DOI:
10.1039/C7QM00211D
(Research Article)
Mater. Chem. Front., 2017,
1, 2243-2255
Photovoltaic performances of type-II dye-sensitized solar cells based on catechol dye sensitizers: retardation of back-electron transfer by PET (photo-induced electron transfer)†
Received
12th May 2017
, Accepted 26th June 2017
First published on 27th June 2017
Abstract
Catechol dyes (CAT-PET), possessing PET (photo-induced electron transfer) characteristics as well as dye-to-TiO2 charge-transfer (DTCT) characteristics, which are composed of a catechol unit linked to a diethylamino group as an electron donor moiety via a methylene spacer, have been designed and developed as dye sensitizers for type-II dye-sensitized solar cells (DSSCs). The PET takes place from the nitrogen atom of the amino moiety to the photoexcited catechol unit, that is, the CAT-PET dyes make it possible to retard the back-electron transfer rate from the electrons injected into the TiO2 electrode to the oxidized catechol unit by the PET characteristic in type-II DSSCs. The CAT-PET dyes exhibited a broad absorption band corresponding to DTCT upon binding to TiO2 films, and the DTCT band for the CAT-PET dyes broadened and showed a red-shift compared to that for CAT dyes without PET characteristics. In addition, our results demonstrate that the introduction of moderately electron-withdrawing substituents on the catechol unit for the CAT dyes, but strongly electron-withdrawing substituents as well as moderately electron-withdrawing substituents on the catechol unit for the CAT-PET dyes leads to enhancement of the DTCT characteristics. It was found that the photovoltaic performances of DSSCs based on the CAT-PET dyes are higher than those based on CAT dyes. This indicates that the PET from the diethylamino group to the oxidized catechol dye adsorbed on the TiO2 electrode can efficiently retard the back-electron-transfer, leading to favorable conditions for the type-II electron-injection pathway from the ground state of the catechol unit to the conduction band (CB) of the TiO2 electrode by the photoexcitation of DTCT bands. Consequently, this work proposes that introducing PET characteristics to the catechol dye with a moderately electron-withdrawing substituent is an effective molecular design for type-II dye sensitizers to lead to not only an increase in the DTCT efficiency, but also the retardation of back electron transfer.
Introduction
Dye-sensitized solar cells (DSSCs) employing dye-adsorbed TiO2 electrodes have received considerable attention as one of the most promising new renewable photovoltaic cell alternatives to conventional solar cells based on silicon,1–10 but TiO2-based DSSCs can be actually classified into two types, I and II, depending on the electron-injection pathway from the dye to the conduction band (CB) of the TiO2 electrode.3 The pathway for type-I DSSCs is photoexcitation of the local band of the dye adsorbed on the TiO2 surface followed by an electron injection from the photoexcited dye to the CB of the TiO2 electrode (Fig. 1: i.e., an electron is excited from the HOMO to the LUMO level of the dye by sunlight illumination, followed by injection into the CB of the TiO2 electrode). Thus, the pathway for type-I DSSCs can also be called a ‘‘two-step’’ or ‘‘indirect’’ electron-injection pathway. Much effort has been made on the development of various types of D–π–A organic dye sensitizers for type-I DSSCs which are constructed of a diphenyl- or dialkylamino group as the electron donor (D) and a carboxyl group,11–17 2-(1,1-dicyanomethylene)rhodanine,18 an azine ring such as pyridine, pyrazine and triazine,19–22 or 8-hydroxylquinoline23 as the electron acceptor (A) and anchor linked by π-conjugated bridges, and there has been a gradual accumulation of information about the relationship between the chemical structures and the photovoltaic performances of DSSCs. In particular, the photoabsorption properties of a D–π–A dye are associated with intramolecular charge transfer (ICT) excitation from the D to the A moiety of the dye, resulting in efficient electron transfer from the photoexcited dye through the acceptor moiety (carboxyl group) into the CB of the TiO2 electrode, according to the type-I pathway. As a result, type-I DSSCs have achieved a solar energy-to-electricity conversion yield (η) of up to 13%.12 However, to achieve efficient electron injection from the excited dye to the CB of the TiO2 electrode, the LUMO energy level of the dye sensitizers for type-I DSSCs must be higher than the energy level (Ecb) of the CB of the TiO2 electrode (some researchers have proposed that an energy gap of over 0.2–0.3 V is necessary for efficient electron injection).3–5 Thus, the restriction of the LUMO levels results in low light harvesting efficiency (LHE).
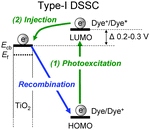 |
| | Fig. 1 Electron-injection pathway from the dye to the CB of the TiO2 electrode for a type-I DSSC, in which an electron is excited from the HOMO to the LUMO level of the dye, followed by injection into the CB of the TiO2 electrode. | |
In contrast, the electron-injection pathway for type-II DSSCs is direct, that is, ‘‘one-step’’ electron injection from the ground state of the dye to the CB of TiO2 by photoexcitation of the dye-to-TiO2 charge transfer (DTCT) bands (Fig. 2a: i.e., an electron is injected directly from the HOMO of the dye into the CB of the TiO2 electrode upon photoexcitation).3 Catechol (Cat) dyes such as dopamine and fluorine, which are classified as type-II dye sensitizers, show strong new absorption bands in the longer wavelength region corresponding to the DTCT upon binding to the TiO2 surface through a bidentate mononuclear chelating linkage and/or a bidentate binuclear bridging linkage at the Brønsted acid site on TiO2.24–31 The most advantageous aspect of type-II DSSCs over type-I DSSCs is their light-harvesting capabilities over the wide spectral range of sunlight because the direct electron injection in type-II DSSCs can lead to the creation of a broad DTCT band and the easing of restrictions on the LUMO levels of the dye sensitizers relative to type-I DSSCs with an indirect electron-injection pathway, although the absorption coefficient of the DTCT band for type-II dye sensitizers is much lower than in the absorption band based on the π–π* transition for conventional dye sensitizers due to lower overlap of the ground (molecular-localized) and the excited state (Ti-localized) wave functions.31d On the other hand, fundamental studies on the interfacial electron transfer dynamics of Cat dyes adsorbed on TiO2 nanoparticles have revealed that the back-electron transfer (the charge recombination from the electrons injected into the TiO2 electrode to the oxidized dye) of electrons injected into the TiO2 electrode for the type-II pathway takes place in a picosecond timescale (Fig. 2a), which was significantly faster than in the type-I pathway (μs–ms). The photovoltaic performance of type-II DSSCs based on Cat dyes is significantly lower than in type-I DSSCs based on D–π–A organic dye sensitizers. In fact, the η values of type-II DSSCs based on Cat dyes had never exceeded 2.5%.31e Thus, to achieve a breakthrough in the development of new and efficient Cat dye sensitizers for type-II DSSCs, an epoch-making molecular design by the effective chemical modification of the Cat structure, leading to the retardation of the back-electron transfer, is required.
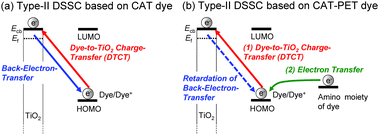 |
| | Fig. 2 Electron-injection pathway from the dye to the CB of the TiO2 electrode for (a) a type-II DSSC based on CAT dye and (b) a type-II DSSC based on CAT-PET dye, in which an electron is injected directly from the HOMO of the dye into the CB of the TiO2 electrode upon photoexcitation, and for the CAT-PET dye, subsequently, PET takes place from the nitrogen atom of the amino moiety to the photoexcited catechol unit. | |
In our previous work, to gain insight into the molecular design of Cat dye sensitizers for type-II DSSCs, we investigated the DTCT characteristics of CAT dyes (Fig. 3) with various substituents and their photovoltaic performance in DSSCs.30b It was found that the introduction of a strongly electron-donating substituent such as a diphenylamino group on the Cat unit can lead to a high HOMO energy level, resulting in thermodynamically unfavorable regeneration of the oxidized dye by electron transfer from the I3−/I− redox couple in the electrolyte. Moreover, the introduction of a strongly electron-withdrawing substituent such as a tricyanoethenyl group and nitro/formyl groups on the Cat unit may lead to a decrease in the DTCT efficiency due to the stabilization of the LUMO energy level and the facilitation of the back transfer of electrons injected into the TiO2 electrode to the oxidized Cat dye due to the strong electron-accepting ability; this, in turn, results in low photovoltaic performance. On the other hand, the introduction of a moderately electron-withdrawing substituent such as an acetyl group or a cyano group onto the Cat unit leads to not only an increase in the DTCT efficiency, but also the retardation of the back electron transfer, resulting in favorable conditions for the type II electron-injection pathway from the ground state of the Cat dye to the CB of the TiO2 electrode by the photoexcitation of DTCT bands. Our results also show that the interaction between the nitro group on the Cat unit and I− ions in the electrolyte might cause the destabilization of the LUMO energy level, resulting in an increase in the DTCT efficiency in the DSSC and/or the retardation of the back electron transfer.
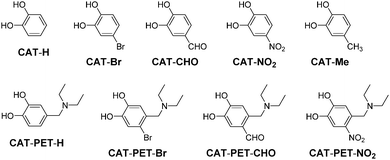 |
| | Fig. 3 Chemical structures of CAT and CAT-PET dyes with various substituents. | |
In this work, as an effective strategy to cause not only the retardation of the back-electron-transfer rate in type-II DSSCs but also an increase in the DTCT efficiency of Cat dyes, we have designed and developed catechol dye sensitizers (CAT-PET) possessing PET (photo-induced electron transfer) characteristics which are composed of a catechol unit linked to a diethylamino group as an electron donor moiety via a methylene spacer. For the PET-type fluorescence sensors, the PET takes place from the nitrogen atom of the amino moiety to the photoexcited fluorophore skeleton,32 leading to fluorescence quenching of the fluorophore. When the nitrogen atom of the amino moiety is protonated or strongly interacts with a cation, a drastic enhancement in fluorescence is observed because of the retardation of PET. Thus, as shown in Fig. 2b, the oxidized CAT-PET dye resulting from the DTCT is subsequently reduced back to its original neutral state through electron donation from the nitrogen atom of the amino moiety, that is, PET takes place from the nitrogen atom of the amino moiety to the photoexcited catechol unit. Therefore, it is expected that the CAT-PET dyes will make it possible to retard the back-electron transfer rate from the electrons injected into the TiO2 electrode to the oxidized Cat dye by the PET characteristic in type-II DSSCs. Herein we report the effect of PET characteristics as well as substituents (H, Br, CHO, NO2) in the CAT-PET dye sensitizers on the DTCT characteristics and the photovoltaic performance of type-II DSSCs (Fig. 3). It was found that the photovoltaic performances of DSSCs based on the CAT-PET dyes are higher than those for the CAT dyes without PET characteristics. This work reveals that introducing PET characteristics to the catechol dye with a moderately electron-withdrawing substituent is an effective molecular design for type-II dye sensitizers to lead to not only an increase in the DTCT efficiency, but also the retardation of back-electron transfer.
Results and discussion
Synthesis
In this work, we used unsubstituted CAT dye (CAT-H) and CAT-PET dye (CAT-PET-H), and CAT and CAT-PET dyes derivatized with a bromo group as a weak electron-withdrawing substituent (CAT-Br and CAT-PET-Br), an aldehyde group as a moderately electron-withdrawing group (CAT-CHO and CAT-PET-CHO) and a nitro group as a strongly electron-withdrawing substituent (CAT-NO2 and CAT-PET-NO2) (Fig. 3). All these CAT dyes were commercially available. A series of CAT-PET dyes with a (dimethylamino)methyl group were newly synthesized according to the stepwise synthetic protocol illustrated in Scheme 1. For the preparation of CAT-PET-H, CAT-PET-Br and CAT-PET-NO2, the corresponding methoxymethyl (MOM)-protected catechol derivatives with an aldehyde group were treated with ethylamine to give the intermediate imine which was then reduced with sodium borohydride to give the amines (2, 5 and 9). CAT-PET-H, CAT-PET-Br and CAT-PET-NO2 were prepared by the reaction of the amines with bromoethane, respectively, followed by treatment with acid. The formylation of MOM-protected CAT-PET-Br precursor 6 yielded the compound 7, which is then treated with acid to provide CAT-PET-CHO.
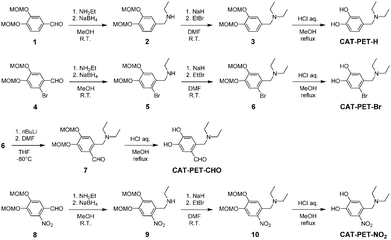 |
| | Scheme 1 Synthesis of CAT-PET dyes. | |
Photophysical properties
The photoabsorption of 4-methylbenzene-1,2-diol (Fig. 1; for CAT-Me see Fig. S12a, ESI†) as a reference to demonstrate the effect of the amino moiety on the PET characteristics as well as CAT and CAT-PET dyes in acetonitrile and adsorbed on TiO2 film is shown in Fig. 4a–d, and their optical data are summarized in Table 1. As with a series of CAT dyes,30b the CAT-PET dyes in acetonitrile show an absorption band at around 280–350 nm, which is assigned to the π–π* transition. The absorption bands of the CAT-PET dyes appear at slightly longer wavelengths than those of the CAT dyes. For both the CAT and CAT-PET dyes the absorption band shows red-shift upon increasing the electron-withdrawing ability of the substituents from an unsubstituted group (H) to a NO2 group. Moreover, CAT-CHO, CAT-PET-CHO, CAT-NO2 and CAT-PET-NO2 exhibit broad absorption bands centered at around 340, 380, 440 and 450 nm, respectively, which is attributed to the formation of a phenoxide ion by the deprotonation of the hydroxyl group.33 This is demonstrated by the fact that these absorption bands are not observed for the dye solution containing 6 wt% HCl (Fig. 4e and f). In addition to the absorption band observed in acetonitrile, the CAT and CAT-PET dyes adsorbed on the TiO2 film show a broad absorption band corresponding to DTCT upon binding to the TiO2 film, that is, the DTCT band appears in the region of 350 to 550 nm. It is worth noting that the DTCT bands for the CAT-PET dyes broaden and show a red-shift compared to those for the CAT dyes. CAT-Me in acetonitrile and adsorbed on a TiO2 film shows similar photoabsorption spectra to CAT-PET-H. This indicates that the CAT-Me and CAT-PET dyes having an alkyl group (methylene group) on the catechol unit exhibit a relatively strong DTCT profile upon binding to the TiO2 film, compared to CAT dyes without the alkyl group. In addition, our results also demonstrate that the introduction of the moderately electron-withdrawing substituents on the catechol unit for the CAT dyes but the strongly electron-withdrawing substituents as well as moderately electron-withdrawing substituents on the catechol unit for the CAT-PET dyes leads to enhancement of the DTCT characteristics, such that the red-shift of the absorption bands for CAT-CHO, CAT-PET-CHO and CAT-PET-NO2 upon binding to the TiO2 film is large compared to those of the other CAT and CAT-PET dyes. As shown in Fig. 4g and h, the solutions of the CAT and CAT-PET dyes in acetonitrile are colorless for CAT-H, CAT-PET-H, CAT-Br, CAT-PET-Br, CAT-CHO and CAT-PET-CHO, and light yellow for CAT-NO2 and CAT-PET-NO2 due to the formation of phenoxide ions.33 In contrast, the colors of the CAT and CAT-PET dyes adsorbed on the TiO2 film are yellow, which indicates a decrease in the transition energy by the appearance of the DTCT band that is associated with the formation of a surface-bound Ti–catechol complex.24–31
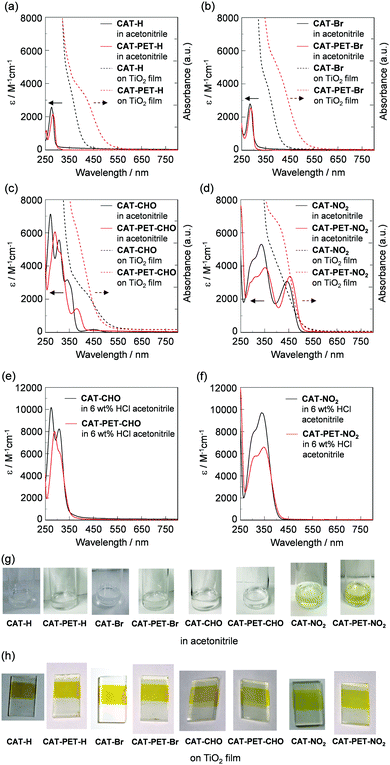 |
| | Fig. 4 (a–d) Photoabsorption spectra of the CAT and CAT-PET dyes in acetonitrile and adsorbed on TiO2 film. Photoabsorption spectra of (e) CAT-CHO and CAT-PET-CHO, and (f) CAT-NO2 and CAT-PET-NO2 in acetonitrile containing 6 wt% HCl. Photographs of the CAT and CAT-PET dyes (g) in acetonitrile and (h) adsorbed on TiO2 film. | |
Table 1 Optical data, HOMO and LUMO energy levels, and DSSC performance parameters of the CAT and CAT-PET dyes
| Dye |
λ
absmax/nm (ε/M−1 cm−1) |
HOMO (HOMO−1)/eV |
LUMOd/eV |
Moleculese/cm−2 |
J
sc
/mA cm−2 |
V
oc
/mV |
fff |
η
(%) |
|
In acetonitrile.
π → π* transition.
In acetonitrile containing 6 wt% HCl.
The LUMO energy level of the catechol moiety was obtained through eq. [HOMO + E0–0] for the CAT dyes and eq. [(HOMO−1) + E0–0] for the CAT-PET dyes, where the E0–0 transition energy is the onset of the photoabsorption spectra corresponding to the energy gap between the HOMO or HOMO−1 and LUMO.
Adsorption amount per unit area of TiO2 electrode.
Under simulated solar light (AM 1.5, 100 mW cm−2).
4-Methylbenzene-1,2-diol.
|
|
CAT-H
|
276 (2570)a,b |
−5.83 |
−1.65 |
3.46 × 1017 |
1.26 |
384 |
0.60 |
0.29 |
|
CAT-Br
|
285 (2790)a,b |
−5.81 |
−1.76 |
3.78 × 1017 |
1.65 |
399 |
0.58 |
0.38 |
|
CAT-CHO
|
307 (5560),a,b 308 (8230)b,c |
−5.99 |
−2.53 |
1.54 × 1017 |
2.26 |
440 |
0.60 |
0.60 |
|
CAT-NO2
|
335 (5300),a,b 338 (9700)b,c |
−6.06 |
−3.05 |
3.64 × 1017 |
2.84 |
488 |
0.53 |
0.73 |
|
CAT-PET-H
|
282 (2150)a,b |
−5.67 (−5.93) |
−1.88 |
1.31 × 1017 |
1.77 |
408 |
0.60 |
0.43 |
|
CAT-PET-Br
|
289 (2550)a,b |
−5.69 (−6.00) |
−1.98 |
1.36 × 1017 |
2.06 |
419 |
0.57 |
0.49 |
|
CAT-PET-CHO
|
309 (4840),a,b 308 (6180)b,c |
−5.73 (−6.08) |
−2.50 |
1.82 × 1017 |
2.40 |
452 |
0.55 |
0.60 |
|
CAT-PET-NO2
|
351 (3900),a,b 348 (6590)b,c |
−5.73 (−6.13) |
−3.15 |
1.74 × 1017 |
2.39 |
456 |
0.52 |
0.57 |
|
CAT-Me
|
282 (2350)a,b |
−5.71 |
−1.60 |
1.32 × 1017 |
0.96 |
372 |
0.55 |
0.20 |
Electrochemical properties
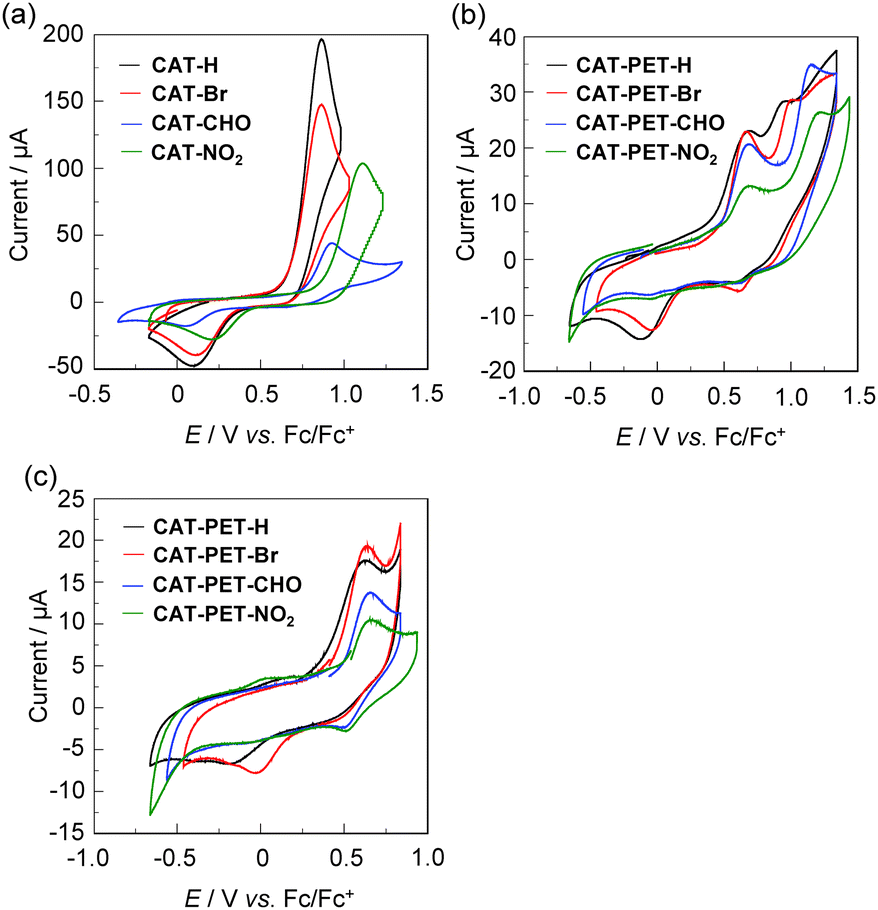
The electrochemical properties of the CAT and CAT-PET dyes were determined using cyclic voltammetry (CV) in acetonitrile containing 0.1 M tetrabutylammonium perchlorate (Bu4NClO4). The potentials were internally referenced to ferrocene/ferrocenium (Fc/Fc+). The cyclic voltammograms of the CAT and CAT-PET dyes are shown in Fig. 5. For CAT dyes the one-electron oxidation wave appeared at 0.8–1.1 V, whereas the corresponding reduction wave was observed at 0–0.2 V, which is related to the transformation of the catechol to the o-benzoquinone (Fig. 5a and Fig. S12c, ESI,† for CAT-Me). On the other hand, for the CAT-PET dyes two oxidation waves were observed at around 0.6 V and 0.8–1.1 V, corresponding to the oxidations of the diethylamino group and the catechol moiety, respectively (Fig. 5b). As with the CAT dyes, the corresponding reduction wave to the second oxidation wave appeared at −0.2 to 0 V which is due to the transformation of the catechol to the o-benzoquinone, whereas the corresponding reduction wave to the first oxidation wave was not clearly observed. It is worth noting that the reversible first oxidation wave for the CAT-PET dyes was clearly observed at around 0.65 V versus Fc/Fc+ in the potential scan range of −0.8 to 0.9 V, that is, the corresponding reduction wave appeared at around 0.6 V (Fig. 5c), although for CAT-PET-H and CAT-PET-Br the reduction wave due to the transformation of the catechol to the o-benzoquinone was also observed because the second oxidation wave is close to the first oxidation wave. Consequently, the oxidation potential of the CAT dyes and the second oxidation potential of the CAT-PET dyes increases upon increasing the electron-withdrawing ability of the substituents from an unsubstituted group (H) to a NO2 group. On the other hand, the first oxidation potential of the CAT-PET dyes is almost constant regardless of the type of substituent. This result indicates that the first and the second oxidation waves for the CAT-PET dyes are attributed to the oxidations of the diethylamino group and the catechol moiety, respectively. Indeed, the result from CV is strongly supported by the DFT calculations, which are described in the next section (Theoretical calculations).
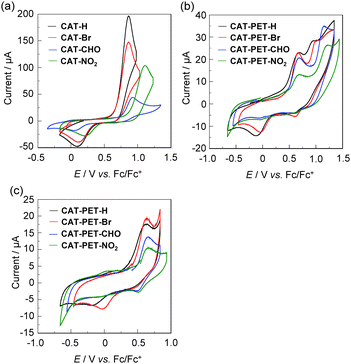 |
| | Fig. 5 Cyclic voltammograms of the (a) CAT and (b) CAT-PET dyes in acetonitrile containing 0.1 M Bu4NClO4. (c) The cyclic voltammograms of the CAT-PET dyes in the potential scan range of −0.8 to 0.9 V. | |
For the CAT dyes, the HOMO energy level (versus the vacuum level) corresponding to the catechol moiety was estimated from the onset of the oxidation wave (Table 1). For the CAT-PET dyes, on the other hand, the HOMO and HOMO−1 energy levels (versus the vacuum level) corresponding to the diethylamino group and the catechol moiety, respectively, are estimated from the half-wave potential (E1st1/2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ox (versus Fc/Fc+) = 0.54 V for CAT-PET-H, 0.56 V for CAT-PET-Br, 0.60 V for both CAT-PET-CHO and CAT-PET-NO2) of the first oxidation wave and the onset of the second oxidation wave, respectively. The HOMO energy level of the CAT dyes and the HOMO−1 energy level of the CAT-PET dyes slightly decreases upon increasing the electron-withdrawing ability of the substituents from an unsubstituted group (H) to a NO2 group, which is due to the stabilization of the HOMO energy level. In contrast, the HOMO energy level of the CAT-PET dyes is ca. −5.7 eV, that is, the HOMO energy level is almost constant regardless of the type of substituent. This indicates that the HOMO energy levels corresponding to the diethylamino group are independent of the type of substituent (H, Br, CHO, NO2) on the CAT-PET structure. This result also shows that the HOMO energy levels of the CAT and CAT-PET dyes are lower (more negative) than the I3−/I− redox potential (−4.9 eV). Thus, for both the CAT and CAT-PET dyes this ensures the efficient regeneration of the oxidized catechol dyes by electron transfer from the I3−/I− redox couple in the electrolyte. Consequently, the CV and the photoabsorption spectral analysis indicates that as shown in Fig. 2b the oxidized CAT-PET dyes resulting from the DTCT upon binding to the TiO2 film is subsequently reduced back to its original neutral state through electron donation from the nitrogen atom of the amino moiety, that is, the PET takes place from the nitrogen atom of the amino moiety to the photoexcited catechol unit. The LUMO energy levels of the CAT and CAT-PET dyes were estimated from the HOMO and the onset of the photoabsorption spectra (306 nm, 4.05 eV for CAT-PET-H; 308 nm, 4.02 eV for CAT-PET-Br; 346 nm, 3.58 eV for CAT-PET-CHO; 416 nm, 2.98 eV for CAT-PET-NO2; ref. 30b for CAT dyes) in the solution, that is, the LUMO energy level of the catechol moiety was obtained through eq. [HOMO + E0–0] for the CAT dyes and eq. [(HOMO−1) + E0–0] for the CAT-PET dyes, where the E0–0 transition energy is the onset of the photoabsorption spectra corresponding to the energy gap between the HOMO or HOMO−1 and the LUMO. Evidently, the LUMO energy levels of the CAT and CAT-PET dyes are higher (more positive) than the energy level (ECB = −4.0 eV) of the CB of the TiO2 electrode. For both the CAT and CAT-PET dyes it was found that the LUMO energy level decreases with increasing electron-withdrawing ability of the substituents from the unsubstituted group (H) to the NO2 group, which is due to the stabilization of the LUMO energy level by the introduction of electron-withdrawing groups to the catechol moiety. Thus, this result shows that from CAT-H and CAT-PET-H to CAT-NO2 and CAT-PET-NO2, that is, by replacing the unsubstituted group (H) with an electron-withdrawing group, the lowering of the LUMO energy level is larger than that of the HOMO energy level, resulting in the red-shift of the photoabsorption band in the solution due to a decrease in the HOMO–LUMO band gap.
ox (versus Fc/Fc+) = 0.54 V for CAT-PET-H, 0.56 V for CAT-PET-Br, 0.60 V for both CAT-PET-CHO and CAT-PET-NO2) of the first oxidation wave and the onset of the second oxidation wave, respectively. The HOMO energy level of the CAT dyes and the HOMO−1 energy level of the CAT-PET dyes slightly decreases upon increasing the electron-withdrawing ability of the substituents from an unsubstituted group (H) to a NO2 group, which is due to the stabilization of the HOMO energy level. In contrast, the HOMO energy level of the CAT-PET dyes is ca. −5.7 eV, that is, the HOMO energy level is almost constant regardless of the type of substituent. This indicates that the HOMO energy levels corresponding to the diethylamino group are independent of the type of substituent (H, Br, CHO, NO2) on the CAT-PET structure. This result also shows that the HOMO energy levels of the CAT and CAT-PET dyes are lower (more negative) than the I3−/I− redox potential (−4.9 eV). Thus, for both the CAT and CAT-PET dyes this ensures the efficient regeneration of the oxidized catechol dyes by electron transfer from the I3−/I− redox couple in the electrolyte. Consequently, the CV and the photoabsorption spectral analysis indicates that as shown in Fig. 2b the oxidized CAT-PET dyes resulting from the DTCT upon binding to the TiO2 film is subsequently reduced back to its original neutral state through electron donation from the nitrogen atom of the amino moiety, that is, the PET takes place from the nitrogen atom of the amino moiety to the photoexcited catechol unit. The LUMO energy levels of the CAT and CAT-PET dyes were estimated from the HOMO and the onset of the photoabsorption spectra (306 nm, 4.05 eV for CAT-PET-H; 308 nm, 4.02 eV for CAT-PET-Br; 346 nm, 3.58 eV for CAT-PET-CHO; 416 nm, 2.98 eV for CAT-PET-NO2; ref. 30b for CAT dyes) in the solution, that is, the LUMO energy level of the catechol moiety was obtained through eq. [HOMO + E0–0] for the CAT dyes and eq. [(HOMO−1) + E0–0] for the CAT-PET dyes, where the E0–0 transition energy is the onset of the photoabsorption spectra corresponding to the energy gap between the HOMO or HOMO−1 and the LUMO. Evidently, the LUMO energy levels of the CAT and CAT-PET dyes are higher (more positive) than the energy level (ECB = −4.0 eV) of the CB of the TiO2 electrode. For both the CAT and CAT-PET dyes it was found that the LUMO energy level decreases with increasing electron-withdrawing ability of the substituents from the unsubstituted group (H) to the NO2 group, which is due to the stabilization of the LUMO energy level by the introduction of electron-withdrawing groups to the catechol moiety. Thus, this result shows that from CAT-H and CAT-PET-H to CAT-NO2 and CAT-PET-NO2, that is, by replacing the unsubstituted group (H) with an electron-withdrawing group, the lowering of the LUMO energy level is larger than that of the HOMO energy level, resulting in the red-shift of the photoabsorption band in the solution due to a decrease in the HOMO–LUMO band gap.
Theoretical calculations
To examine the electronic structures of the CAT and CAT-PET dyes and their titanium complexes (Ti-CAT and Ti-CAT-PET), the molecular orbitals of CAT, CAT-PET, Ti-CAT and Ti-CAT-PET were calculated using density functional theory (DFT) at the B3LYP/6-31G(d,p) level (Fig. 6 and Fig. S13, ESI,† for CAT-Me). The DFT calculations indicate that the HOMOs of the CAT dyes are mostly localized on the catechol moiety. On the other hand, the HOMOs of the CAT-PET dyes, with the exception of CAT-PET-H, are localized on the diethylamino group, but the HOMO−1 is mostly localized on the catechol moiety. In contrast, for CAT-PET-H, the HOMO is mostly localized on the Catechol moiety, and the HOMO−1 is mostly localized on the diethylamino group, but the benzene ring slightly contributes to the HOMO−1. The LUMOs of CAT-H, CAT-Br, CAT-PET-H and CAT-PET-Br are mostly localized on the benzene ring, but for CAT-CHO, CAT-NO2, CAT-PET-CHO and CAT-PET-NO2 with moderately or strongly electron-withdrawing substituents, the formyl and nitro groups greatly contribute to the LUMO. The HOMO energy levels of the CAT dyes and the HOMO−1 energy levels (the HOMO energy level for CAT-PET-H) of the CAT-PET dyes decrease upon increasing the electron-withdrawing ability of the substituents from an unsubstituted group (H) to a NO2 group. On the other hand, the HOMO energy level of the CAT-PET dyes (the HOMO−1 energy level for CAT-PET-H) is almost constant regardless of the kind of substituent. In addition, the LUMO energy levels of CAT-CHO, CAT-NO2, CAT-PET-CHO and CAT-PET-NO2 are significantly lower than those of the other CAT and CAT-PET dyes. Thus, these calculation results are consistent with the experimental results from the CV and the photoabsorption spectral analysis. For the Ti-CAT complexes, the HOMOs are mostly localized on almost the same moiety as those of the CAT dyes. For the CAT-PET dye the HOMO−1 of the CAT-PET complexes is also localized on the catechol moiety. Interestingly, the HOMOs of CAT-PET-CHO and CAT-PET-NO2 are totally localized on the diethylamino group. On the other hand, for CAT-PET-H and CAT-PET-Br the benzene ring slightly contributes to the HOMO, although the HOMOs are mostly localized on the diethylamino group. For both the Ti-CAT and Ti-CAT-PET complexes the LUMOs are localized on the metal center. Consequently, the DFT calculations reveal that when the CAT-PET dyes were adsorbed on the TiO2 surface, the dye excitations upon light irradiation induce DTCT, and subsequently, PET takes place from the nitrogen atom of the amino moiety to the photoexcited catechol unit. The HOMO or the HOMO−1 energy levels of the Ti-CAT and Ti-CAT-PET complexes are almost the same as those of the CAT and CAT-PET dyes. However, the LUMO energy levels of the Ti-CAT and Ti-CAT-PET complexes are lower than those of the CAT and CAT-PET dyes. Accordingly, the DFT calculations also show that the large red-shift of the photoabsorption bands upon binding to TiO2 film can be attributed to the stabilization of the LUMO level of the Ti-CAT and Ti-CAT-PET complexes.
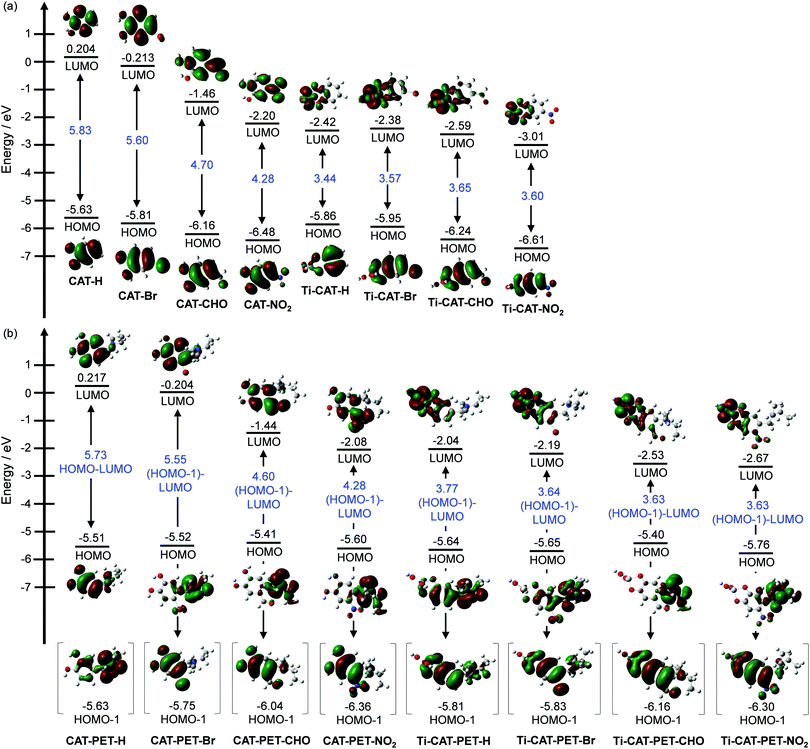 |
| | Fig. 6 Energy level diagram, HOMO and LUMO of the (a) CAT and (b) CAT-PET dyes and their Ti-complexes (Ti-CAT and Ti-CAT-PET), derived from DFT calculations. | |
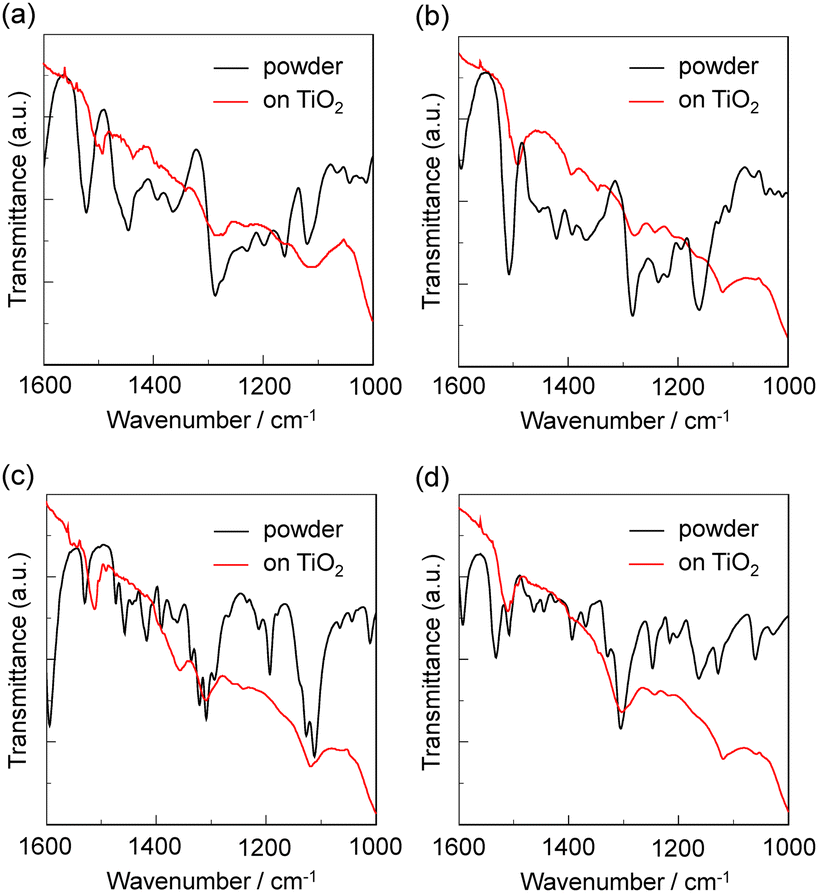
FTIR spectra
To elucidate the adsorption states of the CAT and CAT-PET dyes on TiO2 nanoparticles, we measured the FTIR spectra of the dye powders and the dye-adsorbed TiO2 film. For the powders of CAT and CAT-PET dyes (Fig. 7; ref. 30b for CAT dyes), the stretching vibrations of the phenolic group (C–OH) were observed at around 1280–1310 and 1220–1250 cm−1. In addition, the bending vibrations of the phenolic group were observed at around 1360–1400 cm−1. However, in the FTIR spectra of the dye-adsorbed TiO2 film, the stretching vibrations merge into one prominent signal or lose their hyperfine structure, and the bending vibrations almost disappear or are of lesser intensity. Thus, these observations indicate that the CAT and CAT-PET dyes are adsorbed on the TiO2 surface through the formation of a bidentate mononuclear chelating linkage and/or a bidentate binuclear bridging linkage between the catechol unit and the TiO2 surface.34
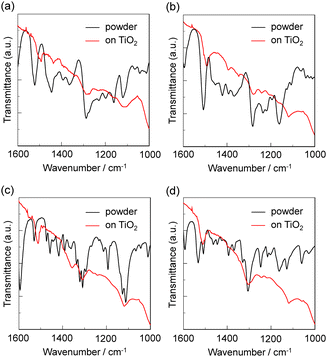 |
| | Fig. 7 FTIR spectra of the powders and the dyes adsorbed on TiO2 nanoparticles. (a) CAT-PET-H, (b) CAT-PET-Br, (c) CAT-PET-CHO and (d) CAT-PET-NO2. | |
Photovoltaic performances
DSSCs based on the CAT and CAT-PET dyes were prepared by using the dye-adsorbed TiO2 electrode (9 mm), Pt-coated glass as a counter electrode, and an acetonitrile solution with iodine (0.05 M), lithium iodide (0.1 M), and 1,2-dimethyl-3-propylimidazolium iodide (0.6 M) as the electrolyte. The photocurrent–voltage (I–V) characteristics were measured under simulated solar light (AM 1.5, 100 mW cm−2). The incident photon-to-current conversion efficiency (IPCE) spectra and the I–V curves are shown in Fig. 8. The photovoltaic performance parameters are collected in Table 1. It is worth mentioning here that the adsorption amount of the dyes adsorbed on the TiO2 electrode is approximately 1–4 × 1017 molecules per cm2, which is comparable to that of dye sensitizers with carboxyl groups for type-I DSSCs. The IPCE spectra demonstrate that for both the CAT and CAT-PET dyes the DTCT bands contribute to the IPCE spectra. However, the contribution to the IPCE originating from the DTCT band is different between the CAT and CAT-PET dyes, that is, the IPCEs of the CAT-PET dyes, with the exception of CAT-PET-NO2, are higher than those of the CAT dyes (Fig. 8a and b). In addition, the maximum IPCE values of the CAT and CAT-PET dyes with moderately electron-withdrawing substituents such as CAT-CHO (42%) and CAT-PET-CHO (45%) are much higher than those of the unsubstituted CAT-H (12%), CAT-PET-H (21%) and CAT-Me (10%, Fig. S12e, ESI†). Interestingly, the maximum IPCE value of CAT-NO2 (60%) is relatively high compared to the other CAT and CAT-PET dyes, although CAT-NO2 exhibits relatively weak DTCT. This result may be associated with the hypothesis that the interaction between the nitro group and I− ions in the electrolyte causes destabilization of the LUMO energy level, resulting in an increase in the DTCT efficiency in the DSSC and/or the retardation of the back electron transfer. The I–V curves (Fig. 8c, d and Fig. S12f, ESI,† for CAT-Me) show that the short circuit photocurrent density (Jsc) and η increase in the order of CAT-Me (0.96 mA cm−2, 0.20%) < CAT-H (1.26 mA cm−2, 0.29%) < CAT-Br (1.65 mA cm−2, 0.38%) < CAT-PET-H (1.77 mA cm−2, 0.43%) < CAT-PET-Br (2.06 mA cm−2, 0.49%) < CAT-CHO (2.26 mA cm−2, 0.60%) < CAT-PET-NO2 (2.39 mA cm−2, 0.57%) ≈ CAT-PET-CHO (2.40 mA cm−2, 0.60%) < CAT-NO2 (2.84 mA cm−2, 0.73%). Thus, this result indicates that the photovoltaic performance of the DSSCs based on the CAT-PET dyes, with the exception of CAT-PET-NO2, are higher than those based on the CAT dyes without PET characteristics. Moreover, the photovoltaic performances of the DSSCs based on the CAT and CAT-PET dyes with electron-withdrawing substituents are higher than those based on the unsubstituted CAT-H, CAT-PET-H and CAT-Me. This indicates that the PET from the diethylamino group to the oxidized catechol dye adsorbed on the TiO2 electrode can efficiently retard the back-electron-transfer, leading to favorable conditions for the type-II electron-injection pathway from the ground state of the catechol unit to the CB of the TiO2 electrode by the photoexcitation of DTCT bands. Consequently, this work proposes that introducing PET characteristics to the catechol dye with moderately electron-withdrawing substituent is an effective molecular design for type-II dye sensitizers to lead to not only an increase in the DTCT efficiency due to the PET moiety ((dimethylamino)methyl group) and the electron-withdrawing substituent, but also the retardation of back electron transfer due to the PET characteristics.
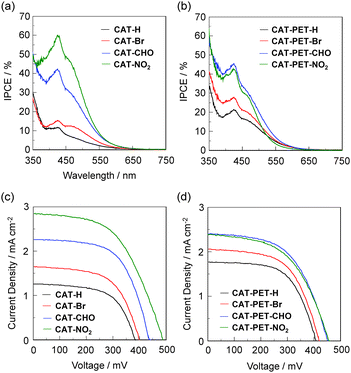 |
| | Fig. 8 (a and b) IPCE spectra and (c and d) I–V curves of DSSCs based on the CAT and CAT-PET dyes. | |
On the other hand, it is also worth mentioning here that the open-circuit photovoltage (Voc) values of the DSSCs based on the CAT-PET dyes, with the exception of CAT-PET-NO2, are higher than those for the CAT dyes. Moreover, the Voc values of the DSSCs based on CAT-CHO (440 mV), CAT-NO2 (488 mV), CAT-PET-CHO (452 mV) and CAT-PET-NO2 (456 mV) with an electron-withdrawing substituent are higher than those for the other CAT and CAT-PET dyes (ca. 400 mV). Thus, electrochemical impedance spectroscopy (EIS) analysis was performed to study the electron recombination process between the injected electrons into the TiO2 electrode and I3− ions in the electrolyte. Unfortunately, there is no good correlation between the Voc values and the electron recombination lifetimes (τe). However, it was assumed that the charge recombination between the injected electrons into the TiO2 electrode and I3− ions in the electrolyte may be one of the major reasons for the difference in Voc value between the DSSCs based on these catechol dyes, that is, the CAT-PET dyes with a diethylamino group as a relatively bulky group (sterically hindered substituent) can effectively retard the charge recombination compared with the CAT dyes. In addition, the difference in Voc values among these catechol dyes may be attributed to the number of injected electrons in the CB of TiO2 and/or the shift of the ECB of TiO2 by differences in the direction and the magnitude of the dipole moment of each substituent.3,4 Thus, the lowering of the Ecb of the TiO2 electrode due to the dipole moment of the catechol dyes adsorbed on the TiO2 surface and the decrease in the number of electrons injected into the CB of the TiO2 electrode result in a lower Voc value for the DSSCs based on the catechol dyes such as CAT-H and CAT-Br.
Conclusions
To provide an effective strategy to not only promote the retardation of the back-electron-transfer rate in type-II DSSCs but also increase in the dye-to-TiO2 charge transfer (DTCT) efficiency of Cat dyes, we have designed and developed catechol dye sensitizers (CAT-PET) possessing PET (photo-induced electron transfer) characteristics which are composed of a catechol unit linked to a diethylamino group as an electron donor moiety via a methylene spacer. It was found that the photovoltaic performances of the DSSCs based on the CAT-PET dyes, with the exception of CAT-PET-NO2, are higher than those for CAT dyes without PET characteristics. We demonstrate that introducing PET characteristics to the catechol dye with a moderately electron-withdrawing substituent is an effective molecular design for type-II dye sensitizers to lead to not only an enhancement of the DTCT efficiency, but also the retardation of back electron transfer from the electrons injected into the TiO2 electrode to the oxidized catechol dye.
Experimental
General
Melting points were measured with a Yanaco micro melting point apparatus MP model. IR spectra were recorded on a Perkin Elmer Spectrum One FT-IR spectrometer using the ATR method. High-resolution mass spectral data were acquired on a Thermo Fisher Scientific LTQ Orbitrap XL. 1H NMR and 13C NMR spectra were recorded on a Varian-400 (400 MHz) or a Varian-500 (500 MHz) FT NMR spectrometer. Absorption spectra were observed with a HITACHI U-2910 spectrophotometer. Cyclic voltammetry (CV) curves were recorded in acetonitrile/Bu4NClO4 (0.1 M) solution with a three-electrode system consisting of Ag/Ag+ as a reference electrode, Pt plate as a working electrode, and Pt wire as a counter electrode by using an Electrochemical Measurement System HZ-7000 (HOKUTO DENKO). The potentials were referenced to ferrocene/ferrocenium (Fc/Fc+) as the internal reference.
Synthesis
N-(3,4-Bis(methoxymethoxy)benzyl)ethanamine (2).
A methanol solution (100 mL) of 1 (ref. 35 and 36) (2.34 g, 10.33 mmol) under an argon atmosphere was bubbled with vaporized ethylamine by adding ethylamine aqueous solution (20 mL) to NaOH for 2 h at room temperature. The excess ethylamine was removed under reduced pressure. The reaction mixture was cooled to 0 °C. Next, sodium borohydride (0.21 g, 5.12 mmol) was added to the reaction mixture and the reaction mixture was stirred at room temperature for 4 h. After concentrating under reduced pressure, the resulting residue was dissolved in dichloromethane and washed with water. The dichloromethane extract was dried over MgSO4, filtrated, and concentrated. The residue was chromatographed on reverse-phased silica gel (methanol as eluent) to give 2 (2.41 g, yield 91%) as a yellow oil; 1H NMR (400 MHz, CDCl3) δ = 1.08 (t, J = 7.2 Hz, 3H), 2.63 (q, 2H), 3.46 (s, 3H), 3.47 (s, 3H), 3.67 (s, 2H), 5.16 (s, 2H), 5.19 (s, 2H), 6.87 (dd, J = 2.0 and 8.3 Hz, 1H), 7.06 (d, J = 8.3 Hz, 1H), 7.08 (d, J = 2.0 Hz, 1H) ppm; 13C NMR (125 MHz, CDCl3) δ = 15.18, 43.50, 53.43, 55.98, 56.07, 95.31, 95.41, 116.54, 116.65, 121.93, 135.04, 146.01, 147.10 ppm; HRMS (ESI): m/z (%): [M + H+] calcd for C13H22NO4, 256.15433; found 256.15427.
N-(3,4-Bis(methoxymethoxy)benzyl)-N-ethylethanamine (3).
A solution of 2 (2.25 g, 8.79 mmol) in DMF (90 mL) under an argon atmosphere was treated with sodium hydride (60%, 1.05 g, 26.37 mmol) and stirred for 1 h at room temperature. Bromoethane (3.5 mL, 44 mmol) was added dropwise and the solution was stirred at room temperature for 12 h. Water was added to the reaction mixture and the aqueous layer was extracted with a mixture of ethyl acetate and n-hexane. After concentrating under reduced pressure, the residue was chromatographed on silica gel (methanol as eluent) to give 3 (1.88 g, yield 76%) as an orange oil; 1H NMR (500 MHz, acetone-d6) δ = 1.00 (t, J = 7.2 Hz, 6H), 2.47 (q, 4H), 3.46 (s, 6H), 3.47 (s, 2H), 5.15 (s, 2H), 5.17 (s, 2H), 6.91 (dd, J = 1.9 and 8.2 Hz, 1H), 7.04 (d, J = 8.2 Hz, 1H), 7.14 (d, J = 1.9 Hz, 1H) ppm; 13C NMR (125 MHz, CDCl3) δ = 11.73, 46.58, 56.16, 56.22, 57.09, 95.47, 95.57, 116.49, 117.42, 122.80, 134.41, 146.09, 147.00 ppm; HRMS (ESI): m/z (%): [M + H+] calcd for C15H26NO4, 284.18563; found 284.18570.
4-((Diethylamino)methyl)benzene-1,2-diol (CAT-PET-H).
A solution of 3 (0.80 g, 2.81 mmol) and 1 M HCl (19 mL) in methanol (20 mL) was stirred under reflux for 12 h. After concentrating under reduced pressure, the residue was chromatographed on reverse-phased silica gel (methanol as eluent) to give CAT-PET-H (0.49 g, yield 89%) as a brown solid; m.p. 127–130 °C; IR (ATR): ṽ = 1602, 1522, 1445 cm−1; 1H NMR (400 MHz, CD3OD) δ = 1.33 (t, J = 7.4 Hz, 6H), 3.16 (q, 4H), 4.15 (s, 2H), 6.87 (dd, J = 2.0 and 8.0 Hz, 1H), 6.84 (d, J = 8.0 Hz, 1H), 6.89 (d, J = 2.0 Hz, 1H) ppm; 13C NMR (125 MHz, CD3OD) δ = 9.05, 47.57, 57.17, 116.76, 118.78, 121.66, 123.80, 147.19, 148.31 ppm; HRMS (ESI): m/z (%): [M + H+] calcd for C11H18NO2, 196.13321; found 196.13356.
N-(2-Bromo-4,5-bis(methoxymethoxy)benzyl)ethanamine (5).
A methanol solution (50 mL) of 4 (ref. 36 and 37) (1.47 g, 4.82 mmol) under an argon atmosphere was bubbled with vaporized ethylamine by adding ethylamine aqueous solution (20 mL) to NaOH for 2 h at room temperature. The excess ethylamine was removed under reduced pressure. The reaction mixture was cooled to 0 °C. Next, sodium borohydride (0.21 g, 5.12 mmol) was added to the reaction mixture and the reaction mixture was stirred at room temperature for 12 h. After concentrating under reduced pressure, the resulting residue was dissolved in dichloromethane and washed with water. The dichloromethane extract was dried over MgSO4, filtrated, and concentrated. The residue was compound 5 (1.5 g, yield 94%) as an orange oil; 1H NMR (400 MHz, CDCl3) δ = 1.13 (t, J = 7.2 Hz, 3H), 2.67 (q, 2H), 3.51 (s, 6H), 3.77 (s, 2H), 5.20 (s, 2H), 5.22 (s, 2H), 7.19 (s, 1H), 7.34 (s, 1H) ppm; 13C NMR (125 MHz, CDCl3) δ = 15.39, 43.52, 53.46, 56.39, 56.40, 95.61, 95.68, 115.68, 118.35, 120.86, 133.51, 146.61, 146.83 ppm; HRMS (APCI): m/z (%): [M + H+] calcd for C13H21NO4Br, 334.06485; found 334.06583.
N-(2-Bromo-4,5-bis(methoxymethoxy)benzyl)-N-ethylethanamine (6).
A solution of 5 (0.21 g, 0.64 mmol) in DMF (6 mL) under an argon atmosphere was treated with sodium hydride (60%, 0.077 g, 1.92 mmol) and stirred for 1 h at room temperature. Bromoethane (0.26 mL, 3.21 mmol) was added dropwise and the solution was stirred at room temperature for 12 h. Water was added to the reaction mixture and the aqueous layer was extracted with a mixture of ethyl acetate and n-hexane. After concentrating under reduced pressure, the residue was chromatographed on silica gel (methanol as eluent) to give 6 (0.12 g, yield 52%) as a colorless oil; 1H NMR (400 MHz, CDCl3) δ = 1.04 (t, J = 7.2 Hz, 6H), 2.55 (q, 4H), 3.50 (s, 3H), 3.51 (s, 3H), 3.54 (s, 2H), 5.20 (s, 2H), 5.22 (s, 2H), 7.31 (s, 1H), 7.39 (s, 1H) ppm; 13C NMR (125 MHz, acetone-d6) δ = 12.49, 47.77, 56.28, 56.35, 57.58, 96.27, 96.36, 115.57, 120.03, 121.73, 134.29, 147.77, 147.89 ppm; HRMS (APCI): m/z (%): [M + H+] calcd for C15H25NO4Br, 362.09615; found 362.09723.
4-Bromo-5-((diethylamino)methyl)benzene-1,2-diol (CAT-PET-Br).
A solution of 6 (0.12 g, 0.34 mmol) and 1 M HCl (2.3 mL) in methanol (2 mL) was stirred under reflux for 16 h. After concentrating under reduced pressure, the residue was chromatographed on reverse-phased silica gel (methanol as eluent) to give CAT-PET-Br (0.93 g, quantitative yield) as a brown solid; m.p. 171–172 °C; IR (ATR): ṽ = 1611, 1507, 1425 cm−1; 1H NMR (400 MHz, CD3OD) δ = 1.34 (t, J = 7.2 Hz, 6H), 3.16 (q, 4H), 4.23 (s, 2H), 6.98 (br, 1H), 7.06 (s, 1H) ppm; 13C NMR (125 MHz, CD3OD) δ = 9.06, 48.29, 56.84, 115.41, 120.17, 120.36, 120.46, 120.66, 121.01, 147.15, 149.71 ppm; HRMS (APCI): m/z (%): [M + H+] calcd for C11H17NO2Br, 274.04372; found 274.04416.
2-((Diethylamino)methyl)-4,5-bis(methoxymethoxy)benzaldehyde (7).
To a THF solution (7 mL) of 6 (0.53 g, 1.47 mol) under an argon atmosphere was added 1.6 M hexane solution of nBuLi (1 mL, 1.6 mmol) at −80 °C. After stirring for 30 min, DMF (0.23 mL, 1.94 mol) was added, and then the solution was stirred for 12 h at room temperature. The reaction was quenched with water. The solution was washed with NaHCO3 aq and then extracted with ethyl acetate. The residue was dissolved in toluene, and recycling gel permeation chromatography (GPC) was performed to give 7 (0.045 g, yield 10%) as a yellow oil; 1H NMR (400 MHz, acetone-d6) δ = 1.00 (t, J = 7.1 Hz, 6H), 2.52 (q, 4H), 3.48 (s, 6H), 3.87 (s, 2H), 5.24 (s, 2H), 5.32 (s, 2H), 7.30 (s, 1H), 7.60 (s, 1H), 10.35 (s, –CHO) ppm; 13C NMR (125 MHz, acetone-d6) δ = 11.91, 47.04, 55.27, 56.35, 56.51, 95.69, 96.17, 117.57, 118.72, 130.05, 139.49, 147.20, 152.43, 190.87 ppm; HRMS (APCI): m/z (%): [M + H+] calcd for C16H26NO5, 312.18055; found 312.18127.
2-((Diethylamino)methyl)-4,5-dihydroxybenzaldehyde (CAT-PET-CHO).
A solution of 7 (0.05 g, 0.14 mmol) and 1 M HCl (1 mL) in methanol (1 mL) was stirred under reflux for 12 h. After concentrating under reduced pressure, the residue was chromatographed on reverse-phased silica gel (methanol as eluent) to give CAT-PET-CHO (0.032 g, quantitative yield) as a green solid; m.p. 197–198 °C; IR (ATR): ṽ = 1686, 1593, 1457 cm−1; 1H NMR (400 MHz, CD3OD) δ = 1.36 (t, J = 7.3 Hz, 6H), 3.23 (q, 4H), 4.42 (s, 2H), 6.95 (s, 1H), 7.39 (s, 1H), 9.76 (br, –CHO) ppm; 13C NMR (125 MHz, CD3OD) δ = 8.74, 48.16, 55.35, 122.12, 123.93, 124.79, 128.88, 148.29, 152.79, 195.06 ppm; HRMS (APCI): m/z (%): [M + H+] calcd for C12H18NO3, 224.12812; found 224.12833.
N-(4,5-Bis(methoxymethoxy)-2-nitrobenzyl)ethanamine (9).
A methanol solution (120 mL) of 8 (ref. 36) (2.05 g, 7.55 mmol) under an argon atmosphere was bubbled with vaporized ethylamine by adding ethylamine aqueous solution (20 mL) to NaOH for 1 h at room temperature. The excess ethylamine was removed under reduced pressure. The reaction mixture was cooled to 0 °C. Next, sodium borohydride (0.15 g, 3.78 mmol) was added to the reaction mixture and the reaction mixture was stirred at room temperature for 2 h. Water was added to the reaction mixture and the aqueous layer was extracted with dichloromethane. After concentrating under reduced pressure, the resulting residue was dissolved in dichloromethane and washed with water. The dichloromethane extract was dried over MgSO4, filtrated, and concentrated. The residue was compound 9 (1.27 g, yield 56%) as a black oil; 1H NMR (400 MHz, CDCl3) δ = 1.14 (t, J = 7.2 Hz, 3H), 2.69 (q, 2H), 3.52 (s, 6H), 4.01 (s, 2H), 5.28 (s, 2H), 5.34 (s, 2H), 7.37 (s, 1H), 7.91 (s, 1H) ppm; 13C NMR (125 MHz, CDCl3) δ = 15.30, 43.87, 51.17, 56.63, 56.79, 95.18, 95.66, 113.64, 117.32, 131.80, 142.21, 145.64, 151.57 ppm; HRMS (APCI): m/z (%): [M + H+] calcd for C13H21N2O6, 301.13941; found 301.13992.
N-(4,5-Bis(methoxymethoxy)-2-nitrobenzyl)-N-ethylethanamine (10).
A solution of 9 (1.0 g, 3.34 mmol) in DMF (33 mL) under an argon atmosphere was treated with sodium hydride (60%, 0.4 g, 10 mmol) and stirred for 1 h at room temperature. Bromoethane (3.98 mL, 50 mmol) was added dropwise and the solution was stirred at room temperature for 12 h. Water was added to the reaction mixture and the aqueous layer was extracted with a mixture of ethyl acetate and n-hexane. After concentrating under reduced pressure, the residue was chromatographed on silica gel (methanol as eluent) to give 10 (0.68 g, yield 61%) as a black oil; 1H NMR (400 MHz, CDCl3) δ = 1.02 (t, J = 7.2 Hz, 6H), 2.52 (q, 4H), 3.52 (s, 3H), 3.53 (s, 3H), 3.85 (s, 2H), 5.27 (s, 2H), 5.33 (s, 2H), 7.30 (s, 1H), 7.83 (s, 1H) ppm; 13C NMR (125 MHz, CDCl3) δ = 12.30, 47.61, 55.04, 56.60, 56.63, 95.08, 95.71, 113.35, 116.60, 133.22, 142.37, 145.07, 151.23 ppm; HRMS (APCI): m/z (%): [M + H+] calcd for C15H25N2O6, 329.17071; found 329.17136.
4-((Diethylamino)methyl)-5-nitrobenzene-1,2-diol (CAT-PET-NO2).
A solution of 10 (0.26 g, 0.78 mmol) and 1 M HCl (5.4 mL) in methanol (5 mL) was stirred under reflux for 12 h. After concentrating under reduced pressure, the residue was chromatographed on reverse-phased silica gel (methanol as eluent) to give CAT-PET-NO2 (0.19 g, quantitative yield) as a brown solid; m.p. 213–214 °C; IR (ATR): ṽ = 1592, 1532, 1305 cm−1; 1H NMR (500 MHz, CD3OD) δ = 1.40 (t, J = 7.3 Hz, 6H), 3.27–3.30 (m, 4H, overlapping peak of residual proton in CD3OD), 4.52 (s, 2H), 7.01 (s, 1H), 7.81 (s, 1H) ppm; 13C NMR (125 MHz, CD3OD) δ = 8.71, 48.41, 55.82, 114.23, 119.58, 121.76, 141.41, 148.66, 153.74 ppm; HRMS (APCI): m/z (%): [M + H+] calcd for C11H17N2O4, 241.11828; found 241.11858.
Preparation of DSSCs
TiO2 paste (JGC Catalysts and Chemicals Ltd, PST-18NR) was deposited on a fluorine-doped-tin-oxide (FTO) substrate by doctor-blading, and sintered for 50 min at 450 °C. The 9 μm thick TiO2 electrode was immersed into 0.1 mM dye solution in acetonitrile for 15 hours, which was long enough to adsorb the dye sensitizers (CAT and CAT-PET). The DSSCs were fabricated by using the TiO2 electrode (0.5 × 0.5 cm2 in photoactive area) thus prepared, Pt-coated glass as a counter electrode, and a solution of 0.05 M iodine, 0.1 M lithium iodide, and 0.6 M 1,2-dimethyl-3-propylimidazolium iodide in acetonitrile as electrolyte. The photocurrent–voltage characteristics were measured using a potentiostat under simulated solar light (AM 1.5, 100 mW cm−2). IPCE spectra were measured under monochromatic irradiation with a tungsten-halogen lamp and a monochromator. The amount of dye molecules adsorbed on the TiO2 film was determined from the calibration curve by absorption spectral measurement of the concentration change of the dye solution before and after adsorption. The quantification of the dye was performed based on the molar extinction coefficient for λabsmax of the dye in the above solution. The photoabsorption spectra of the dyes adsorbed on TiO2 film (9 μm) were recorded on the dye-adsorbed TiO2 film in transmission mode with a calibrated integrating sphere system. Electrochemical impedance spectroscopy (EIS) for the DSSCs in the dark under a forward bias of −0.6 V with a frequency range of 0.1 Hz to 100 kHz was performed with an AMETEK Versa STAT 3.
Acknowledgements
This work was supported by Grants-in-Aid for Scientific Research (B) from the Japan Society for the Promotion of Science (JSPS) KAKENHI Grant Number 15H03859, by Matching Planner Program (MP27115659061) from Japan Science and Technology Agency (JST) and by Yazaki Memorial Foundation for Science and Technology.
Notes and references
- B. O’Regan and M. Grätzel, Nature, 1991, 353, 737 CrossRef.
- A. Mishra, M. K. R. Fischer and P. Bäuerle, Angew. Chem., Int. Ed., 2009, 48, 2474 CrossRef CAS PubMed.
-
(a) Y. Ooyama and Y. Harima, Eur. J. Org. Chem., 2009, 2903 CrossRef CAS;
(b) Y. Ooyama and Y. Harima, ChemPhysChem, 2012, 13, 4032 CrossRef CAS PubMed.
-
(a) Z. Ning and H. Tian, Chem. Commun., 2009, 5483 RSC;
(b) Z. Ning, Y. Fu and H. Tian, Energy Environ. Sci., 2010, 3, 1170 RSC.
- A. Hagfeldt, G. Boschloo, L. Sun, L. Kloo and H. Pettersson, Chem. Rev., 2010, 110, 6595 CrossRef CAS PubMed.
- N. Manfredi, B. Cecconi and A. Abbotto, Eur. J. Org. Chem., 2014, 7069 CrossRef CAS.
- L. Zhang and J. M. Cole, ACS Appl. Mater. Interfaces, 2015, 7, 3427 CAS.
- C.-P. Lee, R. Y.-Y. Lin, L.-Y. Lin, C.-T. Li, T.-C. Chu, S.-S. Sun, J. T. Lin and K.-C. Ho, RSC Adv., 2015, 5, 23810 RSC.
- B. Pashaei, H. Shahroosvand, M. Graetzel and M. K. Nazeeruddin, Chem. Rev., 2016, 116, 9485 CrossRef CAS PubMed.
- C.-T. Li, R. Y.-Y. Lin and J. T. Lin, Chem. – Asian J., 2017, 12, 486 CrossRef CAS PubMed.
-
(a) H. Imahori, T. Umeyama and S. Ito, Acc. Chem. Res., 2009, 42, 1809 CrossRef CAS PubMed;
(b) L.-L. Li and E. W.-G. Diau, Chem. Soc. Rev., 2013, 42, 291 RSC;
(c) K. Ladomenou, T. N. Kitsopoulos, G. D. Sharma and A. G. Coutsolelos, RSC Adv., 2014, 4, 21379 RSC;
(d) T. Higashino and H. Imahori, Dalton Trans., 2015, 44, 448 RSC.
-
(a) T. Bessho, S. M. Zakeeruddin, C.-Y. Yeh, E. W.-G. Diau and M. Grätzel, Angew. Chem., Int. Ed., 2010, 49, 6646 CrossRef CAS PubMed;
(b) A. Yella, H.-W. Lee, H. N. Tsao, C. Yi, A. K. Chandiran, M. K. Nazeeruddin, E. W.-G. Diau, C.-Y. Yeh, S. M. Zakeeruddin and M. Grätzel, Science, 2011, 334, 629 CrossRef CAS PubMed;
(c) S. Mathew, A. Yella, P. Gao, R. Humphry-Baker, B. F. E. Curchod, N. Ashari-Astani, I. Tavernelli, U. Rothlisberger, Md. K. Nazeeruddin and M. Grätzel, Nat. Chem., 2014, 6, 242 CrossRef CAS PubMed;
(d) A. Yella, C.-L. Mai, S. M. Zakeeruddin, S.-N. Chang, C.-H. Hsieh, C.-Y. Yeh and M. Grätzel, Angew. Chem., Int. Ed., 2014, 53, 2973 CrossRef CAS PubMed;
(e) T. Higashino, Y. Fujimori, K. Sugiura, Y. Tsuji, S. Ito and H. Imahori, Angew. Chem., Int. Ed., 2015, 54, 9052 CrossRef CAS PubMed;
(f) J. P. Hill, Angew. Chem., Int. Ed., 2016, 55, 2976 CrossRef CAS PubMed;
(g) G. Copley, D. Hwang, D. Kim and A. Osuka, Angew. Chem., Int. Ed., 2016, 55, 10287 CrossRef CAS PubMed.
-
(a) Y. Xie, Y. Tang, W. Wu, Y. Wang, J. Liu, X. Li, H. Tian and W.-H. Zhu, J. Am. Chem. Soc., 2015, 137, 14055 CrossRef CAS PubMed;
(b) S. Mathew, N. A. Astani, B. F. E. Curchod, J. H. Delcamp, M. Marszalek, J. Frey, U. Rothlisberger, M. K. Nazeeruddina and M. Grätzel, J. Mater. Chem. A, 2016, 4, 2332 RSC;
(c) L. Zeininger, F. Lodermeyer, R. D. Costa, D. M. Guldi and A. Hirsch, Chem. Commun., 2016, 52, 8842 RSC;
(d) J. Luo, J. Zhang, K.-W. Huang, Q. Qi, S. Dong, J. Zhang, P. Wang and J. Wu, J. Mater. Chem. A, 2016, 4, 8428 RSC;
(e) C. Li, L. Luo, D. Wu, R. Jiang, J. Lan, R. Wang, L. Huang, S. Yang and J. You, J. Mater. Chem. A, 2016, 4, 11829 RSC;
(f) Y.-C. Liu, H.-H. Chou, F.-Y. Ho, H.-J. Wei, T.-C. Wei and C.-Y. Yeh, J. Mater. Chem. A, 2016, 4, 11878 RSC.
-
(a) J.-J. Cid, J.-H. Yum, S.-R. Jang, M. K. Nazeeruddin, E. Martínez-Ferrero, E. Palomares, J. Ko, M. Grätzel and T. Torres, Angew. Chem., Int. Ed., 2007, 46, 8358 CrossRef CAS PubMed;
(b) M. Kimura, H. Nomoto, N. Masaki and S. Mori, Angew. Chem., Int. Ed., 2012, 51, 4371 CrossRef CAS PubMed;
(c) M.-E. Ragoussi, J.-J. Cid, J.-H. Yum, G. De La Torre, D. D. Censo, M. Grätzel, M. K. Nazeeruddin and T. Torres, Angew. Chem., Int. Ed., 2012, 51, 4375 CrossRef CAS PubMed;
(d) T. Ikeuchi, H. Nomoto, N. Masaki, M. J. Griffith, S. Mori and M. Kimura, Chem. Commun., 2014, 50, 1941 RSC;
(e) T. Ikeuchi, S. Agrawal, M. Ezoe, S. Mori and M. Kimura, Chem. – Asian J., 2015, 10, 2347 CrossRef CAS PubMed;
(f) V. A. Chiykowski, B. Lam, C. Du and C. P. Berlinguette, Chem. Commun., 2017, 53, 2367 RSC.
-
(a) K. Kakiage, Y. Aoyama, T. Yano, T. Otsuka, T. Kyomen, M. Unno and M. Hanaya, Chem. Commun., 2014, 50, 6379 RSC;
(b) A. Amacher, C. Yi, J. Yang, M. P. Bircher, Y. Fu, M. Cascella, M. Grätzel, S. Decurtins and S.-X. Liu, Chem. Commun., 2014, 50, 6540 RSC;
(c) W.-I. Hung, Y.-Y. Liao, T.-H. Lee, Y.-C. Ting, J.-S. Ni, W.-S. Kao, J. T. Lin, T.-C. Wei and Y.-S. Yen, Chem. Commun., 2015, 51, 2152 RSC;
(d) X. Li, Z. Zheng, W. Jiang, W. Wu, Z. Wang and H. Tian, Chem. Commun., 2015, 51, 3590 RSC;
(e) K. Kakiage, Y. I. Aoyama, T. Yano, K. Oya, T. Kyomen and M. Hanaya, Chem. Commun., 2015, 51, 6315 RSC;
(f) L. Yang, Z. Zheng, Y. Li, W. Wu, H. Tian and Z. Wang, Chem. Commun., 2015, 51, 4842 RSC;
(g) N. Shibayama, Y. Inoue, M. Abe, S. Kajiyama, H. Ozawa, H. Miura and H. Arakawa, Chem. Commun., 2015, 51, 12795 RSC;
(h) K. Kakiage, Y. Aoyama, T. Yano, K. Oya, J. Fujisawa and M. Hanaya, Chem. Commun., 2015, 51, 15894 RSC;
(i) D. Joly, M. Godfroy, L. Pellejà, Y. Kervella, P. Maldivi, S. Narbey, F. Oswald, E. Palomares and R. Demadrille, J. Mater. Chem. A, 2017, 5, 6122 RSC.
-
(a) J.-S. Ni, Y.-C. Yen and J. T. Lin, Chem. Commun., 2015, 51, 17080 RSC;
(b) S. S. Soni, K. B. Fadadu, J. V. Vaghasiya, B. G. Solanki, K. K. Sonigara, A. Singh, D. Das and P. K. Iyer, J. Mater. Chem. A, 2015, 3, 21664 RSC;
(c) X. Li, Y. Hu, I. Sanchez-Molina, Y. Zhou, F. Yu, S. A. Haque, W. Wu, J. Hua, H. Tian and N. Robertson, J. Mater. Chem. A, 2015, 3, 2173 Search PubMed;
(d) Y. Hu, A. Ivaturi, M. Planells, C. L. Boldrini, A. O. Biroli and N. Robertson, J. Mater. Chem. A, 2016, 4, 2509 RSC;
(e) H. Wu, L. Yang, Y. Li, M. Zhang, J. Zhang, Y. Guo and P. Wang, J. Mater. Chem. A, 2016, 4, 519 RSC;
(f) J. Wu, G. Li, L. Zhang, G. Zhou and Z.-S. Wang, J. Mater. Chem. A, 2016, 4, 3342 RSC;
(g) J.-S. Ni, Y.-C. Yen and J. T. Lin, J. Mater. Chem. A, 2016, 4, 6553 RSC;
(h) A. J. Huckaba, A. Yella, P. Brogdon, J. S. Murphy, M. K. Nazeeruddin, M. Grätzel and J. H. Delcamp, Chem. Commun., 2016, 52, 8424 RSC;
(i) T.-Y. Li, C. Su, S. B. Akula, W.-G. Sun, H.-M. Chien and W.-R. Li, Org. Lett., 2016, 18, 3386 CrossRef CAS PubMed;
(j) Y. Gao, X. Li, Y. Hu, Y. Fan, J. Yuan, N. Robertson, J. Hua and S. R. Marder, J. Mater. Chem. A, 2016, 4, 12865 RSC;
(k) X. Li, B. Xu, P. Liu, Y. Hu, L. Kloo, J. Hua, L. Sun and H. Tian, J. Mater. Chem. A, 2017, 5, 3157 RSC.
-
(a) Z. Yao, M. Zhang, H. Wu, L. Yang, R. Li and P. Wang, J. Am. Chem. Soc., 2015, 137, 3799 CrossRef CAS PubMed;
(b) N. Zhou, K. Prabakaran, B. Lee, S. H. Chang, B. Harutyunyan, P. Guo, M. R. Butler, A. Timalsina, M. J. Bedzyk, M. A. Ratner, S. Vegiraju, S. Yau, C.-G. Wu, R. P. H. Chang, A. Facchetti, M.-C. Chen and T. J. Marks, J. Am. Chem. Soc., 2015, 137, 4414 CrossRef CAS PubMed;
(c) K. Matsumura, S. Yoshizaki, M. M. Maitani, Y. Wada, Y. Ogomi, S. Hayase, T. Kaiho, S. Fuse, H. Tanaka and T. Takahashi, Chem. – Eur. J., 2015, 21, 9742 CrossRef CAS PubMed;
(d) B. Liu, F. Giordano, K. Pei, J.-D. Decoppet, W.-H. Zhu, S. M. Zakeeruddin and M. Grätzel, Chem. – Eur. J., 2015, 21, 18654 CrossRef CAS PubMed;
(e) Z. Yao, M. Zhang, R. Li, L. Yang, Y. Qiao and P. Wang, Angew. Chem., Int. Ed., 2015, 54, 5994 CrossRef CAS PubMed;
(f) P. Brogdon, F. Giordano, G. A. Puneky, A. Dass, S. M. Zakeeruddin, M. K. Nazeeruddin, Mi. Grätzel, G. S. Tschumper and J. H. Delcamp, Chem. – Eur. J., 2016, 22, 694 CrossRef CAS PubMed;
(g) S. Irie, S. Fuse, M. M. Maitani, Y. Wada, Y. Ogomi, S. Hayase, T. Kaiho, H. Masui, H. Tanaka and T. Takahashi, Chem. – Eur. J., 2016, 22, 2507 CrossRef CAS PubMed;
(h) F.-L. Guo, Z.-Q. Li, X.-P. Liu, L. Zhou, F.-T. Kong, W.-C. Chen and S.-Y. Dai, Adv. Funct. Mater., 2016, 26, 5733 CrossRef CAS.
- J. Mao, N. He, Z. Ning, Q. Zhang, F. Guo, L. Chen, W. Wu, J. Hua and H. Tian, Angew. Chem., Int. Ed., 2012, 51, 9873 CrossRef CAS PubMed.
-
(a) Y. Ooyama, S. Inoue, R. Asada, G. Ito, K. Kushimoto, K. Komaguchi, I. Imae and Y. Harima, Eur. J. Org. Chem., 2010, 92 CrossRef CAS;
(b) Y. Ooyama, S. Inoue, T. Nagano, K. Kushimoto, J. Ohshita, I. Imae, K. Komaguchi and Y. Harima, Angew. Chem., Int. Ed., 2011, 50, 7429 CrossRef CAS PubMed;
(c) Y. Ooyama, T. Nagano, S. Inoue, I. Imae, K. Komaguchi, J. Ohshita and Y. Harima, Chem. – Eur. J., 2011, 17, 14837 CrossRef CAS PubMed;
(d) Y. Ooyama, N. Yamaguchi, I. Imae, K. Komaguchi, J. Ohshita and Y. Harima, Chem. Commun., 2013, 49, 2548 RSC;
(e) Y. Ooyama, Y. Hagiwara, T. Mizumo, Y. Harima and J. Ohshita, New J. Chem., 2013, 37, 2479 RSC;
(f) Y. Ooyama, T. Sato, Y. Harima and J. Ohshita, J. Mater. Chem. A, 2014, 2, 3293 RSC;
(g) Y. Ooyama, K. Uenaka and J. Ohshita, Eur. J. Org. Chem., 2015, 3713 CrossRef CAS;
(h) Y. Ooyama, K. Uenaka, T. Kamimura, S. Ozako, M. Kanda, T. Koide and F. Tani, RSC Adv., 2016, 6, 16150 RSC;
(i) Y. Ooyama, K. Furue, T. Enoki, M. Kanda, Y. Adachi and J. Ohshita, Phys. Chem. Chem. Phys., 2016, 18, 30662 RSC;
(j) Y. Ooyama, N. Yamaguchi, J. Ohshita and Y. Harima, Phys. Chem. Chem. Phys., 2016, 18, 32992 RSC;
(k) Y. Ooyama, M. Kanda, T. EnoKi, Y. Adachi and J. Ohshita, RSC Adv., 2017, 7, 13072 RSC.
-
(a) Y. Harima, T. Fujita, Y. Kano, I. Imae, K. Komaguchi, Y. Ooyama and J. Ohshita, J. Phys. Chem. C, 2013, 117, 16364 CrossRef CAS;
(b) N. Shibayama, H. Ozawa, M. Abe, Y. Ooyama and H. Arakawa, Chem. Commun., 2014, 50, 6398 RSC;
(c) Y. Ooyama, K. Uenaka, T. Sato, N. Shibayama and J. Ohshita, RSC Adv., 2015, 5, 2531 RSC;
(d) J. Ohshita, Y. Adachi, D. tanaka, M. Nakashima and Y. Ooyama, RSC Adv., 2015, 5, 36673 RSC;
(e) Y. Harima, Y. Kano, T. Fujita, I. Imae, Y. Ooyama and J. Ohshita, RSC Adv., 2015, 5, 71387 RSC;
(f) Y. Adachi, Y. Ooyama, N. Shibayama and J. Ohshita, Dalton Trans., 2016, 45, 13817 RSC;
(g) Y. Ooyama, K. Uenaka, Y. Harima and J. Ohshita, RSC Adv., 2014, 4, 30225 RSC;
(h) Y. Ooyama, K. Uenaka and J. Ohshita, RSC Adv., 2015, 5, 21012 RSC;
(i) Y. Ooyama, K. Uenaka and J. Ohshita, Org. Chem. Front., 2015, 2, 552 RSC.
-
(a) D. Daphnomili, G. D. Sharma, S. Biswas, T. K. R. Justin and A. G. Goutsolelos, J. Photochem. Photobiol., A, 2013, 253, 88 CrossRef CAS;
(b) J. Lu, X. Xu, Z. Li, k. Cao, J. Cui, Y. Zhang, Y. Shen, Y. Li, J. Zhu, S. Dai, W. Chjen, Y. Cheng and M. Wang, Chem. – Asian J., 2013, 8, 956 CrossRef CAS PubMed;
(c) M.-D. Zhang, H.-X. Xie, X.-H. Ju, L. Qin, Q.-X. Yang, H.-G. Zheng and X.-F. Zhou, Phys. Chem. Chem. Phys., 2013, 15, 634 RSC;
(d) D. Daphnomili, G. Landrou, P. Singh, A. Thomas, K. Yesudas, B. K. G. D. Sharma and A. G. Goutsolelos, RSC Adv., 2012, 2, 12899 RSC;
(e) L. Wang, X. yang, S. Li, M. Cheng and L. Sun, RSC Adv., 2013, 3, 13677 RSC;
(f) T. Sakurada, Y. Arai and H. Segawa, RSC Adv., 2014, 4, 13201 RSC;
(g) J. Mao, D. Wang, S.-H. Liu, Y. Hang, Y. Xu, Q. Zhang, W. Wu, P.-T. Chou and J. Hua, Asian J. Org. Chem., 2014, 3, 153 CrossRef CAS;
(h) T. Ikeuchi, S. Agrawal, M. Ezoe, S. Mori and M. Kimura, Chem. – Asian J., 2015, 10, 2347 CrossRef CAS PubMed.
-
(a) C. Stangel, A. Bagaki, P. A. Angaridis, G. Charalambidis, G. D. Sharma and A. G. Coutsolelos, Inorg. Chem., 2014, 53, 11871 CrossRef CAS PubMed;
(b) M. N. K. P. Bolisetty, C.-T. Li, K. R. J. Thomas, G. B. Bodedla and K.-C. Ho, Tetrahedron, 2014, 70, 4203 Search PubMed;
(c) D. Franchi, M. Calamante, G. Reginato, L. Zani, M. Peruzzini, M. Taddei, F. F. de Biani, R. Basosi, A. Sinicropi, D. Colonna, A. Di Carlo and A. Mordin, Tetrahedron, 2014, 70, 6285 CrossRef CAS;
(d) E. V. Verbitskiy, E. M. Cheprakova, J. O. Subbotina, A. V. Schepochkin, P. A. Slepukhin, G. L. Rusinov, V. N. Charushin, O. N. Chupakhin, N. I. Makarova, A. V. Metelitsa and V. I. Minkin, Dyes Pigm., 2014, 100, 201 CrossRef CAS;
(e) C.-L. Mai, T. Moehl, C.-H. Hsieh, J.-D. Décoppet, S. M. Zakeeruddin, M. Grätzel and C.-Y. Yeh, ACS Appl. Mater. Interfaces, 2015, 7, 14975 CrossRef CAS PubMed;
(f) H. Jia, K. Shen, X. I. Ju, M. Zhanga and H. Zheng, New J. Chem., 2016, 40, 2799 RSC;
(g) P. A. Angaridis, E. Ferentinos, G. Charalambidis, K. Ladomenou, V. Nikolaou, S. Biswas, G. D. Sharma and A. G. Coutsolelos, RSC Adv., 2016, 6, 22187 RSC;
(h) U. Meinhardt, F. Lodermeyer, T. A. Schaub, A. Kunzmann, P. O. Dral, A. C. Sale, F. Hampel, D. M. Guldi, R. D. Costa and M. Kivala, RSC Adv., 2016, 6, 67372 RSC.
- H. He, A. Gurung and L. Si, Chem. Commun., 2012, 48, 5910 RSC.
-
(a) R. Huber, S. Spcrlein, J. E. Moser, M. Grätzel and J. Wachtveitl, J. Phys. Chem. B, 2000, 104, 8995 CrossRef CAS;
(b) P. Persson, R. Bergström and S. Lunell, J. Phys. Chem. B, 2000, 104, 10348 CrossRef CAS;
(c) E. Hao, N. A. Anderson, J. B. Asbury and T. Lianl, J. Phys. Chem. B, 2002, 106, 10191 CrossRef CAS;
(d) Y. Wang, K. Hang, N. A. Anderson and T. Lian, J. Phys. Chem. B, 2003, 107, 9434 CrossRef CAS;
(e) S.-C. Li, J.-G. Wang, P. Jacobson, X.-Q. Gong, A. Selloni and U. Dieboldi, J. Am. Chem. Soc., 2009, 131, 980 CrossRef CAS PubMed;
(f) L.-M. Liu, S.-C. Li, H. Cheng, U. Diebold and A. Selloni, J. Am. Chem. Soc., 2011, 133, 7816 CrossRef CAS PubMed;
(g) S. A. Agarkar, R. R. Kulkarni, V. V. Dhas, A. A. Chinchansure, P. Hazra, S. P. Joshi and S. B. Ogale, ACS Appl. Mater. Interfaces, 2011, 3, 2440 CrossRef CAS PubMed;
(h) S. Li, X. Yang, D. Qu, W. Wang, Y. Wang and L. Sun, Chin. J. Chem., 2012, 30, 2315 CrossRef CAS.
-
(a) R. Sánchez-de-Armas, J. O. López, M. A. San-Miguel and J. F. Sanz, J. Chem. Theory Comput., 2010, 6, 2856 CrossRef PubMed;
(b) R. Sánchez-de-Armas, J. Oviedo, M. A. San-Miguel and J. F. Sanz, J. Phys. Chem. C, 2011, 115, 11293 CrossRef;
(c) R. Sánchez-de-Armas, M. A. San-Miguel, J. Oviedo, A. Márquez and J. F. Sanz, Phys. Chem. Chem. Phys., 2011, 13, 1506 RSC;
(d) R. Sánchez-de-Armas, M. A. San-Miguel, J. Oviedo and J. F. Sanz, Comput. Theor. Chem., 2011, 975, 99 CrossRef.
-
(a) L. G. C. Rego and V. S. Batista, J. Am. Chem. Soc., 2003, 125, 7989 CrossRef CAS PubMed;
(b) S. G. Abusabara, L. G. C. Rego and V. S. Batista, J. Am. Chem. Soc., 2005, 127, 18234 CrossRef PubMed;
(c) S. G. Abuabara, G. W. Cady, J. B. Baxter, C. A. Schmuttenmaer, R. H. Crabtree, G. W. Brudvig and V. S. Batsista, J. Phys. Chem. C, 2007, 111, 11982 CrossRef CAS;
(d) L. G. C. Rego, R. de Silva, J. A. Freire, R. C. Snoeberger and V. S. Batsista, J. Phys. Chem. C, 2010, 114, 1317 CrossRef CAS;
(e) W. Macyk, K. Szaciłowski, G. Stochel, M. Buchalska, J. Kuncewicz and P. Łabuz, Coord. Chem. Rev., 2010, 254, 2687 CrossRef CAS;
(f) H. J. Nam, B. Kim, M. J. Ko, M. Jin, J. M. Kim and D.-Y. Jung, Chem. – Eur. J., 2012, 18, 14000 CrossRef CAS PubMed.
-
(a) W. R. Duncan, W. M. Stier and O. V. Prezhdo, J. Am. Chem. Soc., 2005, 127, 7941 CrossRef CAS PubMed;
(b) T. Lana-Villarreal, A. Rodes, J. M. Pérez and R. Gómez, J. Am. Chem. Soc., 2005, 127, 12601 CrossRef CAS PubMed;
(c) W. R. Duncan, W. M. Stier and O. V. Prezhdo, J. Phys. Chem. B, 2005, 109, 365 CrossRef CAS PubMed;
(d) W. R. Duncan, C. F. Craig and O. V. Prezhdo, J. Am. Chem. Soc., 2007, 129, 8528 CrossRef CAS PubMed;
(e) S. Verma, P. Kar, A. Das, D. K. Palit and H. N. Ghosh, Chem. – Eur. J., 2010, 16, 611 CrossRef CAS PubMed;
(f) S. Verma, P. Kar, A. Das and H. N. Ghosh, Chem. – Eur. J., 2011, 17, 1561 CrossRef CAS PubMed.
-
(a) E. L. Tae, S. H. Lee, J. K. Lee, S. S. Yoo, E. J. Kang and K. B. Yoon, J. Phys. Chem. B, 2005, 109, 22513 CrossRef CAS PubMed;
(b) B.-K. An, W. Hu, P. L. Burn and P. Meredith, J. Phys. Chem. C, 2010, 114, 17964 CrossRef CAS;
(c) M. Adineh, P. Tahay, M. Ameri, N. Safari and E. Mohajerani, RSC Adv., 2016, 6, 14512 RSC.
- I. A. Janković, Z. V. Šaponjic, M. I. Čomor and J. M. Nedeljković, J. Phys. Chem. C, 2009, 113, 12645 Search PubMed.
-
(a) Y. Ooyama, T. Yamada, T. Fujita, Y. Harima and J. Ohshita, J. Mater. Chem. A, 2014, 2, 8500 RSC;
(b) Y. Ooyama, M. Kanda, K. Uenaka and J. Ohshita, ChemPhysChem, 2015, 16, 3049 CrossRef CAS PubMed;
(c) Y. Ooyama, K. Uenaka, M. Kanda, T. Yamada, N. Shibayama and J. Ohshita, Dyes Pigm., 2015, 122, 40 CrossRef CAS.
-
(a) G. Ramakrishna, D. A. Jose, D. K. Kumar, A. Das, D. K. Palit and H. N. Ghosh, J. Phys. Chem. B, 2005, 109, 15445 CrossRef CAS PubMed;
(b) F. de Angelis, Chem. Phys. Lett., 2010, 493, 323 CrossRef CAS;
(c) S. Namuangruk, R. Fukuda, M. Ehara, J. Meeprasert, T. Khanasa, S. Morada, T. Kaewin, S. Jungsuttiwong, T. Sudyoadsuk and V. Promarak, J. Phys. Chem. C, 2012, 116, 25653 CrossRef CAS;
(d) S. Manzhos and K. Kotsis, Chem. Phys. Lett., 2016, 660, 69 CrossRef CAS;
(e) J. Fujiwara and M. Nagata, Chem. Phys. Lett., 2015, 619, 180 CrossRef.
-
(a) A. P. de Silva, H. Q. N. Gunaratne, T. Gunnlaugsson, A. J. M. Huxley, C. P. McCoy, J. T. Rademacher and T. E. Rice, Chem. Rev., 1997, 97, 1515 CrossRef CAS PubMed;
(b) A. P. de Silva, T. S. Moody and G. D. Wright, Analyst, 2009, 134, 2385 RSC;
(c) T. D. James, K. R. A. S. Sandanayake and S. Shinkai, Nature, 1995, 374, 345 CrossRef CAS;
(d) T. D. James, K. R. A. S. Sandanayake, R. Iguchi and S. Shinkai, J. Am. Chem. Soc., 1995, 117, 8982 CrossRef CAS;
(e) S. Franzen, W. Ni and B. Wang, J. Phys. Chem. B, 2003, 107, 12942 CrossRef CAS;
(f) W. Ni, G. Kaur, G. Springsteen, B. Wang and S. Franzen, Bioorg. Chem., 2004, 32, 571 CrossRef CAS PubMed;
(g) G. Kaur, H. Fang, X. Gao, H. Li and B. Wang, Tetrahedron, 2006, 62, 2583 CrossRef CAS.
- K. Takagi, A. Mizuno, H. Yoshimura and M. Matsuoka, Dyes Pigm., 1990, 14, 203 CrossRef CAS.
- I. A. Janković, Z. V. Šaponjic, M. I. Čomor and J. M. Nedeljković, J. Phys. Chem. C, 2009, 113, 12645 Search PubMed.
- W. Yang, X. Gao, G. Springsteen and B. Wang, Tetrahedron, 2002, 43, 6339 CrossRef CAS.
- O. S. Fedorova, V. V. Orlovskaya, V. I. Maleev, Y. N. Belokon, T. F. Saveleva, Ch. V. Chang, Ch. L. Chen, R. Sh. Liu and R. N. Krasikova, Russ. Chem. Bull., 2014, 63, 1169 CrossRef CAS.
-
(a) A. Reitz, M. A. Avery, M. S. Verlander and M. Goodman, J. Org. Chem., 1981, 46, 4859 CrossRef CAS;
(b) K. D. Gardner and D. F. Wiemer, J. Org. Chem., 2016, 81, 1585 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7qm00211d |
|
| This journal is © the Partner Organisations 2017 |

 *,
Kosuke
Yamaji
and
Joji
Ohshita
*
*,
Kosuke
Yamaji
and
Joji
Ohshita
*