu, Q. Dong, G. Xu, L. Zhang and J. Zhang, J. Mater. Chem. A, 2015, 3, 139–147 CAS.
- B. Liu, S. Yu, Q. Wang, W. Hu, P. Jing, Y. Liu, W. Jia, Y. Liu, L. Liu and J. Zhang, Chem. Commun., 2013, 49, 3757–3759 RSC.
- M. Pérez-Lorenzo, B. Vaz, V. Salgueiriño and M. A. Correa-Duarte, Chem. – Eur. J., 2013, 19, 12196–12211 CrossRef PubMed.
- J. Qi, X. Lai, J. Wang, H. Tang, H. Ren, Y. Yang, Q. Jin, L. Zhang, R. Yu, G. Ma, Z. Su, H. Zhao and D. Wang, Chem. Soc. Rev., 2015, 44, 6749–6773 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7qm00244k |
| This journal is © the Partner Organisations 2017 |