DOI:
10.1039/C7QM00217C
(Research Article)
Mater. Chem. Front., 2017,
1, 2065-2077
Thermal-induced surface defective Co/Fe–Co planar hybrid composite nanosheet with enhanced catalytic activity in the Fenton-like reaction†
Received
13th May 2017
, Accepted 21st June 2017
First published on 21st June 2017
Abstract
Surface defective or heterojunction sites of catalysts generally afford remarkable catalytic activity thanks to their high-surface-energy. Regularly-generating multiple defective sites on the surface of a catalyst is exceedingly promising in many reactions. In this study, we report an unexpected Co/Fe–Co planar hybrid composite nanosheet with serried surface defects including surface biphasic junction sites or defective holes. A facile thermal-induced process was applied to trigger the generation of defective sites on planar Co/Fe–Co hydroxides. Through a controlled thermal process, massive serried CoO nanocrystals were in situ dissolved out of the surface of cobalt hydroxide, while defective surface holes were formed on the Fe-doped Co(OH)2 nanosheets under a prolonged thermal procedure. The morphology and microscale structure of the resulting Co/Fe–Co hybrid 2-dimensional (2D) composites were systematically examined by virtue of various characterization techniques. As expected, the obtained surface defective Co/Fe–Co planar composites showed obviously enhanced catalytic activity in the Fenton-like reaction. Based on a catalytic study, we proved that the CoO nanocrystals densely-distributed on thinlayer Co(OH)2. Uniformly-introduced Fe heteroatoms in the inter-structure of Co(OH)2, along with the formed surface defective holes on Fe–Co hybrid composites, are synergistically responsible for increased catalytic removal efficiency of MB in the presence of peroxomonosulfate.
Introduction
Two-dimensional (2D) materials like graphene have shown interesting applied prospects in various fields.1–5 Homoplastically, 2-dimensional transition-metal hydroxides (TMHs) possessing several nanoscale thicknesses afford crescent attention for scientists thanks to some of their unique physicochemical features, such as inherent abundantly-exposed active atom proportions, large surface areas, and low diffusion limitations.6–11 These advantages surpass the physical capabilities of some counterparts having other nanostructures, which paves the way for their applications with catalysis,12–15 energy storage,9,16–18 and environmental solutions.19–21
Recently, we noticed that planar TMHs have been widely applied for energy-conversion, especially in electrochemical fields due to their outstanding electrical conduction efficiency, and certain interesting performance has been received accordingly. Actually, planar nanomaterials have more potential superiorities in heterogeneous catalysis because of their almost completely exposed surface atoms at molecular-scale thickness. However, to the best of our knowledge, edges, corners, terraces, and rugged sites on a catalyst are regarded as significant catalytic active sites in heterogeneous catalysis. Continuous and completely planar materials lack the aforementioned defective active features. Therefore, further exploring enhancement of catalytic activity on planar nanomaterials is desirable and expected. As mentioned, such highly active sites tend to come from surface special defects such as a wrinkle or heteropical interface. However, these special defects seem sensitive to being created by certain ingenious skills. Therefore, some researchers proposed certain tactics through physically introducing heteroatoms on the surface of 2-dimensional transition metal hydroxides to produce more catalytic active sites.9,12,22 However, the introduced heteroatoms fail to afford a synergetic function between heteroatoms and TMHs owing to their infirm physicochemical interactions. Whereas, the existence of mixed phases on the surface of a catalyst is extremely beneficial to catalytic reactions due to emerging highly-active interphase junctions. Withal, a structural imperfection also has a certain influence on catalytic activity, for example, highly-stressed defects produced by structural changing promotes atoms around the defect centres to be more active compared to other homogeneous environmental platforms of 2D materials. Therefore, it is very desirable for 2D materials to create highly defective sites to further enhance catalytic activity.
Toxic organics involved in industrial manufacturing is a serious issue in water pollution which could be efficiently resolved by catalytic decomposition using specific 2D materials. Cobaltous mediated homogeneous decomposition of a peroxomonosulfate (Co/PMS) system triggering degrading organic contaminants has been proposed as an advanced oxidation process (AOP); it demonstrated preponderance over Fenton reagents (Fe/H2O2) by demonstrating high efficiency in a wide pH range and required smaller amounts of cobalt catalyst.23–27 Recently, several investigators attempted using cobalt oxides and supported cobalt oxides as heterogeneous catalysts for activating PMS to degrade organic pollutants. Although these catalysts are effective in initiating sulfate radical generation from PMS, low catalytic performance is still a major limitation for practical applications in water treatment.
Herein, we propose a facile thermal-triggered modified processing for 2-dimensional transition-metal hydroxides including pure Co(OH)2 and a heteroatom Fe-doped Co(OH)2 nanosheet to create certain special structural defects for enhancing catalytic properties. 2-Dimensional thin-layer CoO/Co(OH)2 composites were directly achieved and many heterogeneous CoO nanocrystals in situ dissolved from the surfaces of TMHs. Accordingly, serried heterojunctions existed among the CoO/Co(OH)2 present on surfaces of Co(OH)2 nanosheets after thermal-treatment. Additionally, we ulteriorly prepared Fe-doped Co(OH)2 with controlled Fe concentrations. The succedent thermal treatment process induced the presence of surface defective holes on the Fe–Co hybrid composites. We systematically characterized physical and chemical properties of the resulting nanocomposites. We also affirmed the 200 °C thermal treatment procedure could efficiently trigger formation of massive surface defects on the surface of a co-based nanosheet and do no harm to the overall morphology of materials. Through catalytic degradation of methylene blue (MB), we systematically evaluated catalytic properties of these resulting catalysts and studied catalytic correlationships between defective sites and catalytic performance. Results revealed that formation of heterogeneous CoO nanocrystals and the introduction of Fe heteroatom, along with generated defective holes, efficiently promoted catalytic degradation efficiency of methylene blue in the presence of peroxymonosulfate.
Experimental section
Chemicals
All chemicals were used as received without further purification. Cobalt chloride (CoCl2·6H2O, AR), ferrous sulfate (FeSO4·7H2O, AR), sodium borohydride (NaBH4, AR), and PVP (polyvinylpyrrolidone, Mw = 40![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000) were purchased from Sigma-Aldrich (USA). Methylene blue (MB) and oxone (peroxymonosulfate, PMS, 2KHSO5·KHSO4·K2SO4) were purchased from Aladdin (China).
000) were purchased from Sigma-Aldrich (USA). Methylene blue (MB) and oxone (peroxymonosulfate, PMS, 2KHSO5·KHSO4·K2SO4) were purchased from Aladdin (China).
Synthesis of Co(OH)2 ultrathin nanosheets
Pure α-Co(OH)2 planar nanosheets were synthesized according to a reported reference11 with few modifications. In a typical synthesis, CoCl2·6H2O (85 mg) and PVP (1 g) were first dissolved in 215 mL deionized water in a 500 mL round-bottomed flask and purged with N2 for 10 min. Subsequently, a freshly prepared NaBH4 aqueous solution (0.2 g, 10 mL) was added dropwise. After 10 min, the resulting black solution was exposed to air under stirring for 12 h. Resulting products were collected by centrifugation, washed several times with water and ethanol, and finally dried at 60 °C.
Synthesis of CoO/Co(OH)2 hybrid composites nanosheets
The obtained Co(OH)2 ultrathin nanosheets were further thermally-treated at 200 °C, 300 °C, and 400 °C for 1 h, respectively in a 5% hydrogen atmosphere (95% nitrogen balance gas). Finally, the obtained samples were labeled as Co-200, Co-300, and Co-400, accordingly.
Synthesis of Fe-doped Co(OH)2 hybrid composite nanosheets
Synthesis of Fe-doped Co(OH)2 ultrathin nanosheets is similar with above; typically, CoCl2·6H2O (85 mg), a certain amount of FeSO4·7H2O (molar ratio of Fe/Co = 0.2, 0.4, 0.6, and 0.8) and PVP (1 g) were first dissolved in 215 mL deionized water in a 500 mL round-bottomed flask and purged with N2 for 10 min. Subsequently, a freshly prepared NaBH4 aqueous solution (0.2 g, 10 mL) was added dropwise. After 10 min, the resulting black solution was exposed to air for 12 h. With prolonged stirring time, the mixed solution finally transformed to orange. The resulting products were collected by centrifugation, washed several times with water and ethanol, and finally dried at 60 °C. These samples were labeled as 0.2Fe–Co(OH)2, 0.4Fe–Co(OH)2, 0.6Fe–Co(OH)2, and 0.8Fe–Co(OH)2, accordingly.
Synthesis of surface defective Fe–Co hybrid composite 2D nanosheets
The obtained serial Fe-doped Co(OH)2 ultrathin nanosheets were further thermally-treated at 200 °C for 2 h, respectively in a 5% hydrogen atmosphere (95% nitrogen balance gas). Finally, the obtained samples were labeled as 0.2Fe–Co-200, 0.4Fe–Co-200, 0.6Fe–Co-200, and 0.8Fe–Co-200. The comparative catalysts including 0.2Fe–Co-200(1 h), 0.4Fe–Co-200(1 h), 0.6Fe–Co-200(1 h), and 0.8Fe–Co-200(1 h) were prepared by treating the serial Fe-doped Co(OH)2 ultrathin nanosheets at 200 °C for 1 h, respectively in 5% hydrogen atmosphere (95% nitrogen balance gas).
Characterizations
Transmission electron microscopy (TEM) was performed with a JEM 2100F (JEOL, Japan) microscope operated at 200 kV, equipped with EDX elemental mapping accessories. X-ray diffraction (XRD) analysis was carried out using a Bruker D8 Advance X-ray diffractometer (Germany), at a scan rate of 2° min−1 and operated at 40 kV applying a potential current of 30 mA. Textural properties of samples were collected by measuring the physical adsorption of nitrogen at liquid-nitrogen temperature of 77 K using an automatic volumetric sorption analyzer (NOVA3200e, Quantachrome (USA)). X-ray photoelectron spectra (XPS) were recorded on a VG Escalab MKII spectrometer (England), using a mono Al Kα X-ray source (hν = 1486.71 eV, 5 mA, 15 kV), and calibrated by setting the C1s peak at 284.6 eV. AFM was conducted by testing samples deposited on a glass sheet with a Bruker Dimension Icon atomic force microscope (Germany).
TGA was performed using a Shimadzu DTG-60AH thermal analyzer (Japan) under flowing nitrogen (40 mL min−1) with a desirable heating rate. FE-SEM was conducted on a JEOL JSM-7610F scanning electron microscope (Japan). Attenuated total reflectance infrared (ATR-IR) spectroscopy analysis was carried out on a Thermo Scientific Nicolet iS50 FT-IR spectrometer (USA) integrated with a diamond ATR accessory, and IR spectra were collected in the spectral range 4000–525 cm−1.
Metal content in the catalyst was determined by iCAP 6000 series inductively coupled plasma optical emission spectrometry (ICP-OES) (USA). The catalysts were digested in hydrochloric acid and then diluted with deionized water to a certain volume before analysis. The concentrations of the MB solution were measured on a Biochrom Libra UV-Vis spectrometer (England).
Catalytic assessment
Catalytic degradation of MB was carried out in a 200 mL reactor containing 100 mL of 200 ppm MB solution. Incipiently, precisely weighed 5 mg of catalysts were added into a MB solution and followed by ultrasonic treatment for several minutes. The reaction solution was stirred continuously to maintain a homogeneous solution at ambient temperature. Subsequently, 0.25 g of PMS was added to the mixture solution and the reaction was started. At predetermined time intervals, 1 mL of reaction liquid was withdrawn into an HPLC vial using a syringe filter, and 1 mL of methanol was added to quench the reaction. Concentrations of MB were analyzed using an ultraviolet spectrophotometer based on the Lambert–Beer's law. For selected samples of Co-200, total organic carbon (TOC) was determined using an ASI Shimadzu TOC-5000A analyzer, in which a 5 mL sample was extracted from the reaction solution, quenched with 5 mL of 0.3 M sodium nitrite solution, and then analyzed for TOC.
Results and discussion
Characterizations of morphology and structure
The overall synthetic procedure to produce defective Co/Fe–Co metal planar hybrid composite nanosheets is schematically presented in Scheme 1 and minutely described in the Experimental section. The 2D TMHs thinlayer composite was transformed from the metal–boron nanosphere composites exposed to air.11 To trigger formation of surface defective sites, the 2D TMHs thinlayer composites were further treated under 5% H2/N2 forming gas atmosphere. The as-obtained incipient α-Co(OH)2 shows a dark green state, and the color of the thermal-resulted samples Co-200, Co-300, and Co-400 turned dark under an ascending temperature thermal process (Scheme 2), indicating certain physicochemical variations occurring during the thermal-induced process. As such, it can be observed that the color of as-prepared Fe-doped samples turned to orange compared to as-obtained α-Co(OH)2 due to introduction of Fe atoms. After thermal treatment for Fe-doped Co(OH)2, the orange powder transformed into a brownish-black state, signifying a changing physicochemical property (Scheme 2). Microscopic morphology and microscale structure of the controlled samples involved were identified through TEM and SEM techniques. As explicitly displayed in Fig. 1, where representative TEM images of various samples are presented, a typical 2-dimensional ultrathin nanosheet morphology for as-obtained Co(OH)2 and different loads of as-prepared Fe-doped Co(OH)2 is proved (Fig. 1a and 2a). High transparency under the electron beam ulteriorly demonstrates their ultrathin nature. As evidence, we found that the corrugated position presenting on the TEM image (Fig. 1a) shows a thickness of ca. 5.1 nm for the folded layer (marked with yellow line) of incipient α-Co(OH)2, indicating the monolayer thickness of Co(OH)2 is lower than 2.6 nm. This result is consistent with the displayed AFM profiles of α-Co(OH)2 (Fig. S2, ESI†). Additionally, in the case of the crystalline structure state of Co(OH)2, the initial cobalt hydroxide product can be clearly identified by X-ray diffraction as α-Co(OH)2 (JCPDS # 46-0605), which is consistent with the reported one.11
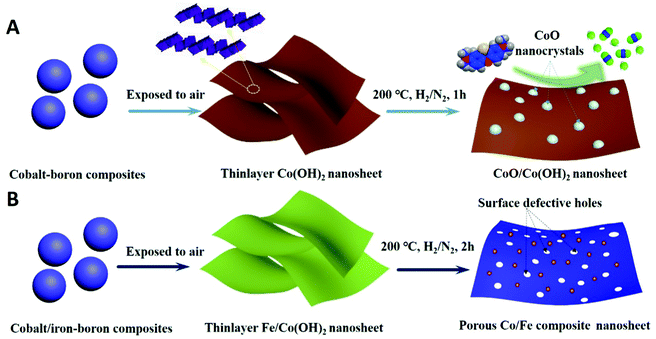 |
| | Scheme 1 Schematic illustration for the synthesis of surface defective 2D Co/Fe–Co hybrid composite nanosheets, (A) CoO nanocrystals automatically dissolve from the surface of Co(OH)2; (B) defective surface holes form on the surface of Co/Fe hybrid 2D composites. | |
 |
| | Scheme 2 Photographs for the various controlled samples. | |
 |
| | Fig. 1 TEM images of controlled thermal procedure induced samples including (a) Co(OH)2, (b) Co-200, (c) Co-300, and (d) Co-400. | |
When exerting a specific thermal treated procedure (200 °C, 1 h) on an ultrathin Co(OH)2 nanosheet, interestingly, we observed emerging serried blackspots on the surface of the Co(OH)2 nanosheet, attributable to heterogeneous cobalt oxide nanocrystals in situ dissolved from the Co(OH)2 phase during the thermal treatment procedure. Also, elemental mapping results (Fig. 1b, inset) still indicate a uniform distribution of elemental cobalt on the materials from thermal-resulting Co-200. As evidence for the presence of CoO, the corresponding crystalline lattice fringes (d111 = 0.246 nm) detected on the TEM image (Fig. 1b and Fig. S1, ESI†) for the darker nanocrystals were confirmed and demonstrated to coincide with the CoO phase. Furthermore, wide-angle XRD results (Fig. 3a) present the newly emerging diffraction (200) at 2θ = 42.5° of CoO phase in the curve of Co-200, definitely confirming the presence of CoO nanocrystals after the thermal treatment, which is in agreement with the TEM evidence. However, it should be noted that the dominant diffraction of α-Co(OH)2 in the curve verifies that α-Co(OH)2 is still the main phase and was retained for Co-200 even though massive serried CoO nanocrystals dissolved out and distributed on the Co(OH)2 nanosheets. In addition to the XRD results, it should be noted that the nanosheet morphology of ultrathin Co(OH)2 is still well preserved for Co-200 after being treated in 5% hydrogen at 200 °C for 1 h (Fig. 1b). This result proves that the 200 °C treatment does not cause great detriment to 2D integral morphology structure. Meanwhile, the presence of these serried CoO nanocrystals contributed to the biphasic CoO/Co(OH)2 hybrid composite and undoubtedly to the accompanying heterojunction sites around the CoO nanocrystals.
For the Co-300 sample treated at 300 °C, we noticed that the morphology of Co(OH)2 was damaged to some extent; even though a 2-dimensional structure still faintly is observed, it looks more like the composites stacked with lots of nanoparticles rather than an unbroken 2-dimensional nanosheet. When treatment temperature was sequentially increased to 400 °C, morphology of the 2D thin-layer composite was further destroyed and the 2D morphology almost vanished in Co-400. Accordingly, wide-angle XRD results (Fig. 3a) display the dominant diffraction phase (111), (200) at 36.5° and 42.5° of CoO (JCPDS # 43-1004) accompanied by vanishing of the α-Co(OH)2 diffraction for samples Co-300 and Co-400, signifying the phase transforming from α-Co(OH)2 to CoO under higher treatment temperatures.
On the other hand, with the Fe-doped serial samples, we observed that the as-synthesized Fe-doped Co(OH)2 hybrid nanosheets still afford the benign 2-dimentional structure morphology (Fig. 2a–d) even with different loadings of iron, indicating that introduction of iron hardly influences the morphology of Co(OH)2. To assess distribution of iron in the Co(OH)2 nanosheets, elemental mapping of 0.6Fe–Co(OH)2 was inspected and corresponding results are presented in Fig. 2i. As expected, the elements Co, Fe, and O mainly constitute the 2D Co/Fe hybrid composites, and uniform distribution of element Fe on them also was verified. Further, wide-angle XRD was utilized to inspect the crystalline structure of Co–Fe hybrid composites. Compared to pure α-Co(OH)2, Fe-doped Co(OH)2 shows a similar crystalline diffraction profile based on different proportions of Fe/Co (Fig. 3b). However, obvious shifts of diffraction peaks from Co(OH)2 are observed in the curves, which is evidence for efficient doping of Fe atoms in the Co(OH)2 crystal structure.
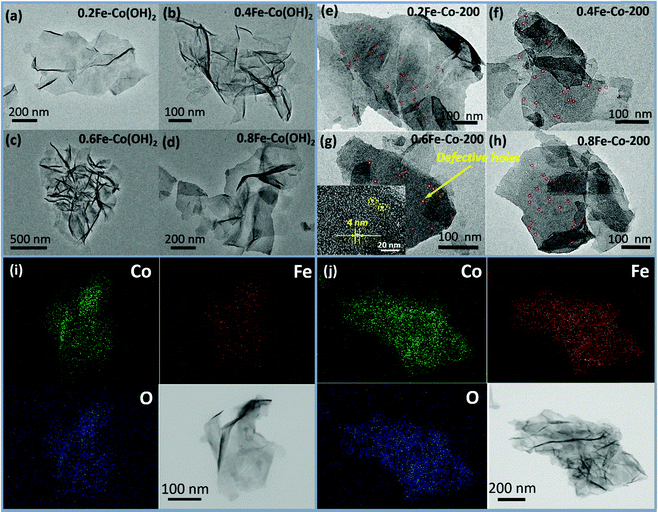 |
| | Fig. 2 TEM images of Fe-doped Co(OH)2 (a–d) and thermal treated Fe–Co hybrid composites nanosheet (e–h) and EDX elemental mapping patterns for 0.6Fe–Co(OH)2 (i) and 0.6Fe–Co-200 (j). | |
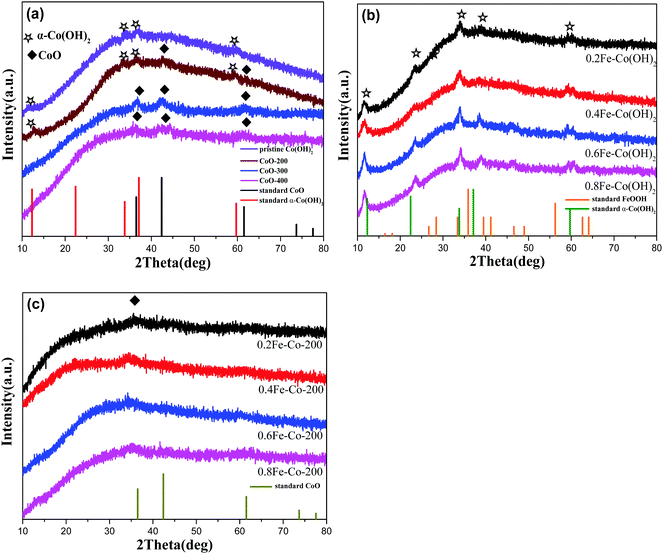 |
| | Fig. 3 Wide-angle XRD patterns of various controlled comparative samples including (a) single metal cobalt-containing series samples, (b) Fe-doping series Co(OH)2 samples, (c) thermal-treated Fe-doping series Co-based samples. | |
For serial samples of xFe–Co-200(1 h) treated with the identical thermal procedure (200 °C, 1 h), corresponding TEM results (Fig. S4, ESI†) affirm almost the same morphology with pristine xFe–Co(OH)2. Meanwhile, wide-angle XRD results (Fig. S5, ESI†) reveal vanishing of the α-Co(OH)2 phase and the presence of a weak mixed metal oxides phase. After a prolonged thermal treatment process (200 °C, 2 h) for the Fe–Co hybrid nanosheets, resulting samples of the Fe-doped 2D hybrid composites were further examined with TEM and XRD. TEM results (Fig. 2e–h) showed that all the samples still retained the structure of a 2D nanosheet after 2 h of thermal processing under 5% of H2/N2; no heterojunction nanocrystals were probed, which differs from the resulting Co-200 sample. Accordingly, EDX elemental mapping (Fig. 2j) reveals that thermal-processing hardly resulted in any change to the distribution of various elements including Co, Fe, and O, thus verifying that the Fe atoms were assuredly stabilized in the material's skeleton rather than being mechanically adsorbed on the surface. However, note that the crystalline structure of the Fe–Co hybrid composites was transformed into mixed metal oxides states with a relatively low crystalline degree, which is evidenced by wide-angle XRD results (Fig. 3c). Apart from that, compared to the as-obtained xFe–Co(OH)2 and the thermal resulting serial xFe–Co-200(1 h), we noticed the obvious surface holes present in each sample with a mesoporous size (Fig. 2e–h). In addition, the surfaces of those samples became much rougher, indicating production of surface defects after the thermal treatment process. We believe that production of these surface defects stems from shrinking and degradation of Fe–Co hydroxide composites.
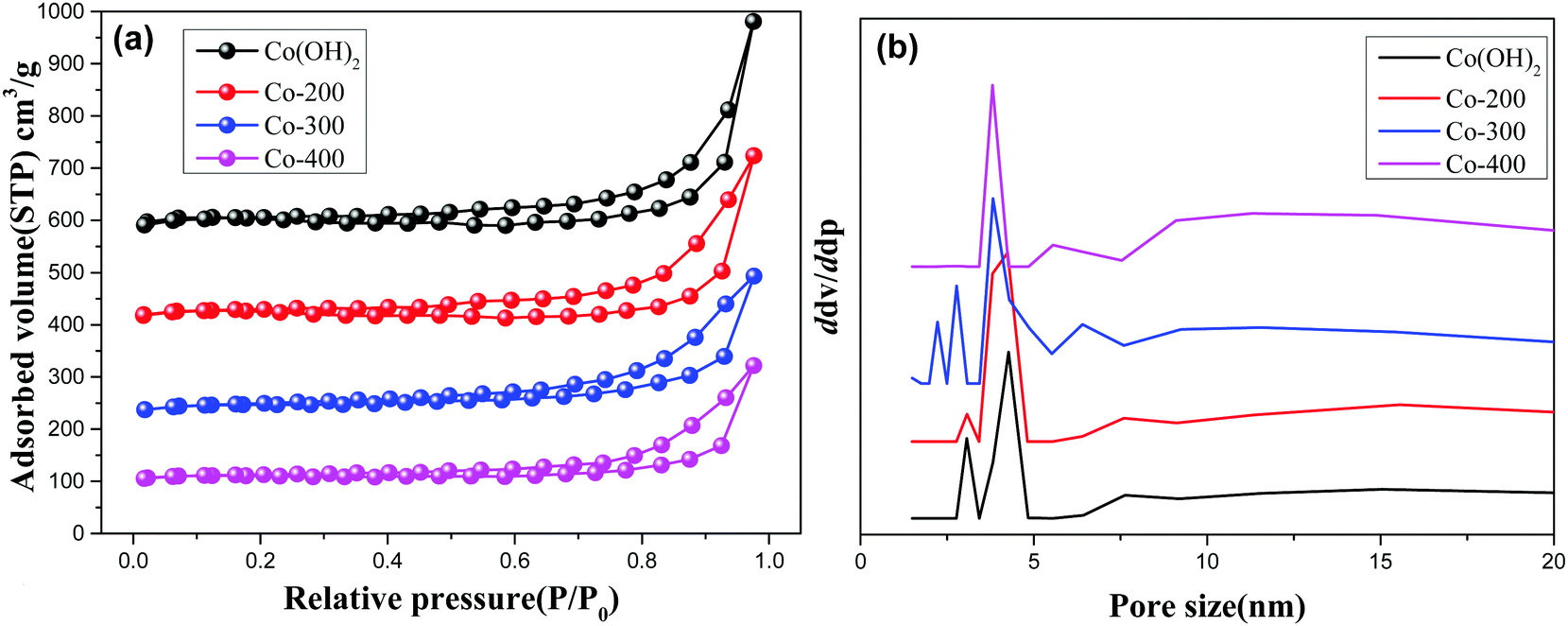
Fig. 4 and Fig. S3 (ESI†) display the N2-adsorption/desorption isotherms and pore size distribution profiles of various controlled samples. As observed, all the present samples afford a type H3 hysteresis loop, indicating typical slit-shaped mesoporous materials.28 These show average pores afford ca. 5 nm size due to inter-nanosheet voids. But, some wide pore size distribution with larger pore size also can be observed in the BJH profiles for various samples. We calculated the detailed textural properties and summarized them in Table 1. A pure Co(OH)2 nanosheet shows 209.7 cm2 g−1 surface area. Increasing treatment temperatures for the Co(OH)2 nanosheet decreases surface areas of Co(OH)2, Co-200, Co-300, and Co-400 from 209.7 cm2 g−1 to 112.9 cm2 g−1 accordingly, indicating the high-temperature treatment likely damaged the morphology and microscale structure of the nanosheets to some extent. In the case of Fe-doped Co(OH)2, inferior textural properties are verified compared to pure α-Co(OH)2, signifying introduction of Fe heteroatoms causes certain variations of cobalt hydroxides. Meanwhile, textural parameters of thermally treated Fe-doped Co(OH)2 are further displayed in Table 1. As observed, marginal decreasing of surface area can be observed for all the samples, indicating the structure of the nanosheet was not gravely damaged, which is in accordance with TEM results. In conjunction with TEM and N2 desorption results, we speculate that introduction of Fe heteroatoms efficiently stabilizes the integrated morphology state of the Fe–Co hybrid 2D composites nanosheet. The given SEM images further support the N2 adsorption/desorption results; as displayed in Fig. 5, the samples of α-Co(OH)2 and Co-200 show a typical plicated and cumulate thinlayer 2D materials morphology state. In contrast, the corresponding higher temperature treated samples Co-300 and Co-400 exhibit a broken and agminated bulk state, and the 2D morphology seems to be invisible, signifying a grievous breakage of morphology and structure.
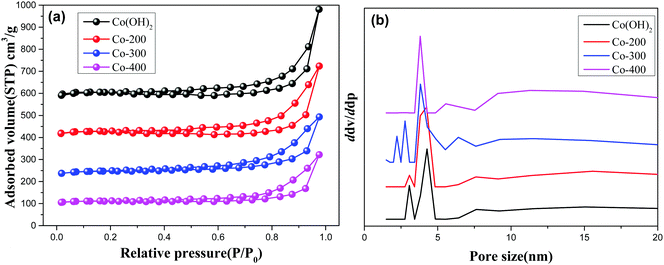 |
| | Fig. 4 N2 adsorption/desorption isotherms (a) and pore size distribution (b) of Co(OH)2, Co-200, Co-300, and Co-400. | |
Table 1 Textural parameters of various comparative samples
| Samples |
S
BET (m2 g−1) |
D
p
(nm) |
V
a (cm3 g−1) |
|
Average pore size.
|
| Co(OH)2 |
209.7 |
6.39 |
0.67 |
| Co-200 |
161.8 |
6.59 |
0.53 |
| Co-300 |
152.6 |
5.75 |
0.44 |
| Co-400 |
112.9 |
6.59 |
0.37 |
| 0.2Fe–Co(OH)2 |
128.4 |
6.70 |
0.43 |
| 0.4Fe–Co(OH)2 |
139.1 |
6.04 |
0.42 |
| 0.6Fe–Co(OH)2 |
120.9 |
6.95 |
0.42 |
| 0.8Fe–Co(OH)2 |
135.8 |
4.86 |
0.33 |
| 0.2Fe–Co-200 |
117.6 |
4.43 |
0.26 |
| 0.4Fe–Co-200 |
125.1 |
4.46 |
0.29 |
| 0.6Fe–Co-200 |
113.2 |
4.24 |
0.24 |
| 0.8Fe–Co-200 |
123.4 |
4.45 |
0.28 |
 |
| | Fig. 5 Representative SEM images of pure α-Co(OH)2 (a), Co-200 (b), Co-300 (c), and Co-400 (d). | |
XPS analyses provide precise cognition on constituent elements in the samples. As displayed in the full-survey-scan X-ray photoelectron spectroscopy (XPS) spectra (Fig. 6d) of Co(OH)2, Co-200, 0.6Fe–Co(OH)2, and 0.6Fe–Co-200, several obvious peaks corresponding to elements of Co2p, O1s, and C1s can be identified in each curve, indicating dominant cobalt-based materials. In the case of the Fe-doped samples, two relatively weak peaks assigned to Fe2p and Fe2s, at 725 eV and 845 eV, respectively, were detected in the full-survey-scan curves, signifying presence of Fe species in them. Fig. 6a–c display high-resolution XPS spectra of the Co2p and Fe2p core levels of various comparative samples. Primarily, binding energy values of Co2p transition splits located at ca. 780.9 and 796.9 eV, for the 2p3/2 and 2p1/2, respectively, along with two shake-up satellite peaks at ca. 787 and 803 eV are observed for each sample. Note that orbital splitting between 2p1/2 and 2p3/2 peaks is ca. 16 eV for the two samples including α-Co(OH)2 and Co-200, which is in accordance with that of Co(II) (16.0 eV) rather than Co(III) compounds (15.0 eV).13,24 This clearly indicates that cobalt atoms mainly are present in a Co(II) oxidation state before and after thermal treatment. However, in the case of Fe-doped Co(OH)2, it should be noted that a certain shift of Co binding energy occurs between the pure α-Co(OH)2 and Fe-doped Co(OH)2, verifying Fe atoms entered the crystal structure of Co(OH)2, and introduction of Fe atoms in the skeleton of Co(OH)2 induced variation in the Co chemical environment.
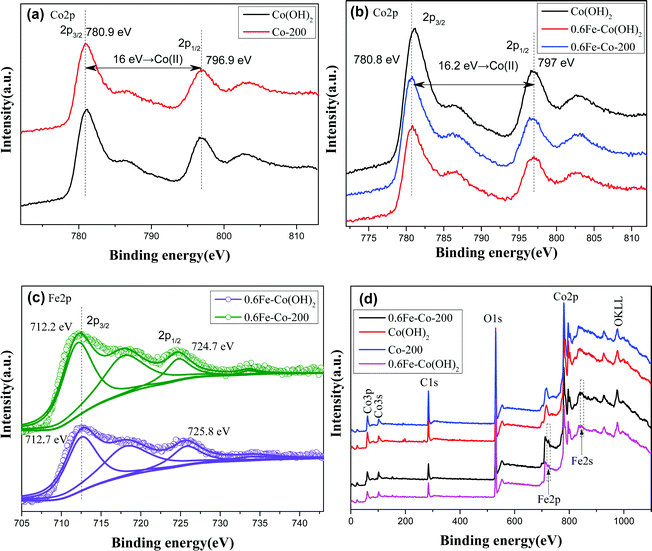 |
| | Fig. 6 High-resolution X-ray photoelectron spectroscopy of Co(OH)2, Co-200, 0.6Fe–Co(OH)2, and 0.6Fe–Co-200, (a) and (b) Co2p core level spectra,(c) Fe2p core level spectra, (d) full-survey-scan curves. | |
Besides, to more clearly identify the state of Fe atoms, the Fe2p spectra of 0.6Fe–Co(OH)2 and 0.6Fe–Co-200 were further fitted. As shown in Fig. 6c, the high-resolution Fe2p spectrum of 0.6Fe–Co(OH)2 shows two peaks at 712.7 and 725.8 eV, with a spin–orbit separation of 13.1 eV, which can be assigned to Fe2p3/2 and Fe2p1/2 of coordinated Fe3+ in Co(OH)2.29 A satellite peak at binding energy of 733.7 eV is further attributable to Fe3+, while the satellite peak at binding energy of 718.5 eV is attributed to isolated Fe3+.30 Compared to the reported binding energy of Fe2p in pure FeOOH,30,31 there is a distinct shift toward higher binding energies for the doped Fe atoms in Co(OH)2. This probably is attributed to Fe cations incorporated into the Co(OH)2 crystal structure and giving electrons to Co2+.30 In addition, there is a certain shift for Fe2p3/2 between 0.6Fe–Co(OH)2 and 0.6Fe–Co-200, indicating a changing chemical environment of Fe atoms before and after thermal treatment because of their phase transformation.
To further confirm the above analytical results, as-prepared Fe–Co hybrid composites were examined by attenuated total reflectance infrared (ATR-IR) spectroscopy in a range of 525–4000 cm−1 as shown in Fig. 7a. The band at 3396 cm−1 corresponds to O–H vibration of hydrogen-bonded hydroxyl groups and intercalated water molecules located in the interlamellar space of Co(OH)2, and the peak around 1648 cm−1 corresponds to bending vibrations of interlayer water molecules.32 Therefore, we noticed that the absorption intensity at 1648 cm−1 became weaker with increased thermal treatment temperature because of the removal of interlayer water molecules. Peaks in the region of 490–540 cm−1 can be assigned to Co–O stretching vibrations and Co–OH bending vibrations in α-cobalt hydroxide. Bands at ca. 965 and 513 cm−1 are assigned to stretching vibrations of the Co–OH bond.33 The strong peak at 652 cm−1 is assigned to the δOH of the hydroxyl group.32 Additionally, phase transformation can be reflected from IR spectra of varied temperature treated samples. As observed, sample Co-200 retains the whole outline of α-Co(OH)2, but the refined changing at 1502 cm−1 and 1285 cm−1 still can be probed, indicating chemical variations occurring after thermal treatment. With higher temperature treatment, a rigorous variation ranging from 525–1800 cm−1 can be observed for Co-300 and Co-400 compared to pure α-Co(OH)2, verifying a nearly-complete chemical phase transformation. With Fe-doped serial samples xFe–Co(OH)2, the semblable IR spectra can be proved (Fig. S6a, ESI†), while corresponding thermal treated samples show a certain changing compared to the incipient one (Fig. S6b, ESI†), signifying occurrence of chemical transformation, which is in agreement with the XRD analysis.
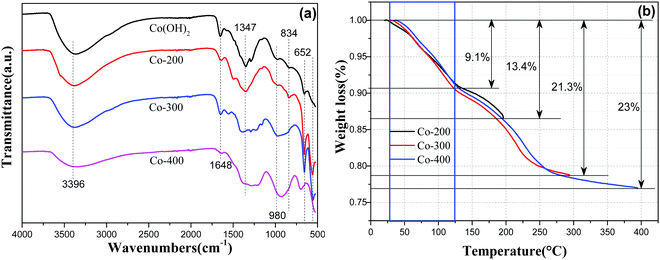 |
| | Fig. 7 ATR-IR spectra (a) of α-Co(OH)2, Co-200, Co-300, and Co-400; TG curves (b) for samples of α-Co(OH)2 at different temperatures. | |
To further identify the thermal treatment process of α-Co(OH)2 at different targeted temperatures, we checked the TG profile of α-Co(OH)2 with identical heating times for different targeted temperatures via thermogravimetric analysis (TGA) under nitrogen atmosphere. The corresponding results are displayed in Fig. 7b; two weight loss steps were detected during the heating procedure. The weight loss of α-Co(OH)2 ranging from room temperature to 125 °C should be attributed to removal of intercalated water molecules, which can be observed in each curve of different temperatures. Therewith, the weight loss of α-Co(OH)2 ranging from 125 °C to 260 °C is ascribable to the dominant chemical transformation of α-Co(OH)2 to phase CoO. Therefore, we can know that sample Co-300 should be the thermal-resulted CoO phase, which is consistent with XRD analysis. Apart from that, analysis of weight loss on removal of water further confirmed a phase transformation during the thermal treatment process. Based on TG results, the 200 °C thermal treatment induced partial transformation of α-Co(OH)2 to CoO; hence, the CoO phase dissolved from the surface of α-Co(OH)2, which is the result producing biphasic CoO/Co(OH)2. However, higher temperatures resulted in presence of a single-phase CoO and breaking of the planar nanosheet. On the other hand, TG profiles of Fe-containing Co(OH)2 show similar weight loss steps at a given temperature (Fig. S7, ESI†), signifying degradation of Co–Fe hydroxides during temperature programmed treatment.
Catalytic analysis
A Co-based catalyst, especially the Co(II)/PMS system, is regarded as a Fenton-like reaction system, which is efficient for stimulating peroxysulphate to produce sulfate radicals (SO4˙−) affording strong oxidability and a higher oxidation potential of sulfate radical [2.5–3.1 eV] compared to hydroxyl radical [2.7 eV].34 In a typical Co(II)/PMS system, Co species can activate PMS to simultaneously generate hydroxyl radicals (OH˙) and sulfate radicals (SO4˙−). During the sulfate radicals oxidizing process, hydroxyl radicals also participated, though at an inferior reaction rate. Meanwhile, during this oxidation cycle, the original state of the catalyst can be recovered accompanied with a further generation of hydroxyl radicals.
First, the purified Co(OH)2 and its resulting thermal-treated samples of Co-200, Co-300, and Co-400, respectively were used for catalytic degradation of methylene blue, and peroxomonosulfate appears as the oxidant. The reaction was conducted without any adjustment of acid–base. As observed, the concentrations of MB decreased with time in the presence of peroxomonosulfate and catalysts (Fig. 8a, c and e). As a contrast, catalytic degradation experiments only in the presence of PMS or Co(OH)2 were conducted to identify the functions of catalyst and oxidant (Fig. 8a). The feeble drop of MB's concentrations with only PMS or Co(OH)2 indicates the real degradation reaction occurs only with the synergetic effect of PMS or Co(OH)2. The MB degradation process was evaluated by a first-order kinetic model depicted as the equation ln(Ct/C0) = kt, in which Ct and C0 represent MB concentrations at different times (t) and t = 0, respectively, and k is the calculated rate constant. Fig. 8b, d, and f show that MB degradation with both catalysts followed a first order kinetic model. Reaction results and the calculated MB degradation rate constants are displayed in Table 2.
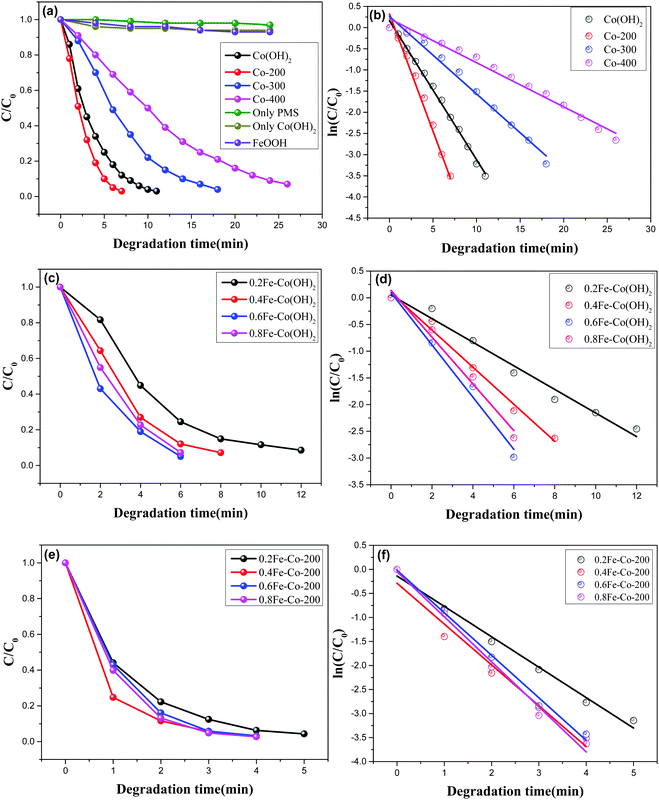 |
| | Fig. 8 Catalytic degradation of MB versus reaction time based on various controlled samples (a), (c) and (e), and their corresponding linear fitting relationship (b), (d) and (f) between ln(C/C0) and reaction time using a pseudo-first-order reaction scheme. | |
Table 2 Catalytic degradation results of MB using various comparative samples
| Samples |
Fe/Coa |
Catalyst dosage (mg) |
Degradation time (min) |
Rate constant K (min−1) |
|
Molar ratio from the ICP results.
|
| Co(OH)2 |
— |
5 |
11 |
0.3284 |
| Co-200 |
— |
5 |
7 |
0.5540 |
| Co-300 |
— |
5 |
18 |
0.1832 |
| Co-400 |
— |
5 |
26 |
0.1042 |
| 0.2Fe–Co(OH)2 |
0.203 |
5 |
12 |
0.2207 |
| 0.4Fe–Co(OH)2 |
0.398 |
5 |
8 |
0.3468 |
| 0.6Fe–Co(OH)2 |
0.594 |
5 |
6 |
0.4371 |
| 0.8Fe–Co(OH)2 |
0.805 |
5 |
6 |
0.4886 |
| 0.2Fe–Co-200 |
0.206 |
5 |
5 |
0.6328 |
| 0.4Fe–Co-200 |
0.401 |
5 |
4 |
0.8500 |
| 0.6Fe–Co-200 |
0.598 |
5 |
4 |
0.8851 |
| 0.8Fe–Co-200 |
0.802 |
5 |
4 |
0.9374 |
With varied samples and different thermal treatment temperatures, Co-200 affords the highest catalytic activity as compared with other two samples Co-300, and Co-400; MB was completely degraded within 7 min in the presence of Co-200. Additionally, TOC measurements show that Co-200 affords a 77% TOC reduction in 7 min, demonstrating the actual degradation process of MB in the presence of catalyst and PMS. Associated with the aforementioned characterization analysis, we found that the sample of Co-200 possesses a better morphology and structure compared to the other two treated at higher temperatures. However, it is worth noting that Co-200 gives a higher catalytic activity than pure α-Co(OH)2 even affording its relatively poor textural properties. Enhanced catalytic activity should derive from these highly active serried surface defects thanks to the existence of biphasic cobalt species. As we know, heterojunction sites existing in biphasic metal composites frequently afford interesting catalytic activity. Herein, the present CoO nanocrystals distributed on planar Co(OH)2 made the 2D Co(OH)2 afford more surface defects, thereby resulting in enhanced catalytic activity in a Fenton-like oxidation reaction.
The Fe-doped Co(OH)2 hybrid composite nanosheets were further examined using the degradation reaction of MB under the same reaction conditions. Before this examination, we evaluated the catalytic degradation efficiency of FeOOH synthesized according to a reported paper11 using the PMS. At the initial reaction step, the concentration of MB showed a feeble drop (Fig. 8a), and then the MB was almost consumed after 3 h reaction time, which was accompanied by a lighter color, indicating the single Fe(III) species failed to efficiently stimulate the PMS. However, as displayed in Fig. 8c, the introduction of Fe atoms obviously influences the efficiency of MB degradation together with Co(OH)2. Wherein, the low concentration of Fe-doped 0.2Fe–Co(OH)2 afforded comparable catalytic efficiency with the thermal-resulted sample of Co-200 but it was superior to pure α-Co(OH)2. With more incorporation of Fe in Co(OH)2, an obviously enhanced efficiency of the degradation of MB occurs. The highest catalytic activity was found for the degradation system using 0.6Fe–Co(OH)2. Associating with the corresponding ref. 35, we speculate the enhanced catalytic activity should contribute from the synergetic effects of cobalt and iron catalytic active sites. Especially, homogeneously incorporated Fe atoms contribute to producing more oxyradicals, which attack the MB to degrade it into CO2 and H2O.
After thermal treatment, the resulting catalysts were then ulteriorly applied to MB degradation under identical conditions. The time-dependent reaction relationship for different Fe-doped samples is shown in Fig. 8e. Compared to the Fe-doped serial samples without thermal treatment, the thermally treated serial samples exhibit an enhanced catalytic activity in the MB degradation process. Degradation time was shortened to less than 5 min for each sample; especially the high Fe-doped concentration samples were consumed in just 4 minutes during the degradation process, which is obviously superior to the serial samples xFe–Co(OH)2. The calculated rate constants are 0.6328, 0.8500, 0.8851, and 0.9374 corresponding to 0.2Fe–Co-200, 0.4Fe–Co-200, 0.6Fe–Co-200, and 0.8Fe–Co-200. Associating with the aforementioned TEM results, we know that, compared to the incipient xFe–Co(OH)2, there are massive defective holes formed on the Fe/Co composite nanosheets, which likely give rise to the high catalytic activity. To verify these results, we checked catalytic removal efficiency of MB using the thermal-treated serial samples xFe–Co-200(1 h); the results (Fig. S8, ESI†) reveal a similar catalytic activity with the as-prepared serial xFe–Co(OH)2, but lower activity than that of serial xFe–Co-200. As mentioned in the characterization section (Fig. S4, ESI†), serial xFe–Co-200(1 h) afford a similar morphology and crystalline structure with the serial xFe–Co-200, but the surface defective holes seem to be undetectable for xFe–Co-200(1 h). These phenomena further favor the surface defects inducing enhancement of the catalytic property. On the other hand, it should be noted that thermally treating Fe-doped samples, including 0.4Fe–Co-200, 0.6Fe–Co-200, and 0.8Fe–Co-200, affords comparable catalytic activity in each other, but higher activity than 0.2Fe–Co-200. This is a compositive catalytic result based on conjunct promotion from the introduced Fe heteroatoms and formed defective surface holes. As observed, among the serial samples of xFe–Co(OH)2, the 0.6Fe–Co(OH)2 affords the highest catalytic activity because of its applicable doping concentration. However, with the thermally treated serial samples of xFe–Co-200, presence of surface defective holes induced a further improvement of catalytic activity, leading to similar catalytic properties of 0.4Fe–Co-200, 0.6Fe–Co-200, and 0.8Fe–Co-200. This result may be attributed to the presence of different densities of defective holes in the various samples. In addition, we list the references of some reported catalysts in catalytic removal efficiency of MB (Table 3). As clearly displayed, our designed 2D composite catalysts exhibit a much higher catalytic activity compared to those references even with a higher initial MB concentration and lower catalyst dosage. This is a comprehensive catalytic result associated with the efficient and unstinted diffusion effect and enhanced catalytic active sites present on the densely defective 2D catalyst.
| | | Co(II) + HSO5− → Co(III) + SO4˙− + OH− | (1) |
| | | Co(II) + HSO5− → Co(III) + ˙OH + SO42− | (2) |
| | | Fe(III) + ˙OH → Fe(III)–˙OH | (3) |
| | | Fe(III)–˙OH + HSO5− → Fe(III)–˙OHOSO3 + OH− | (4) |
| | | Fe(III)–˙OHOSO3 + HSO5− → Fe(II) + 2SO5˙− + 2H+ | (5) |
| | | Fe(II) + HSO5− → Fe(III) + SO4˙− + OH− | (6) |
| | | Co(III) + HSO5− → Co(II) + SO5˙− + H+ | (7) |
| | | 2SO5˙− + 2OH− → 2SO42− + 2˙OH + O2 | (8) |
| | | ˙OH + SO4˙− + MB → MB–SO4˙− → MB˙+ + SO42− → many steps → CO2 + H2O | (9) |
Table 3 Comparison of catalytic degradation efficiency of MB based on reported catalysts
| Samples |
Catalyst dosage (mg) |
MB concentrations (ppm) |
Degradation time (min) |
Ref. |
| Co/MgO |
10 |
40 |
7 |
36
|
| S-Co-DTPMP |
5 |
80 |
25 |
37
|
| Co3O4 NPs/TNWs |
100 |
10 |
8 |
38
|
| Co3O4–Bi2O3 |
50 mg L−1 |
6.4 |
10 |
39
|
| Co/IBA-B |
200 |
32 |
6 |
40
|
| Co-200 |
5 |
200 |
7 |
This work |
| 0.6-Fe–Co-200 |
5 |
200 |
4 |
A possible mechanism process for Fe-doped Fe–Co hybrid composites
Granted, the reaction mechanism for the reduction is not yet fully clear. Associating with the reported reference mechanism35,41–43 for various transition metals in the presence of PMS, we tentatively proposed a possible mechanism for degradation of MB using a Fe-doped Co-based catalyst to stimulate the PMS (Scheme 3). As depicted in above equations, Co species can activate PMS to generate hydroxyl radicals and sulfate radicals (eqn (1) and (2)).24,43 Simultaneously, excitative hydroxyl radicals can bond to surface Fe(III) atoms to produce Fe(III)–˙OH (eqn (4)), which further reacts with PMS, then, in turn, converts to Fe(II) and more sulfate radicals (eqn (3)–(5)). Afterwards, the produced Fe(II) will be recycled to Fe(III) with the assistance of HSO5− (eqn (6)). Note that production of hydroxyl radicals promotes the cycle reaction (Fe(III) ↔ Fe(II)), benefiting stimulation of more radicals. Therefore, introducing the Fe species is extremely serviceable for stimulating sulfate radicals, which serve as a synergetic active species. This hypothesis would account well for enhancing catalytic properties in the presence of certain proportions of iron species during the cobalt stimulated PMS process. The resulting Co(III) will be recycled to Co(II) with assistance of HSO5− (eqn (7)). Then, some of the produced hydroxyl radicals would further stimulate PMS to produce more sulfate radicals (eqn (8)). The produced sulfate radicals and hydroxyl radicals participate together in the oxidation of MB (eqn (9)).
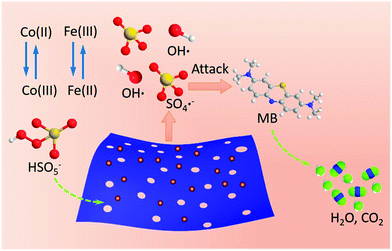 |
| | Scheme 3 Proposed mechanism for PMS activation over the Fe/Co hybrid nanosheet. | |
Conclusions
In summary, we successfully synthesized surface defective Co/Fe–Co planar hybrid composite nanosheets with biphasic heterojunction sites or highly-stressed defective holes. A controlled thermal procedure was employed to trigger production of surface defective sites. The heterojunction phase CoO/Co(OH)2 and defective surface holes on the targeted materials of Fe-doped Fe–Co planar hybrid composites were verified. Among the controlled thermal procedures, the 200 °C thermal treatment was demonstrated to not destroy the morphology of 2D nanosheets for the initial samples, and efficiently triggered formation of highly active defective sites on catalytic surfaces. The introduction of Fe heteroatoms and generation of surface holes consecutively promoted enhancement of catalytic activity for 2D composite nanosheets in a Fenton-like catalytic degradation of MB.
Acknowledgements
The authors acknowledge the project Funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD) and the financial support of National Natural Science Foundations of China (21476108, 20876077, and 21276125). The author Fu Yang thanks China Scholarship Council for supporting his exchange study in National University of Singapore.
Notes and references
- A. C. Ferrari, J. C. Meyer, V. Scardaci, C. Casiraghi, M. Lazzeri, F. Mauri, S. Piscanec, D. Jiang, K. S. Novoselov, S. Roth and A. K. Geim, Phys. Rev. Lett., 2006, 97, 187401 CrossRef CAS PubMed.
- A. K. Geim, Science, 2009, 324, 1530–1534 CrossRef CAS PubMed.
- C. Lee, X. D. Wei, J. W. Kysar and J. Hone, Science, 2008, 321, 385–388 CrossRef CAS PubMed.
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, Y. Zhang, S. V. Dubonos, I. V. Grigorieva and A. A. Firsov, Science, 2004, 306, 666–669 CrossRef CAS PubMed.
- S. Stankovich, D. A. Dikin, R. D. Piner, K. A. Kohlhaas, A. Kleinhammes, Y. Jia, Y. Wu, S. T. Nguyen and R. S. Ruoff, Carbon, 2007, 45, 1558–1565 CrossRef CAS.
- J. P. Liu, Y. Y. Li, X. T. Huang, G. Y. Li and Z. K. Li, Adv. Funct. Mater., 2008, 18, 1448–1458 CrossRef CAS.
- Z. P. Liu, R. Z. Ma, M. Osada, N. Iyi, Y. Ebina, K. Takada and T. Sasaki, J. Am. Chem. Soc., 2006, 128, 4872–4880 CrossRef CAS PubMed.
- R. Mas-Balleste, C. Gomez-Navarro, J. Gomez-Herrero and F. Zamora, Nanoscale, 2011, 3, 20–30 RSC.
- J. H. Zhong, A. L. Wang, G. R. Li, J. W. Wang, Y. N. Ou and Y. X. Tong, J. Mater. Chem., 2012, 22, 5656–5665 RSC.
- Y. Q. Zhu, C. B. Cao, S. Tao, W. S. Chu, Z. Y. Wu and Y. D. Li, Sci. Rep., 2014, 4, 5787–5793 CrossRef CAS PubMed.
- H. Fan, X. Huang, L. Shang, Y. T. Cao, Y. F. Zhao, L. Z. Wu, C. H. Tung, Y. D. Yin and T. R. Zhang, Angew. Chem., Int. Ed., 2016, 55, 2167–2170 CrossRef CAS PubMed.
- J. Wu, Z. Y. Ren, S. C. Du, L. J. Kong, B. W. Liu, W. Xi, J. Q. Zhu and H. G. Fu, Nano Res., 2016, 9, 713–725 CrossRef CAS.
- Y. M. Jiang, X. Li, T. X. Wang and C. M. Wang, Nanoscale, 2016, 8, 9667–9675 RSC.
- S. Z. Deng, C. T. Cherian, X. I. Liu, H. R. Tan, L. H. Yeo, X. J. Yu, A. Rusydi, B. V. R. Chowdari, H. M. Fan and C. H. Sow, Chem. – Eur. J., 2014, 20, 12444–12452 CrossRef CAS PubMed.
- W. L. Ong, S. W. L. Ng, C. Zhang, M. H. Hong and G. W. Ho, J. Mater. Chem. A, 2016, 4, 13307–13315 CAS.
- C. M. Zhao, X. Wang, S. M. Wang, Y. Y. Wang, Y. X. Zhao and W. T. Zheng, Int. J. Hydrogen Energy, 2012, 37, 11846–11852 CrossRef CAS.
- G. Chen, S. S. Liaw, B. S. Li, Y. Xu, M. Dunwell, S. G. Deng, H. Y. Fan and H. M. Luo, J. Power Sources, 2014, 251, 338–343 CrossRef CAS.
- J. Tang, D. Q. Liu, Y. X. Zheng, X. W. Li, X. H. Wang and D. Y. He, J. Mater. Chem. A, 2014, 2, 2585–2591 CAS.
- Y. Yang, H. J. Wang, L. L. Wang, Y. L. Ge, K. Kan, K. Y. Shi and J. H. Chen, New J. Chem., 2016, 40, 4678–4686 RSC.
- F. Zhang, Y. J. Liu, Y. Cai, H. Li, X. Y. Cai, I. Djerdj and Y. D. Wang, Powder Technol., 2013, 235, 121–125 CrossRef CAS.
- C. Peng, B. W. Jiang, Q. Liu, Z. Guo, Z. J. Xu, Q. Huang, H. J. Xu, R. Z. Tai and C. H. Fan, Energy Environ. Sci., 2011, 4, 2035–2040 CAS.
- J. P. Cheng, J. H. Fang, M. Li, W. F. Zhang, F. Liu and X. B. Zhang, Electrochim. Acta, 2013, 114, 68–75 CrossRef CAS.
- Y. J. Yao, C. Xu, S. D. Miao, H. Q. Sun and S. B. Wang, J. Colloid Interface Sci., 2013, 402, 230–236 CrossRef CAS PubMed.
- F. Yang, S. Zhou, H. Wang, S. Long, X. Liu and Y. Kong, Dalton Trans., 2016, 45, 6371–6382 RSC.
- S. Muhammad, E. Saputra, H. Q. Sun, H. M. Ang, M. O. Tade and S. B. Wang, Ind. Eng. Chem. Res., 2012, 51, 15351–15359 CrossRef CAS.
- H. Q. Sun, H. W. Liang, G. L. Zhou and S. B. Wang, J. Colloid Interface Sci., 2013, 394, 394–400 CrossRef CAS PubMed.
- Y. J. Yao, Z. H. Yang, H. Q. Sun and S. B. Wang, Ind. Eng. Chem. Res., 2012, 51, 14958–14965 CrossRef CAS.
- H. B. M. Sidek, Y. K. Jo, I. Y. Kim and S. J. Hwang, J. Phys. Chem. C, 2016, 120, 23421–23429 Search PubMed.
- Z. Z. Yang, Z. Zhang, Y. Z. Jiang, M. Q. Chi, G. D. Nie, X. F. Lu and C. Wang, Rsc Adv., 2016, 6, 33636–33642 RSC.
- Y. N. Cao, L. J. Shen, X. L. Hu, Z. J. Du and L. L. Jiang, Chem. Eng. J., 2016, 306, 124–130 CrossRef CAS.
- J. X. Feng, H. Xu, Y. T. Dong, S. H. Ye, Y. X. Tong and G. R. Li, Angew. Chem., Int. Ed., 2016, 55, 3694–3698 CrossRef CAS PubMed.
- Q. Wang, S. W. Liu, H. Y. Sun and Q. F. Lu, Rsc Adv., 2015, 5, 48181–48186 RSC.
- Z. G. Zhao, F. X. Geng, J. B. Bai and H. M. Cheng, J. Phys. Chem. C, 2007, 111, 3848–3852 CAS.
- P. Shukla, H. Q. Sun, S. B. Wang, H. M. Ang and M. O. Tade, Catal. Today, 2011, 175, 380–385 CrossRef CAS.
- H. W. Liang, H. Q. Sun, A. Patel, P. Shukla, Z. H. Zhu and S. B. Wang, Appl. Catal., B, 2012, 127, 330–335 CrossRef CAS.
- W. Zhang, H. L. Tay, S. S. Lim, Y. S. Wang, Z. Y. Zhong and R. Xu, Appl. Catal., B, 2010, 95, 93–99 CrossRef CAS.
- Y. P. Zhu, T. Z. Ren and Z. Y. Yuan, Nanoscale, 2014, 6, 11395–11402 RSC.
- Z. L. Chen, S. H. Chen, Y. H. Li, X. L. Si, J. Huang, S. Massey and G. L. Chen, Mater. Res. Bull., 2014, 57, 170–176 CrossRef CAS.
- Y. B. Ding, L. H. Zhu, A. Z. Huang, X. R. Zhao, X. Y. Zhang and H. Q. Tang, Catal. Sci. Technol., 2012, 2, 1977–1984 CAS.
- Y. B. Wang, Y. Xie, S. M. Yin, R. Xu and R. Lau, J. Taiwan Inst. Chem. Eng., 2016, 68, 246–253 CrossRef CAS.
- Y. X. Wang, H. Q. Sun, H. M. Ang, M. O. Tade and S. B. Wang, ACS Appl. Mater. Interfaces, 2014, 6, 19914–19923 CAS.
- H. Q. Sun, G. L. Zhou, S. Z. Liu, H. M. Ang, M. O. Tade and S. B. Wang, ACS Appl. Mater. Interfaces, 2012, 4, 6235–6241 CAS.
- Y. X. Wang, L. Zhou, X. G. Duan, H. Q. Sun, E. L. Tin, W. Q. Jin and S. B. Wang, Catal. Today, 2015, 258, 576–584 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7qm00217c |
|
| This journal is © the Partner Organisations 2017 |
Click here to see how this site uses Cookies. View our privacy policy here.