
Self-oriented TiO2 nanosheets in films for enhancement of electron transport in nanoporous semiconductor networks†
M. M.
Maitani‡
 *,
C.
Xu
,
K.
Hashimoto
,
Y.
Ueda
and
Y.
Wada
*
*,
C.
Xu
,
K.
Hashimoto
,
Y.
Ueda
and
Y.
Wada
*
School of Chemical Science and Engineering, Tokyo Institute of Technology, Meguro, Tokyo 152-8552, Japan. E-mail: mmaitani@dsc.rcast.u-tokyo.ac.jp; yuji-w@apc.titech.ac.jp
First published on 28th June 2017
Abstract
Self-oriented anisotropic nanoparticles in nanoporous films were obtained by controlling the surface functionality induced by the organic amino acid derivatives adsorbed onto TiO2 nanosheets. In particular, tyrosine-functionalized TiO2 nanosheets were self-oriented in a nonparallel orientation with respect to the substrate plane owing to the following surface properties: (1) the ζ-potential, (2) hydrogen bonding, and (3) hydrophobicity due to the collaborative effects of segregative interactions from hydrophilic surroundings, i.e., water and oxide substrates. Although nanoporous films with both parallel and nonparallel orientations of TiO2 nanosheets with respect to the substrate plane exhibited a similar internal surface area and porosity, evaluation of the electrochemical properties of TiO2 films with non-parallel orientation showed a significant improvement in electron transport through the TiO2 nanoporous network, as demonstrated in dye-sensitized solar cell devices.
Introduction
Nanoporous films composed of nanoparticles with large internal surface areas, high porosity, and nanostructured voids have attracted attention recently for their potential applications in photo-electrochemical devices, batteries, thermoelectric and photocatalytic devices, and solar cells.1–6 In the field of nanoparticle engineering, the development of facet-controlled anisotropic oxide nanomaterials, such as nanosheets, nanotubes, nanorods, and nanowires, has resulted in the development of a large variety of anisotropic oxide nanoparticles instead of conventional spherical particles.7,8 Recently, these anisotropic particles have been proposed as building blocks for the construction of novel nanostructured porous films. This is because of the advantage that they offer of superior properties in carrier transport through the nanoporous material network and their ion diffusion properties in electrolytes filled in the void of the nanoporous films. These properties are due to their morphological ordering, in addition to fewer inter-particle connections through the nanoporous network compared with conventional random-networking spherical isotropic nanoparticles. This is especially true for applications in electrochemical and photo-electrochemical devices by assembling the nanoporous network with liquid or quasi-solid electrolytes.7,8 To utilize the morphological advantage of the anisotropic materials, the desired morphology of the nanoporous network would be for their long axis to be perpendicularly orientated in films with respect to the substrate plane (Scheme 1a) rather than the orientation being random. Perpendicular orientation of anisotropic nanomaterials was recently achieved by self-orientation of directly grown particles, nanowires, nanosheets, nanotubes, and others on substrates by utilizing solution phase growth techniques, such as hydrothermal, solvo-thermal, and anodic oxidation processes, which involve immersing the substrates in a precursor solution.9,10 However, these processes are not widely applicable in practical applications due to the drawbacks of limited coating area, less uniformity, and batch-to-batch-based slow processes. In contrast, a practically more feasible coating-based process involving anisotropic nanoparticle suspensions generally results in the parallel orientation of the longer axis of nanoparticles with respect to the substrates11 (Scheme 1b), owing to the minimization of the surface energy of anisotropic particles. Therefore, anisotropic nanoparticles mostly have a parallel orientation with respect to substrates fabricated by the coating process. These parallel-oriented anisotropic nanoparticles sometimes do not show significant advantage of electrochemical properties over conventional spherical nanoparticles possessing a random network. For example, a high aspect ratio of nanorod TiO2 revealed less effective charge transport in the nanoporous films due to their unfavorable parallel orientation with respect to substrates.11,12 A method to control the orientation of nanoparticles in the suspension has therefore been proposed based on electrophoretic deposition under a strong magnetic field.13 However, obtaining a large production area with a uniform film is still a challenge because of the complexity of the system setup. Another method based on convective self-assembly using dispersion suffers from the problem of sensitivity to the environment. Solvent drying and film preparation therefore take a long time.14–18 | ||
| Scheme 1 (a) Self-oriented direct grown NP on substrates, (b) anisotropic NP by the printing process, and (c) self-assembled anisotropic NP for nonparallel orientation by the printing process as the novel proposed concept. | ||
The self-assembly of nanoparticles by chemically functionalizing the surface with organic compounds to control the nano-construction based on the building blocks of nanoparticles by utilizing inter-particle interactions has gained attention recently.19,20 Here, we utilize a coating process with the self-assembly of anisotropic oxide nanoparticles on a TiO2 nanosheet to control their orientation in a nanoporous film by engineering the surface functionality of particles by inter-particle interactions. We demonstrate a novel method to control the orientation of the longer axis of TiO2 nanosheets in parallel (Scheme 1b) and non-parallel (Scheme 1c) orientations in nanoporous films with respect to the substrate plane even with the low-cost coating-based film process, called doctor-blade coating, of the nanoparticle suspension on to the substrates. We also propose a possible mechanism of orientation control of anisotropic TiO2 nanosheets attributed to the surface functionality with organic compounds such as amino acid derivatives, which affect inter-particle interactions, i.e., electrostatic force, segregative hydrophobic interactions, and hydrogen bonding. As the purpose of this study was the uniform coating of nanopros films with nanoparticles, the particles had to be well-suspended as a coating ink. Precise control of secondary aggregation cluster size is important to maintain the well-suspended solution for uniform film creation by printable methods. Therefore, we propose the control of inter-particle interactions based on weak interactions, e.g. hydrophilic–hydrophobic segregative interactions, electrostatic forces, and hydrogen bonding, by means of surface functionalization with amino acid, which could be strongly bonded on TiO2 nanoparticles and provide those weak inter-particle interactions depend on the functional groups under varied pH in the surrounding medium.
Furthermore, the advantages of the selectively orientated TiO2 nanoporous network in thin film are demonstrated in terms of the minority carrier transport in the photovoltaic device of dye-sensitized solar cells (DSSCs). The results exhibit significant enhancement of electron transport properties in nanoporous films, which is reflected by their nano-morphology compared with the non-parallel and parallel orientation of building blocks of nanosheets with respect to the substrate plane.
Experimental
TiO2 nanosheet synthesis
Tetrabutoxytitanium(IV) (Ti(OBu)4, 25 mL) and aqueous hydrofluoric acid solution (43 wt%, 3 mL) were mixed in a dried Teflon-lined autoclave under ambient conditions. The autoclave was tightened and kept at 180 °C for 24 h. The exposed {001} facet was calculated from the geometry of particles observed with SEM (Fig. S1, ESI†).21–24 The products were thus washed with ethanol, distilled water, and aqueous NaOH solution (0.1 M) several times by sonication followed by decantation to remove fluorinated compounds from the TiO2 surface.21–24 They were then rinsed with aqueous HCl (0.1 M) several times to obtain TiO2 suspension in HCl (3 wt%, pH 2).TiO2 film preparation
The TiO2 nanosheet suspension was applied to prepare nanoporous films on FTO or glass substrates for SEM and XRD analyses. Substrates cleaned by consecutive sonication processes in aqueous detergent solution, deionizer water, acetone, and ethanol, followed by UV-ozone dry cleaning, were used for film preparation by means of the doctor-bread technique.The washed TiO2 nanosheets were dispersed (3 wt%) in HCl (0.1 M) solution to provide a well-dispersed suspension (Fig. S6, ESI†) determined by the dynamic light scattering technique, and the resulting TiO2 suspension and a series aqueous amino acid solutions (0.5 equivalent molar, Scheme 2) were mixed at a volume ratio of 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 in each micro vial (∼1.5 mL). The pH of each suspension was then controlled by the addition of aqueous ammonia solution (25 wt%). Each vial was then shaken for 1 h at a frequency of approximately 5 Hz. Each suspension was then coated on clean FTO substrates by means of the doctor blade method.
:1 in each micro vial (∼1.5 mL). The pH of each suspension was then controlled by the addition of aqueous ammonia solution (25 wt%). Each vial was then shaken for 1 h at a frequency of approximately 5 Hz. Each suspension was then coated on clean FTO substrates by means of the doctor blade method.
 | ||
| Scheme 2 Amino acid derivatives used as Phenylalanine (Phe), Glutamic acid (Glu), Arginine (Arg), Lysine (Lys), Cysteine (Cys), Glycine (Gly), Tyrosine (Tyr), and Histidine (His). | ||
The paste with non-functionalized TiO2 was prepared by the addition of α-terpineol and ethylcellulose in ethanol (2 wt% of TiO2) suspension.25 These TiO2 films were successively annealed at 500 °C for 30 min to remove the organic compounds and the solvent. We confirmed the parallel orientation of the TiO2 film with non-functionalized TiO2 on glass or FTO substrates by SEM and XRD (Fig. 1).
 | ||
| Fig. 1 SEM images of (a and b) Tyr-functionalized and (c) non-functionalized TiO2 nanosheets on FTO substrate as plane view, and their (c and d) cross-section view of SEM images. | ||
Dynamic light scattering
The particle aggregation was determined in the TiO2 suspension including amino acids by means of dynamic light scattering (DLS, Nano Partica SZ-100, HORIBA, Japan). To start the DLS measurement, the concentrations of TiO2 and amino acid were reduced to 1 wt% and approximately 8 × 10−4 M, respectively, to acquire adequate incident light exposure. The solution was shaken for 1 h prior to the measurement.ζ-Potential measurement
The ζ-potential of the TiO2 particles with and without amino acid functionalization was measured in the TiO2 suspension, including amino acids of various pH values, by means of the electrophoretic light scattering method. To start the ζ-potential measurement, the concentrations of TiO2 and amino acid were reduced to 0.01 wt% and 1 × 10−3 M (∼8 × 10−4 M only for tyrosine (Tyr)), respectively, to acquire adequate incident light exposure. The solutions containing TiO2 and each amino acid with pH values 2, 3, 4, 5, 7, and 8 were shaken for 1 h prior to the measurement.Contact angle measurement
To characterize the hydrophilicity (hydrophobicity) of the amino acid-functionalized TiO2 surface, atomically flat rutile TiO2(110) substrates (Nb (0.5 wt%) doped, Crystal Base, Japan) were immersed in each aqueous amino acid solution (equivalent 0.05 M) for 20 h immediately after cleaning by sonication in ethanol (5 min), followed by heating in an oven (450 °C, 1 h). The rinsed samples were placed on the contact angle measurement apparatus (CA-D, Kyowa Interfacial Science Japan), and the contact angle of the droplet of water (2 µL) on the sample surface was determined by means of the θ–2θ method.XRD analysis
The orientation of the TiO2 nanosheets in nanoporous films on glass substrates was characterized by op-XRD and AR-XRD using an X-ray diffractometer for powder (MiniFlex, Rigaku) and film (AXS D8 Discover system, Bruker), both with a Cu Kα radiation source (=1.5406 Å) utilized with 2θ scan. This was utilized at the orientation of a plane of source-sample-beam at psi angles of 85° from the surface normal in the AR-XRD measurement (Fig. S3, ESI†).Evaluation of non-parallel TiO2 orientation with self-assembled units
We determined the self-assembly of TiO2 with non-parallel orientation with respect to substrates using the statistical analysis of plane SEM images of the TiO2 nanoporous films. The number of self-assembled units, including more than two TiO2 nanosheets stacked and oriented with the [100] orientation facing perpendicular to the substrate plane, was calculated in the plane view of SEM images for each condition (Fig. S4, ESI†). The number density of counted self-assembled units was summarized as a function of stacked TiO2 nanosheets with [100] orientation facing perpendicular to the substrate plane. The total number density of the self-assembled TiO2 nanosheets in non-parallel orientation with respect to the substrate plane was calculated and summarized for each amino acid and pH condition (Fig. 2).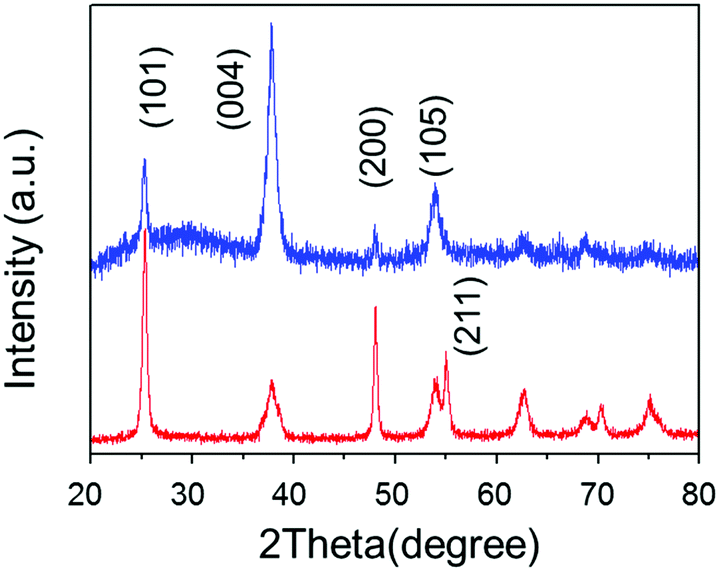 | ||
| Fig. 2 Op-XRD spectra of Tyr-functionalized (red), and non-functionalized (blue) anatine TiO2 nanosheets on glass substrate. | ||
DSSC preparation
The non-parallel and parallel orientated TiO2 nanosheet thin films (2 µm thick) on FTO were applied to the nanoporous TiO2 electrode in DSSC. The TiO2 nanoporous films prepared by the doctor-blade method25,26 with nanoparticle suspensions with or without Tyr were annealed at 500 °C for 30 min in an electric oven to remove organic compounds from the TiO2 surface. They were then immersed in a commercially available conventional ruthenium dye-sensitizer (N719, Solaronix, Switzerland) solution (0.25 mM) in acetonitrile and tert-buthanol (1:1) for 24 h. The films were then rinsed with ethanol, and finally dried under a nitrogen stream. The dye-sensitized TiO2/FTO was then assembled in a sandwich cell configuration with iodine electrolyte (0.1 M LiI, 0.6 M 1-butyl-3-methyl-imethylimidazolium iodide, 0.03 M I2, and 0.5 M 4-tert-butylpyridine in 15/85 (v/v) mixture of valeronitrile and acetonitrile), a platinum-coated FTO counter electrode, and 25 µm thick Surlyn (DuPont) providing 0.4 × 0.4 cm2 as the active cell area. The photocurrent-voltage properties of solar cells were evaluated under an AM1.5 solar simulator (YYS-100, Yamashita Denso).
Analysis of electron transport properties in DSSC
The sealed cells of DSSCs were applied to the analysis of the transient cell responses. This is termed as the stepped light-induced transient measurement method of photocurrent and voltage (SLIM-PCV)27 and the change extraction method28 to determine the diffusion coefficients and life time of electrons in TiO2 by considering the density of electrons as a function of the energy level. The incident laser (λ = 642 nm, cube, Coherent Japan) was modulated by the external analog DC signal and the cell current and voltage responses were monitored by the digital multi-meter (7461A, ADC) as described elsewhere.27 The extracted charge was also counted by the same digital multi-meter with the cell circuit and was controlled in an open or short circuit by a solid-state switching device.28Results and discussion
The anatase TiO2 nanosheets exposing the dominant {001} facet were synthesized by the hydrothermal growth of TiO2 with titaniumtetrabutoxide in a hydrofluoric acid aqueous solution to provide the anisotropic sheet architecture.21–24 The synthesized TiO2 nanosheets exhibited a pure anatase phase exposing a large fraction (about 70%) of the {001} facet with an aspect ratio of approximately 5 in the a-axis against the c-axis, as determined by X-ray diffraction (XRD) and scanning electron microscopic (SEM) images of the dried particles (Fig. S1, ESI†). According to separate crystallographic analyses,21–24 the obtained TiO2 nanosheet possesses the [001] orientation of anatase oriented perpendicularly to the sheet plane of each particle, as discussed later (Fig. S1c, ESI†). This was utilized for the analysis of nanosheet orientations in the films. The TiO2 nanosheets washed first with NaOH aqueous solution (0.1 M) and then with distilled water exhibited no residual fluoride compound on the surface of TiO2.23,24 These particles were consequently dispersed in a HCl aqueous solution (0.01 M). TiO2 suspension in HCl was then mixed with a series of aqueous amino acid solutions (Scheme 2) in each micro vial. Consequently, the pH of each suspension was controlled by the addition of aqueous ammonium hydroxide solution and each vial was mechanically shaken for 1 h. Each suspension was then coated on a clean F:SnO2 on glass (FTO) substrate by means of the doctor blade method. The substrates were cleaned by UV-ozone treatment prior to film coating. Each sample film was then annealed at 500 °C for 30 min to remove the organic solvent and amino acid compounds adsorbed on TiO2.3.1. Nanoporous film morphology
SEM images, taken as part of their morphological analysis, show that a few resulting TiO2 nanoporous films with a certain amino acid, e.g. especially Tyr-functionalization, exhibited non-parallel orientation of nanosheets with respect to the substrate plane (Fig. 1a, b and Fig. S2, ESI†). On the other hand, TiO2 nanosheets without functionalization exhibited parallel orientation of the longer axis with respect to the substrate (Fig. 1c). In addition, the cross-sectional images of each nanoporous film composed of Tyr-functionalized and non-functionalized TiO2 nanosheets clearly support the non-parallel (Fig. 1d and Scheme 1c) and parallel (Fig. 1e and Scheme 1b) orientations of the longer axis of nanosheets with respect to the substrates.The out-plane X-ray diffraction (op-XRD) spectra of the TiO2 nanoporous films also showed each non-parallel and parallel orientation of Tyr-functionalized or non-functionalized TiO2, respectively (Fig. 2). The op-XRD spectra of non-functionalized TiO2 films exhibited an intense (004) peak at 2 theta of about 38° compared with the (101) peak, indicating that most of the anatase (001) plane of nanosheets is oriented in parallel with respect to the substrate. For further analysis of the sheet orientation, angle-resolved XRD (AR-XRD) with a psi-scan of the TiO2 film was applied. In AR-XRD, the psi was scanned with fixed 2θ angles of 38° and 25° exhibiting the diffraction peaks attributed to the (004) and (101) plane, respectively. In the case of non-functionalized TiO2, the intensity of the peaks attributed to the (004) and (101) planes revealed a maximum at psi angles of 0° and 68°, respectively (Fig. S3a, ESI†). It can therefore be seen that the nanosheet without functionalization is deposited parallel to the substrate. Therefore, [001] is oriented perpendicular to the substrate plane, while [101] tilts at an angle of about 68° from the surface normal due to the inter-plane angle of (001) and (101) of 68.3° in the anatase TiO2 crystalline phase in theory.29 On the other hand, the op-XRD of the Tyr-functionalized TiO2 films exhibited a less intense (004) peak compared with the (101) peak, and its ratio was similar to that of randomly oriented anatase TiO2 nanosheets as dried powder (Fig. S1, ESI†). These results indicate that Tyr-functionalized TiO2 films exhibited non-parallel orientation of nanosheets, as described in Scheme 1. The results of AR-XRD also support this result of non-parallel orientation of Tyr-functionalized TiO2 exhibiting no peak in psi-scans (Fig. S3b, ESI†). Therefore, the functionalization with a certain amino acid, such as Tyr or without functionalization successfully achieved the self-orientation of the TiO2 nanosheet, which clearly exhibited significant differences in the nano-morphology of nanoporous TiO2 films with the same component.
3.2. Origin of self-orientation
As the purpose of this study was uniform coating of nanopros films with nanoparticles with changing orientations of the anisotropic nanosheets in the resulting nanoporous films, the particles had to be well-suspended as a coating ink. In our strategy to achieve this purpose, the inter-particle interactions were precisely controlled by functionalization of the TiO2 surface by amino acids and the pH of the surrounding solution. Furthermore, we hypothesized that self-assembly of aggregation occurs in the wet film configuration on the substrate during the drying and annealing process of nanoporous TiO2 films by utilizing the weak inter-particle interactions.Therefore, a series of results of the amino acid functionalization (Scheme 2) after annealing of the TiO2 films are discussed here in detail to reveal the origin of the observed self-orientation of TiO2 nanosheets in nanoporous films. First, to quantitatively evaluate the degree of self-assembly, the number of sheets exhibiting the stacked feature with the presumable [100] direction21–24 of nanosheets facing perpendicular to the substrate plane (Fig. S4, ESI†) was calculated, and the value of the surface density of the corresponding standing TiO2 nanosheets was determined in a specific area of a square micron, as summarized in all conditions with axes of pH and amino acid species in Fig. 3. We presume this evaluated feature was created by the ensemble of weak inter-particle interactions, as discussed in the later section. As a result of the analysis, the precursor suspensions of TiO2 nanosheets containing Tyr under a pH of 4 were the most favorable for the non-parallel orientation of nanosheets with respect to the substrates as the result of nanoporous TiO2 films (Fig. 3 and Fig. S2, ESI†). Therefore, we mainly focused on films prepared with Tyr-functionalized TiO2 under a pH value of 4 in order to investigate the origin of the non-parallel orientation.
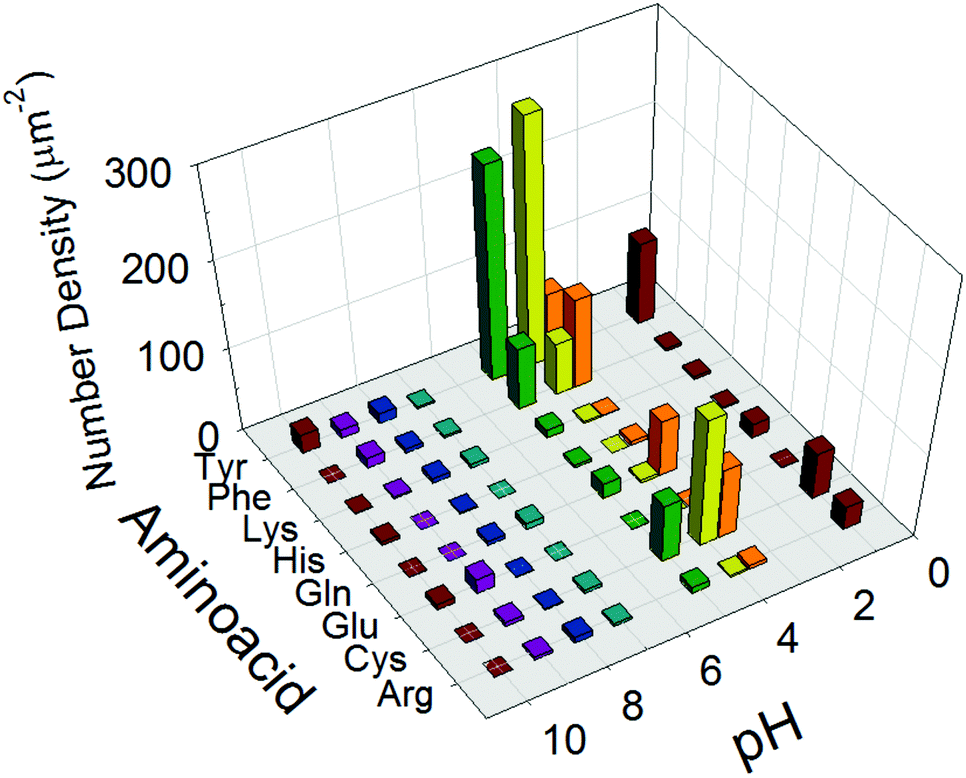 | ||
| Fig. 3 Number density of self-assembled TiO2 nanosheets with nonparallel orientation of the longer axis with respect to the plane of substrates counted in plane view SEM images (Fig. S2, ESI†). | ||
The driving force behind the non-parallel orientation is now discussed with the hypothesized mechanism of particle self-assembly, since many nanosheets with a non-parallel orientation gave the appearance of multiple sheets stacked together in SEM images (Fig. 1 and Fig. S2, S4, ESI†). The adsorbed amount of Tyr on the TiO2 nanosheets was determined to be below the saturated concentration of Tyr in water, ∼8 × 10−4 M, (Fig. S5, ESI†). This is limited by its low solubility in water, while the actual Tyr adsorption is much greater than the measured maximum surface concentration of 0.6 molecule per nm2 determined at a Tyr concentration of ∼8 × 10−4 M. This is because the actual experiment of particle assembly was performed beyond the saturation point of Tyr in water, i.e., Tyr was contained in the TiO2 suspension with an equivalent molar concentration of 0.5 M in the experiment, while Tyr was observed as the solid in the suspension, which indicates that not all of the compound was completely dissolved in the solution. Therefore, the surface concentration of adsorbed Tyr molecules could be much higher than 0.6 molecules per nm2 as a result of the excess amount of Tyr dispersed in aqueous TiO2 suspensions under the experimental conditions of film preparation.
To characterize the surface properties of the TiO2 nanosheets functionalized with several varieties of amino acids in the aqueous suspension, the ζ-potential was measured by the electrophoretic light-scattering method (Fig. 4). The ζ-potential of the oxide surface typically shifted from positive to negative due to the deprotonation of the TiO2 surface, corresponding to an increase in pH of the surrounding solution. Correspondingly the titration process of the hydroxy group on the oxide surface depended on the pH of the surrounding solution. As a consequence, the pH-dependent ζ-potential of the oxide surface significantly influenced the repulsive electrostatic force between the particles.30,31 As reported previously, most of the TiO2 samples, including bare TiO2, revealed an isoelectric point with a pH value of about 7.31 It was, however, clearly observed that the absolute values of the ζ-potential varied with the amino acid, and therefore a few amino acids, including Tyr, exhibited a significant decrease in the ζ-potential. The low values of the ζ-potential presumably resulted in the particles aggregating in the suspension due to a decrease in the electrostatic repulsive force in the stable suspension of oxide nanoparticles. To observe the aggregation of the nanoparticles, dynamic light scattering of the suspension was applied in order to determine the diameter of aggregated nanoparticles in the suspension by utilizing the Stokes–Einstein equation.32 The results revealed that the Tyr-functionalized TiO2 nanoparticles were still well-dispersed in the suspension, while a slight increase in particle size was observed. This is probably an indication of aggregation due to smaller inter-particle repulsive forces compared with a non-functionalized TiO2 suspension (Fig. S6, ESI†).
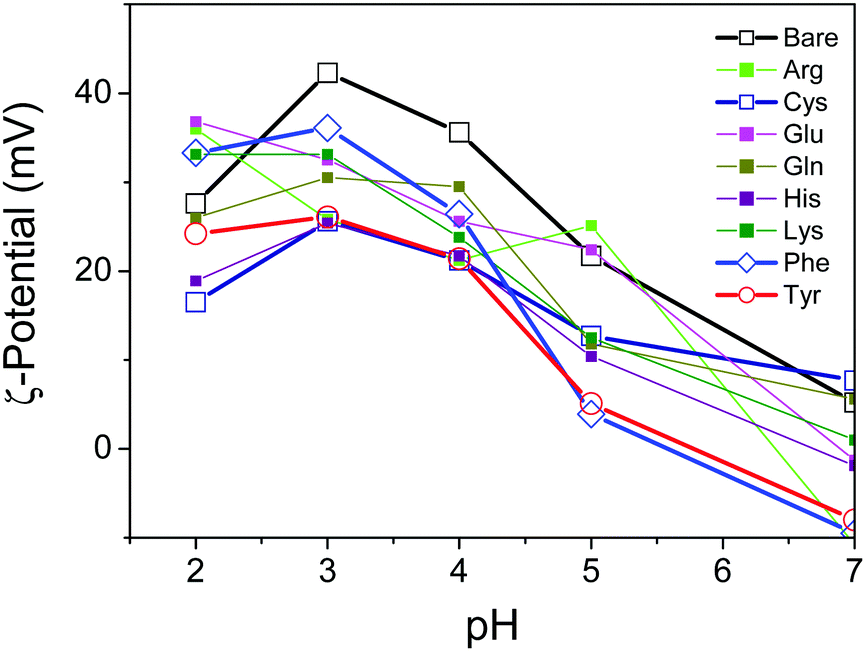 | ||
| Fig. 4 ζ-Potential of TiO2 nanoparticles (0.01 wt%) measured with aqueous suspension in amino acid solution (0.09 mM) under a pH controlled by the addition of NH4OHaq (0.1 M) into acidic suspension containing HCl (pH 2). | ||
We observed that self-assembly with non-parallel orientation of nanosheets with respect to the substrate plane was achieved in most cases with a hydrophobic amino acid of Tyr, Cys, and Phe among the applied conditions. Here, we quantitatively examined the hydrophobicity of the TiO2 surface functionalized by a series of amino acid compounds by means of contact angle measurements of water droplets on the surface of each amino acid-functionalized single-crystal rutile TiO2 (Fig. 5). The substrates of single-crystal rutile TiO2 were cleaned by EtOH under sonication, annealed at 500 °C, and then immersed in an equivalent 0.5 M aqueous solution of each amino acid without ammonia addition for pH control. Although the single-crystal rutile and anatase nanoparticles may have a slight difference in TiO2 surface properties for molecular adsorption,29 we focused on the qualitative examination of the surface of each amino acid-functionalized TiO2. A high contact angle, corresponding to the hydrophobic surface, was consistently observed with the amino acid exhibiting relatively favorable behavior for non-parallel orientation of TiO2 nanosheets with respect to the substrate plane (Fig. 3). These results thus clearly indicate that the mechanism of self-assembly of TiO2 nanosheets resulting in non-parallel orientation is correlated with the hydrophobic surface properties. Although His and Lys also demonstrated a relatively high contact angle, indicating hydrophobicity, the non-parallel orientation of TiO2 nanosheets was not observed, probably due to a certain synergetic mechanism of non-parallel orientation of the TiO2 nanosheets.
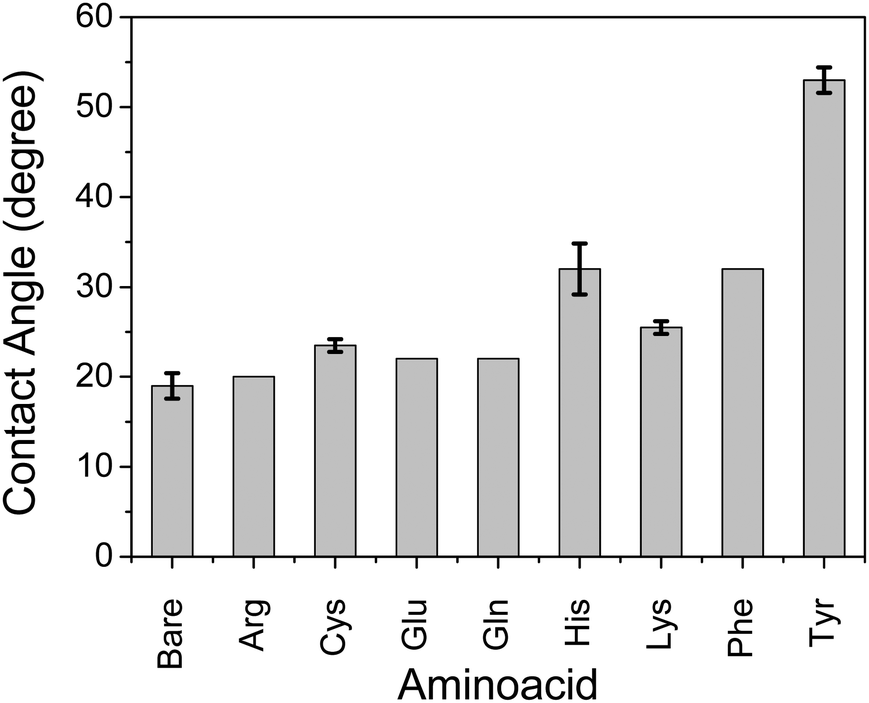 | ||
| Fig. 5 Water contact angle on the amino acid-functionalized single crystalline rutile TiO2(110). Functionalization by amino acids was performed by immersing the substrate in amino acid solution (0.045 M) without pH control for 20 h. | ||
Here, we propose a mechanism for the self-assembly of TiO2 nanosheets with non-parallel orientation with respect to the substrate plane, which was observed the most with Tyr-functionalized TiO2 nanosheets, as schematically described in Scheme 3. In the experimental condition of TiO2 nanosheet coating, a sufficient number of Tyr molecules were adsorbed on the surface of the TiO2 nanosheets with a carboxylic acid moiety in the saturated aqueous solution of Tyr with TiO2 nanosheets. Therefore, the Tyr-adsorbed TiO2 surface revealed the hydrophobic characteristics and a ζ-potential less than 30 mV (Fig. 4 and 5). Lowering the ζ-potential of the TiO2 particles induced by Tyr also resulted in slight aggregation in the suspension, although this was not significant enough to affect the results of dynamic light scattering (Fig. S6, ESI†). The surface hydrophobicity also possibly induced segregative hydrophobic inter-particle interactions due to synergetic interactions with the surrounding polar water as the solvent. Finally, the stacking of anisotropic nanosheets could have been the result of the driving force of the segregative hydrophobic interactions among the TiO2 nanosheets under a low electrostatic repulsive force. The casted suspension became concentrated during the evaporation of the solvent in the film coating process of the TiO2 suspension onto FTO or glass substrates. Furthermore, the segregative hydrophobic inter-particle interaction from the hydrophilic FTO or glass substrates resulted in the non-parallel orientation of the anatase TiO2(001) plane with respect to the substrate plane to avoid non-favorable contact between the hydrophilic substrate and hydrophobic Tyr-functionalized TiO2 surface.
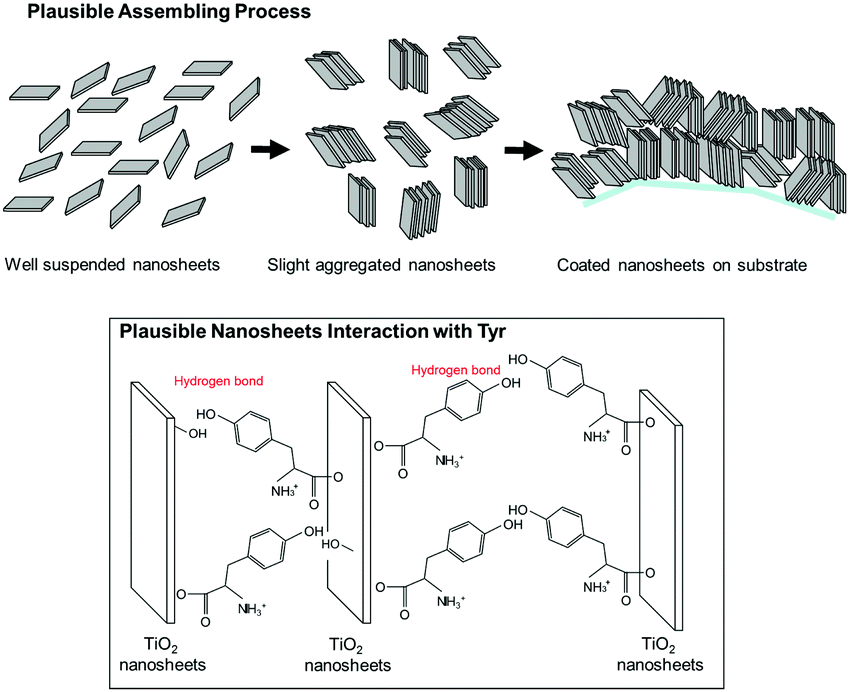 | ||
| Scheme 3 Plausible mechanism of self-oriented TiO2 nanosheets for nonparallel orientation by the printing process proposed in this study. | ||
This mechanism is also supported by a controlled experiment with acetone addition in the suspension at the most favorable condition (Tyr-functionalized TiO2 nanosheets under pH = 4), which significantly degraded the expected non-parallel orientation of the TiO2 nanosheets (Fig. S7, ESI†). This was probably due to a decrease in the segregative hydrophobic interaction among particles, since acetone decreases the polarity of the solvent compared with water. In addition, hydrogen bonding between the hydroxy groups in Tyr moieties or between Tyr and the surface of TiO2, which was typically covered with hydroxy groups especially in aqueous solution, possibly stabilized the stacked nanosheets on the FTO since a controlled experiment with a few simple saturated fatty acid compounds did not give any indication of non-parallel orientation (results are not provided).33
Although His and Lys also demonstrated a relatively high contact angle, indicating hydrophobicity on the TiO2 surface (Fig. 5), non-parallel orientation of the TiO2 nanosheets was not observed. This could be due to the following two factors: (1) a relatively large electrostatic repulsive force in the coating conditions or (2) hydrogen bonding as an inter-particle force. In the study of His and Lys, both molecules possess a protonated ammonium moiety, so each surface is positively charged under the assembling condition (pH < 7), while all of the samples revealed a similar trend of ζ-potential as a function of the pH. This is probably because the ζ-potential was still mainly dominated by the titration of the hydroxy groups on TiO2. Additionally, although we used the high pH region (pH > 7), none of the amino acid-functionalized TiO2 nanosheets revealed non-parallel orientation with respect to the substrate plane. This is probably because the carboxylic acid provides chemical bonding on TiO2 only in an environment possessing low pH regions, so amino acid functionalization is possible only in the low pH region. This is indicated by low coverage of Tyr in the high pH region, as analyzed from the Lineweaver–Burk equation based on Langmuir's adsorption model (Fig. S5, ESI†).34 As a result of (1) low electrostatic repulsive force, (2) hydrogen bonding as inter-particle forces, and (3) segregative interactions between particles and surroundings, such as water and the substrate surface, TiO2 nanosheet functionalized with Tyr, Cys, or Phe at a pH of ∼4 exhibited the most favorable non-parallel orientation of the (001) plane with respect to the substrate plane among the applied conditions.
3.3. Electron transport properties of TiO2 nanoporous films in DSSC
To demonstrate the effect of the nano-morphology of nanoporous films on the electron transport properties of nanoporous TiO2 electrodes, the parallel and non-parallel orientations of TiO2 nanosheets with respect to the FTO substrate plane were examined in the DSSC device configuration composed of a Ru dye-sensitizer (N719), an iodine/iodide redox electrolyte, and a platinum counter electrode. The TiO2 nanoporous network shows non-parallel orientation by Tyr-functionalization, presumably because it possesses fewer inter-particle defects through the nanoporous network in TiO2 films, while the parallel orientation without any functionalization provides more inter-particle defects in the nanoporous TiO2 film in case the films have the same thickness. Therefore, we prepared the two types of nanoporous TiO2 nanosheet films with the same thickness (∼2 µm).A similar loading of dye-sensitizer on both parallel and non-parallel nanoporous TiO2 (Table S1, ESI†) indicates that both nanoporous TiO2 film properties are not affected by the internal surface area and porosity of the nanoporous films. The electron diffusion coefficient in TiO2 was determined by analysis of the transient short-circuit photo-current under modulated incident excitation in the DSSC device configuration.27,28,35 The charge density as a function of the energy level in TiO2, and the electron lifetime representing the recombination exhibits similar with each TiO2 film (Fig. S8, ESI†). On the other hand, the electron diffusion coefficient in TiO2 shows significant improvement (a factor of approximately 4) with non-parallel orientation of nanosheets compared with parallel orientation in TiO2 nanoporous films (Fig. S9, ESI†) in DSSC (Fig. 6).
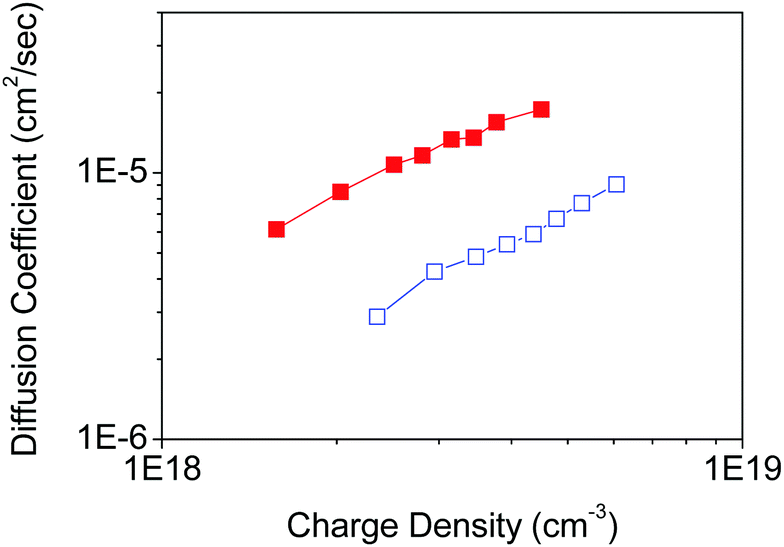 | ||
| Fig. 6 The diffusion coefficient of electrons in TiO2 as a function of the charge density in DSSC with TiO2 porous films composed of Tyr-functionalized (red) and non-functionalized (blue) TiO2 nanosheets on FTO substrates. | ||
Since the orientation of generated electron transport in DSSC with layered film construction is perpendicular to the substrate plane, the characteristics of electron transport towards this direction are the most important. In this case, the orientation of nanosheets in the films would be different in parallel and non-parallel orientations, which we attribute to two potential reasons. (1) Electron transport could have anisotropic characteristics, and the transport in the [001] direction could be slower than other directions,36,37 and (2) the inter-particle connection typically has fewer transport properties due to the local defects and surface trap state locating at the inter-particle region,38,39 and the number of the inter-particle region increases in the parallel orientation compared with non-parallel orientation in the case of the same thickness of nanoporous films, as indicated in Scheme 1. Therefore, with respect to these reasons, the non-parallel orientation of nanosheets in the nanoporous films should result in fewer electron transport properties, which was observed in our results.
As the thickness of the TiO2 film of the DSSC is much less than the diffusion length of the DSSC utilizing commonly used ruthenium complex with iodide redox couples, in the range 10–20 µm,26 the improvement in the diffusion coefficient does not significantly affect the overall conversion efficiency of the solar cell (Fig. S10, ESI†). The effect of the improvement in the diffusion coefficient, however, significantly influences the properties in practical DSSCs with thick TiO2 films of thickness 20–30 µm,26 which is comparable with the diffusion length of electrons in TiO2. The result of the enhanced electron diffusion property with non-parallel orientation of the TiO2 nanosheets with respect to the substrate plane is probably attributed to fewer inter-particle connections through the TiO2 film (Scheme 1).
As a consequence, we demonstrated a novel control method of orientation of anisotropic TiO2 in nanoporous films. This confirms the significant influence of nanomorphology on the electronic properties of nanoporous films constructed with anisotropic nanopaticles. Furthermore, the best efficiency of DSSC was recently achieved by organic-based dye-sensitizers with a cobalt redox couple, which potentially decreased the diffusion length of electrons in nanoporous TiO2 networks.40 Therefore, improvement of the diffusion constant, which extended the diffusion length of the electron in the nanoporous TiO2 network in our study, would contribute to an improvement in the conversion efficiency in those systems. Furthermore, the results we observed here are feasible enough to demonstrate the importance of orientation control improving the carrier transport in the mesoporous semiconductor network.
Conclusions
Self-oriented anisotropic nanoparticles in nanoporous films were obtained by controlling the surface functionality induced by the adsorption of organic amino acid derivatives onto TiO2 nanosheets. Tyrosine-functionalized TiO2 nanosheets were self-assembled in a non-parallel orientation with respect to the substrate plane due to the surface properties of (1) the ζ-potential, (2) hydrogen bonding, and (3) hydrophobicity, providing the synergetic effect of inter-particle aggregations and segregative interactions from the hydrophilic environment of water and oxide substrates. To demonstrate the orientation effects in electrochemical applications, nanoporous TiO2 films constructed with nanosheets with non-parallel and parallel orientations with respect to the substrate plane were applied to the nanoporous semiconductor films in the DSSC. Although both nanoporous films with parallel and non-parallel orientations of TiO2 nanosheets exhibited a similar internal surface area, resulting in the same amount of dye-sensitizer loading, the evaluation of electrochemical properties of TiO2 films showed significant improvement in the electron transport property of the TiO2 film with non-parallel orientation compared with the parallel orientation. The other important factors of charge recombination and charge density were found to remain the same. The proposed process is based on controlling the surface properties of nanoparticles and the surrounding solvent. Furthermore, we demonstrated the importance of the orientation of nanoparticle components in the films to achieve high performance, and therefore we believe that the basic strategy to control the nanoporous films in this work would contribute to a wider range of devices using nano-porous films.Acknowledgements
The authors thank Bunseki Shien Center, Tokyo Inst. Tech., for the crystallographic analysis of TiO2, and Prof. Saji and Dr Ogihara, Tokyo Inst. Tech., for the contact angle measurements. This work was supported by the Murata Science Foundation, General Sekiyu R & D Encouragement Assistance Foundation, and JSPS KAKENHI Grant Numbers 25249113 and 25790013. All authors contributed equally to this work.References
- S. Mathew, A. Yella, G. Gao, R. Humphry-Baker, B. F. E. Curchod, N. Ashari-Astani, I. Tavernelli, U. Rothlisberger, M. K. Nazeeruddin and M. Grätzel, Nat. Chem., 2014, 6, 242 CrossRef CAS PubMed.
- M. M. Lee, J. Teuscher, T. Miyasaka, T. N. Murakami and H. J. Snaith, Science, 2012, 338, 643 CrossRef CAS PubMed.
- NREL chart, http://www.nrel.gov/ncpv/images/efficiency_chart. jpg, accessed Nov., 2016.
- J. Maier, Nat. Mater., 2005, 4, 805 CrossRef CAS PubMed.
- (a) P. Yang, D. Zhao, D. I. Margolese, B. F. Chmelka and G. D. Stucky, Nature, 1998, 396, 152 CrossRef CAS; (b) A. Ivanova, D. Fattakhova-Rohlfing, B. E. Kayaalp, J. Rathousky and T. Bein, J. Am. Chem. Soc., 2014, 136, 5930 CrossRef CAS PubMed; (c) E. J. W. Crossland, N. Noel, V. Sivaram, T. Leijtens, J. A. Alexander Webber and H. J. Snaith, Nature, 2013, 495, 215 CrossRef CAS PubMed.
- T. Wagner, S. Haffer, C. Weinberger, D. Klaus and M. Tiemann, Chem. Soc. Rev., 2013, 42, 4036 RSC.
- Q. Zhang, C. S. Dandeneau, X. Zhou and G. Cao, Adv. Mater., 2009, 21, 4087 CrossRef CAS.
- G. Liu, J. C. Yu, G. Q. Lu and H.-M. Cheng, Chem. Commun., 2011, 47, 6763 RSC.
- X. Chen and S. S. Mao, Chem. Rev., 2007, 107, 2891 CrossRef CAS PubMed.
- A. I. Hochbaum and P. Yang, Chem. Rev., 2010, 110, 527 CrossRef CAS PubMed.
- (a) J. Sheng, L. Hu, S. Xu, W. Liu, L. Mo, H. Tian and S. Dai, J. Mater. Chem., 2011, 21, 5457 RSC; (b) R. Agosta, R. Giannuzzi, L. De Marco, M. Manca, M. R. Belviso, P. D. Cozzoli and G. Gigli, J. Phys. Chem. C, 2013, 117, 2574 CrossRef CAS.
- R. Zhang, S. Kumar, S. Zou and L. L. Kerr, Cryst. Growth Des., 2008, 8, 381 CAS.
- (a) S. Horii, I. Matsubara, M. Sano, K. Fujiie, M. Suzuki, R. Funahashi, M. Shikano, W. Shin, N. Murayama, J. Shimoyama and K. Kishio, Jpn. J. Appl. Phys., 2003, 42, 7018 CrossRef CAS; (b) M. Kawakita, T. Uchikoshi, J. Kawakita and Y. Sakka, J. Am. Ceram. Soc., 2009, 92, 984 CrossRef CAS.
- K. Mimura, F. Dang, K. Kato, H. Imai, S. Wada, H. Haneda and M. Kuwabara, Jpn. J. Appl. Phys., 2011, 50, 09NC09 CrossRef.
- H. Yang, T. Ogawa, D. Hasegawa and M. Takahashi, J. Appl. Phys., 2008, 103, 07D526 CrossRef.
- L. Malaquin, T. Kraus, H. Schmid, E. Delamarche and H. Wolf, Langmuir, 2007, 23, 11513 CrossRef CAS PubMed.
- J.-M. Meijer, F. Hagemans, L. Rossi, D. V. Byelov, S. I. R. Castillo, A. Snigirev, I. Snigireva, A. P. Philipse and A. V. Petukhov, Langmuir, 2012, 28, 7631 CrossRef CAS PubMed.
- Y. Nakagawa, H. Kageyama, Y. Oaki and H. Imai, J. Am. Chem. Soc., 2014, 136, 3716 CrossRef CAS PubMed.
- A. P. Allvisatos, K. P. Johnsson, X. Peng, T. E. Wilson, C. J. Loweth, M. P. Bruchez Jr and P. G. Schultz, Nature, 1996, 382, 609 CrossRef PubMed.
- (a) C. A. Mirkin, R. L. Letsinger, R. C. Mucic and J. J. Storhoff, Nature, 1996, 382, 607 CrossRef CAS PubMed; (b) E. Auyeung, T. I. N. G. Li, A. J. Senesi, A. L. Schmucker, B. C. Pals, M. O. de la Cruz and C. A. Mirkin, Nature, 2014, 505, 73 CrossRef PubMed.
- H. G. Yang, H. S. Sun, S. Z. Qiao, J. Zou, G. Liu, S. C. Smith, H. M. Cheng and G. Q. Lu, Nature, 2008, 453, 638 CrossRef CAS PubMed.
- X. Han, Q. Kuang, M. Jin, Z. Xie and L. Zheng, J. Am. Chem. Soc., 2009, 131, 3152 CrossRef CAS PubMed.
- G. Liu, H. G. Yang, X. Wang, L. Chen, H. Lu, L. Wang, G. Q. Lu and H. M. Cheng, J. Phys. Chem. C, 2009, 113, 21784 CAS.
- M. M. Maitani, K. Tanaka, D. Mochizuki and Y. Wada, J. Phys. Chem. Lett., 2011, 2, 2655 CrossRef CAS.
- S. Ito, in Dye-sensitized Solar Cells, ed. K. Kalyanasundaram, EPFL Press, Lausanne, 2010, ch. 8 Search PubMed.
- A. Hagfeldt, G. Boschloo, L. Sun, L. Kloo and H. Pettersson, Chem. Rev., 2010, 110, 6595 CrossRef CAS PubMed.
- S. Nakade, T. Kanzaki, Y. Wada and S. Yanagida, Langmuir, 2005, 21, 10803 CrossRef CAS PubMed.
- B. C. O'Regan, K. Bakker, J. Kroeze, H. Smit, P. Sommeling and J. R. Durrant, J. Phys. Chem. B, 2006, 110, 17155 CrossRef PubMed.
- U. Diebold, Surf. Sci. Rep., 2003, 48, 53 CrossRef CAS.
- J. B. Peri, Th. F. Tadros, H. P. Boehm, J. Lyklema, J. Hockey, H. Knözinger, J. B. Uytterhoeven, N. D. Parkyns, G. Munuera, A. Zecchina, Z. G. Szabó, C. H. Rochester, Y. A. Eltekov, E. K. Bogacheva, L. Cerruti, E. Guglielminotti, P. W. Schindler, H. Gamsjäger and T. J. Gray, Discuss. Faraday Soc., 1971, 52, 276 RSC.
- B. Ohtani, Y. Okugawa, S. Nishimoto and T. Kagiya, J. Phys. Chem., 1987, 91, 3550 CrossRef CAS.
- P. W. Atkins, Physikalische Chemie, Einstein-stokes equation, VCH, Weinheim, 1996 Search PubMed.
- J. Moser, S. Punchihewa, P. P. Infelta and M. Gräzel, Langmuir, 1991, 7, 3012 CrossRef CAS.
- A. E. Regazzoni, P. Mandelbaum, M. Matsuyoshi, S. Schiller, S. A. Bilmes and M. A. Blesa, Langmuir, 1998, 14, 868 CrossRef CAS.
- N. W. Duffy, L. M. Peter, R. M. G. Rajapakse and K. G. U. Wijayantha, Electrochem. Commun., 2000, 2, 658 CrossRef CAS.
- M. Landmann, E. Rauls and W. G. Schmidt, J. Phys.: Condens. Matter, 2012, 24, 195503 CrossRef CAS PubMed.
- N. A. Deskins and M. Dupuis, J. Phys. Chem. C, 2009, 113, 346 CAS.
- C. J. Barbe, F. Arendse, P. Comte, M. Jirousek, F. Lenzmann, V. Shklover and M. Grätzel, J. Am. Ceram. Soc., 1997, 80, 3157 CrossRef CAS.
- S. Nakade, M. Matsuda, S. Kambe, Y. Saito, T. Kitamura, T. Sakata, Y. Wada, H. Mori and S. Yanagida, J. Phys. Chem. B, 2002, 106, 10004 CrossRef CAS.
- K. Kakiage, Y. Aoyama, T. Yano, K. Oya, J. Fujisawa and M. Hanaya, Chem. Commun., 2015, 51, 15894 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: The experimental details with characterization method, XRD spectra, and EIS. See DOI: 10.1039/c7qm00239d |
| ‡ Present address: Research Center for Advanced Science and Technology, The University of Tokyo, Meguro, Tokyo, 153-8904 Japan. |
| This journal is © the Partner Organisations 2017 |