
Molecular conformation and packing: their critical roles in the emission performance of mechanochromic fluorescence materials
Can
Wang
 and
Zhen
Li
*
and
Zhen
Li
*
Department of Chemistry, Hubei Key Lab on Organic and Polymeric Opto-Electronic Materials, Wuhan University, Wuhan 430072, China. E-mail: lizhen@whu.edu.cn; lichemlab@163.com
First published on 7th June 2017
Abstract
Mechanochromic fluorescence (MCF) materials are a sort of smart material whose photophysical properties are sensitive to mechanical stimulation, such as photoluminescence color, fluorescence quantum yield and emission lifetime. Recently, an increasing number of studies have shown that these photophysical properties can be affected greatly by the molecular packing and conformation, enabling the rapid development of functional materials with mechanochromic fluorescence properties. In this review, we focus on MCF materials with distinct emission properties and various molecular arrangements, especially the inherent correlation between molecular packing modes and emissive behaviors. Many of the selected representative examples possess polymorphism, offering the possibility of exploring different emissions from the exact molecular packing in single crystals. Correspondingly, some remarks are made on the outlook for the next developments in MCF materials and the required thinking about the structure–packing–performance relationship.
 Can Wang | Can Wang received his Bachelor's degree (2012) in the Department of Chemistry at the Wuhan University (China). He is currently a PhD student in the group of Prof. Zhen Li. His research focuses on the design and synthesis of organic functional materials with specific properties in combination with the AIE characteristic, as well as the investigation of corresponding structure–property relationships. |
 Zhen Li | Zhen Li received his BSc and PhD degrees from Wuhan University (WHU) in China in 1997 and 2002, respectively, under the supervision of Prof. Jingui Qin. In 2003–2004, he worked in the Hongkong University of Science and Technology as a Research Associate in the group of Prof. Ben Zhong Tang. In 2010, he worked in Georgia Institute of Technology in the group of Prof. Seth Marder. In 2014, he worked in National University of Singapore as a visiting professor for one month. He has been a full professor at WHU since 2006, and his research interests are in the development of organic molecules and polymers with new structures and new functions for organic electronics and photonics. |
1. Introduction
The design and construction of organic opto-electronic materials have attracted great attention over the past decades.1–8 Although function depends heavily on the electronic nature of the molecular structure, the mode of packing and assembly also affect the performance of a material to a large degree.9–11 Actually, the investigation of the effect of the different molecular assemblies on a property has become a hot research topic in the field of organic opto-electronic materials, including organic field-effect transistor (OFET) devices12,13 and organic photovoltaic (OPV) cells.14–17 To clarify and understand the molecular packing modes in the solid state, many efforts have been made on the preparation and analysis of single crystals, with an attempt to figure out some design rules for the further optimization of material performance.18–22As is well known, fluorescence is very sensitive to subtle changes in luminogens of different status, such as temperature, concentration and viscosity if in solution, in addition to the amorphous/crystalline state as an aggregated form.23–26 For example, as reported by Yamaguchi, hexyl-substituted 1 and ethyl-substituted 2 have the same π-conjugated skeleton but different alkyl substituents (Fig. 1).27 Both exhibit the same high fluorescence quantum yield of 92% in diluted solution; however, in the crystalline state, the quantum yield of 1 was measured to be 64%, while the quantum yield of 2 was lower at 47%. This could be well explained by their different molecular packing modes in the crystalline state. In the crystal of 1, all the π-conjugated aromatic cores are in a parallel arrangement, separated by the long hexyl alkyl chain. Whereas in the crystal of 2, the aromatic cores pack in a herringbone mode because of the shorter ethyl alkyl side chain. The differences in the packing mode cause their different emission efficiencies. Furthermore, in other cases, the different packing modes could even lead to different fluorescent spectra, besides the different efficiencies demonstrated in 1 and 2. Thus, by combining the sensitive fluorescence and the analysis of the single crystal, some important information could be summarized to understand the influence of massive molecular aggregates on their corresponding properties.
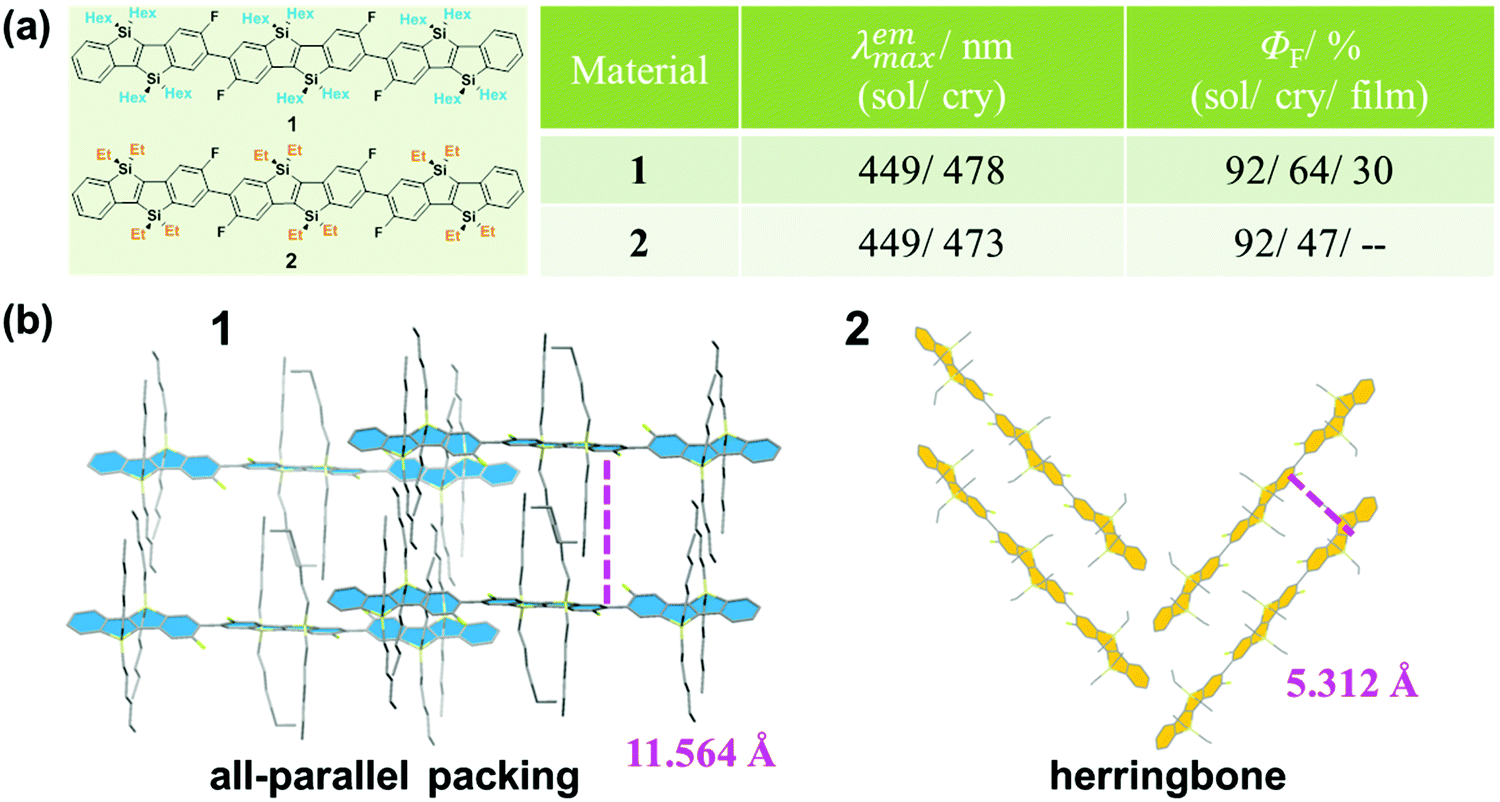 | ||
| Fig. 1 (a) Chemical structures and optical properties of 1 and 2. (b) The packing modes of 1 and 2 in the crystal. | ||
Generally, for conjugated molecules, the molecular packing originates from the weak force balance of π–π interactions and van der Waals interactions.28,29 For some kinds of organic luminogens, named mechanochromic fluorescence (MCF) materials, the molecular packing pattern could be altered reversibly upon external stimuli (e.g. force, heat or solvent), accompanied by a change in fluorescence emission, such as emission color or intensity (Fig. 2).30–32 Research on these stimuli-responsive materials has attracted much attention, because of their great potential in data storage, anti-counterfeiting, sensors and display.33–36 More importantly, investigations of the phase transition during the stimuli-responsive process provide opportunities to figure out how the molecular packing mode conveniently affects the emissive behavior in the aggregated state.37
 | ||
| Fig. 2 Schematic representation of stimuli-responsive fluorescence materials. | ||
In this review, we focus on MCF materials with distinct emission properties as well as different molecular arrangements, especially the inherent correlation between molecular packing modes and emissive behaviors. Thanks to the great efforts of scientists, many of the selected representative examples possess polymorphism, offering the possibility of exploring the inherent reason for the different emissions from the exact molecular packing in single crystals. Correspondingly, some remarks are made on the outlook for the next developments in MCF materials and the required thinking about the structure–packing–performance relationship.
2. Mechanochromic fluorescence materials
2.1 MCF materials with planar π-extended moiety
To the best of our knowledge, the first solid-state organic MCF compound, 4-tert-butyl-1-(4′-dimethylaminobenzylideneamino)pyridinium perchlorate, was reported by Gawinecki et al. in 1993, whose yellow-green luminescence could be quenched by compression.38 However, once the tert-butyl group was replaced by a methyl group, this response disappeared. It was a pity that no exact reason could be explored, without data on their single crystals.Generally, molecules with a planar π-extended structure are liable to form crystals with a relatively orderly molecular arrangement, because of the presence of strong π–π interactions.39,40 When this stable packing mode was disorganized by the stimulus of an external force, the emission property would change. In 2012, Tian et al.41 reported an anthracene derivative with two substituted pyridine units, compound 3, the as-prepared powder of which showed a largely red-shifted emission from green (λmax = 528 nm) to yellow (λmax = 561 nm) upon grinding. Interestingly, under a compressing pressure from 0 to 8 GPa, a more noticeable change was observed from green (λmax = 528 nm) to red (λmax = 652 nm). Fortunately, three crystalline polymorphs of 3 were obtained with corresponding green, yellow and red emissions (Fig. 3), which could be named 3-G, 3-Y and 3-R. In 3-G, molecules adopted a stacking mode of J-type aggregation along the y axis with almost no π–π interaction, since the anthracene planes of two adjacent molecules did not overlap (Fig. 3c). Thus, 3-G exhibited a green emission with λmax = 527 nm, mainly dominated by the basic electronic property of its single molecules like that in diluted THF solution at 77 K. In the case of 3-Y, the molecules adopted a stacking mode of H-type aggregation along the x axis, and the adjacent anthracene planes overlapped with each other by about 40% with a vertical distance of approximately 3.65 Å between them, disclosing the presence of weak π–π interactions between the anthracene planes. Accordingly, 3-Y exhibited an orange emission with λmax = 579 nm. As to 3-R, the adjacent anthracene planes overlapped almost in a face-to-face stack to form a dimer with a closer vertical distance of ∼3.52 Å, directly resulting in red emission (λmax = 618 nm).
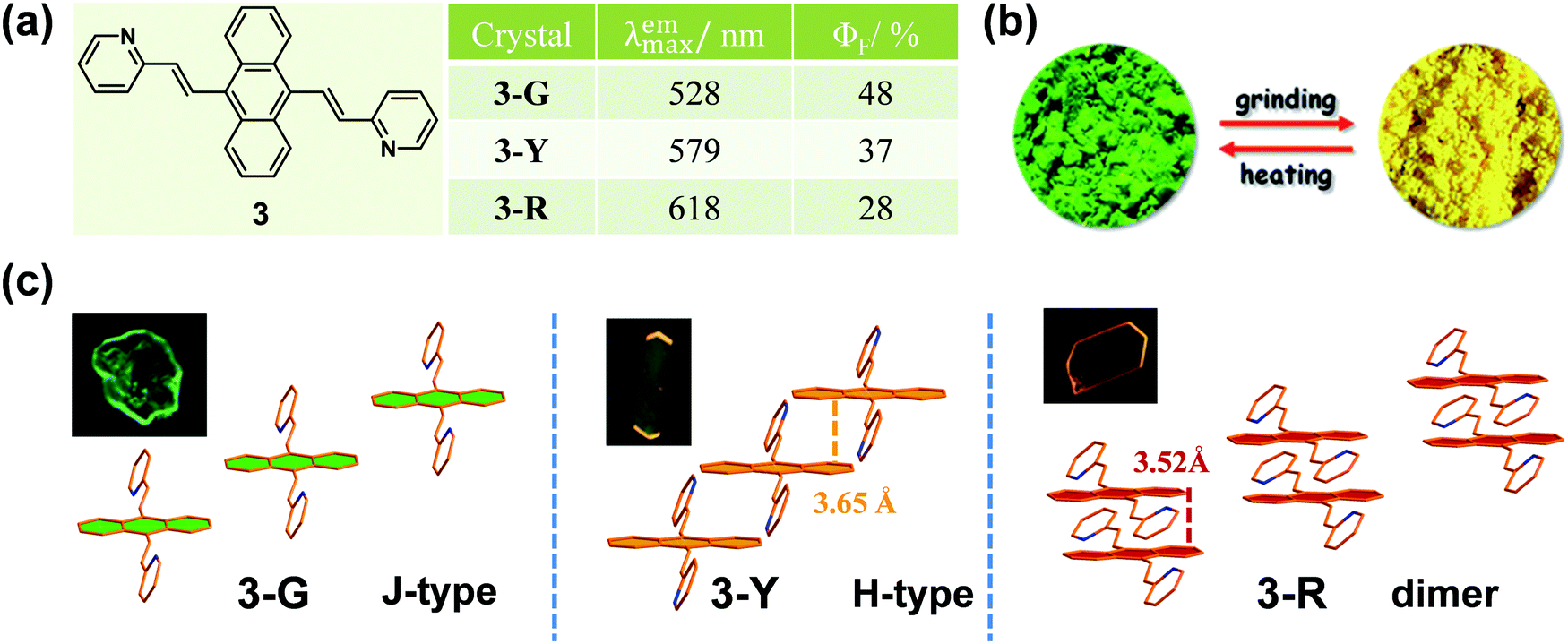 | ||
| Fig. 3 (a) Chemical structures and optical properties of 3. (b) Fluorescent photographs of the ground powder and the heated powder under UV light (365 nm). (c) Stacking modes of the anthracene planes in adjacent molecules in three single crystals. The inset depicts the fluorescence images of crystals under a fluorescence microscope. | ||
The powder X-ray diffraction (PXRD) pattern of as-prepared 3 powder agreed well with that of crystal 3-G, suggesting that the initial samples adopted the same molecular packing mode of J-type aggregation. The ground sample exhibited weak diffraction peaks, indicating that the initial aggregation state was changed by grinding. Furthermore, the orange emission of the ground sample (λmax = 561 nm) was similar to the emission of 3-Y (λmax = 579 nm), which showed red-shifted fluorescence and blue-shifted absorption compared to those of unground sample. These results indicated that the H-type aggregation, similar to the molecular packing in crystal 3-Y, possibly occurred during the grinding process. Upon higher compressing pressure, molecules would further aggregate in more tightly stacked dimers with strong π–π interactions, as in the case of crystal 3-R. These results suggested that the molecular packing changes in the aggregation state upon grinding or under external pressure led to enhanced intermolecular π–π interactions and thus induced the MCF property.
In 2010, Fraser et al. reported the morphology-dependent fluorescence and reversible mechanochromic luminescence of a difluoroboron complex (4) in the solid state (Fig. 4a).42 Later, in 2013, Reddy et al. gained a further insight into the origin of the MCF behavior of 4 by precisely quantifying the mechanical properties of crystals in different forms, and attributed the recoverable MCF property to the presence of slip planes in the molecular packing mode.43 As powerful proofs, two crystalline polymorphs of 4 with cyan (4-Cy, λmax = 470 nm) and green (4-G, λmax = 505 nm) emissions were analyzed in detail. In 4-Cy, molecules adopted parallel packing along the z axis and a head-to-head arrangement in the adjacent 2D plane. As a result, the tert-Bu and -OMe groups were packed together, thus producing two potential slip planes in the structure. The vertical distance of the adjacent layers was measured to be 3.35 Å; however, almost no π–π overlaps were observed, indicating very weak π–π interactions. Due to the steric hindrance between adjacent layers, the whole molecule was not co-planar, but it had torsion angles of φA–B = 7.9° and φB–C = 4.1°, well accounting for its cyan emission. However, in 4-G, the molecules adopted a head-to-tail arrangement in a one-dimensional (1D) chain, and the chains in adjacent layers were packed in an antiparallel arrangement. Thus, the tert-Bu groups from one layer packed on the top of the -OMe groups in the next layer, with a vertical distance between adjacent layers of 3.48 Å and without obvious π–π interactions. However, the whole molecule was perfectly co-planar with φA–B = 0° and φB–C = 0°, indicating better intramolecular conjugation than that in 4-Cy, accompanied by a red-shifted green emission.
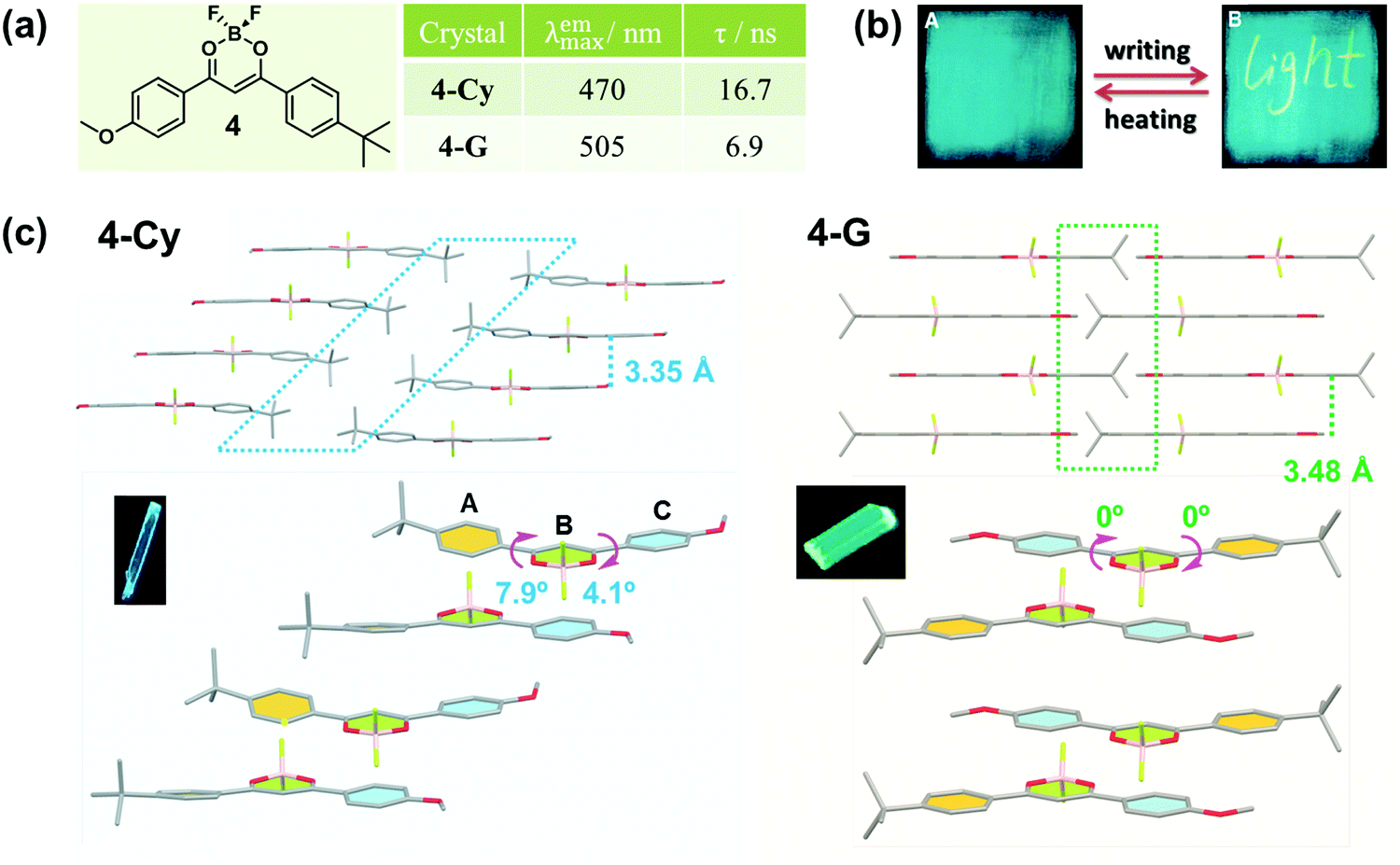 | ||
| Fig. 4 (a) Chemical structures and optical properties of 4. (b) Fluorescent photographs of the film upon thermal annealing and writing. (c) Stacking modes of adjacent molecules in a single crystal. The inset depicts the fluorescence images of crystals under a fluorescence microscope. | ||
In some cases, distinct luminescent responses to different mechanical stimuli, such as compressing, grinding and smashing, could be combined into one organic crystal, which provides in-depth insights into the mechanism and structure–property relationship of MCF materials. In 2013, a tetrathiazolylthiophene compound (5) was reported by Yamaguchi et al., with a distinct luminescent response to mechanical grinding and hydrostatic pressure.44 The crystal of 5 (5-Y) exhibited a yellow emission with a λmax of 556 nm. Interestingly, by grinding the crystals, the emission color was largely blue-shifted to green (5-G) (λmax = 490 nm) with a slight enhancement in the quantum yield from 2% to 5%. In the crystal, as shown in Fig. 5c, the α-positioned thiazole rings A/D and the central thiophene ring adopted an almost coplanar conformation, forming a π-skeleton, while the β-positioned thiazole rings B/C adopted twisted conformations with large dihedral angles of 69.0 and 78.6°, respectively. There were relatively strong C–H⋯N hydrogen bonds in the adjacent molecules, which promoted two π-conjugation skeletons packing in a face-to-face arrangement, with weak π–π stacking interactions (d = 3.96 Å), thus inducing a 3D hydrogen-bond network (Fig. 5d). Upon grinding, the weak 3D hydrogen-bond network could be destroyed and the face-to-face dimeric structure would disaggregate, which eliminated the possibility of excimers, hence leading to the blue-shifted luminescence of 5-Cy. Upon compression, 5-Y showed a continuously red-shifted emission to 609 nm under increasing hydrostatic pressure up to 3.2 GPa (Fig. 5b). As shown in Fig. 5c, the single-crystal structure of 5-R (5-Y under 2.8 GPa pressure) was obtained (red line). There were no 3D hydrogen-bond networks and the two skeletons overlapped with each other to a greater extent with a shortened distance of 3.69 Å between them. These results confirmed that a highly overlapped excimer was formed, which contributed to its red-shifted emission under hydrostatic pressure.
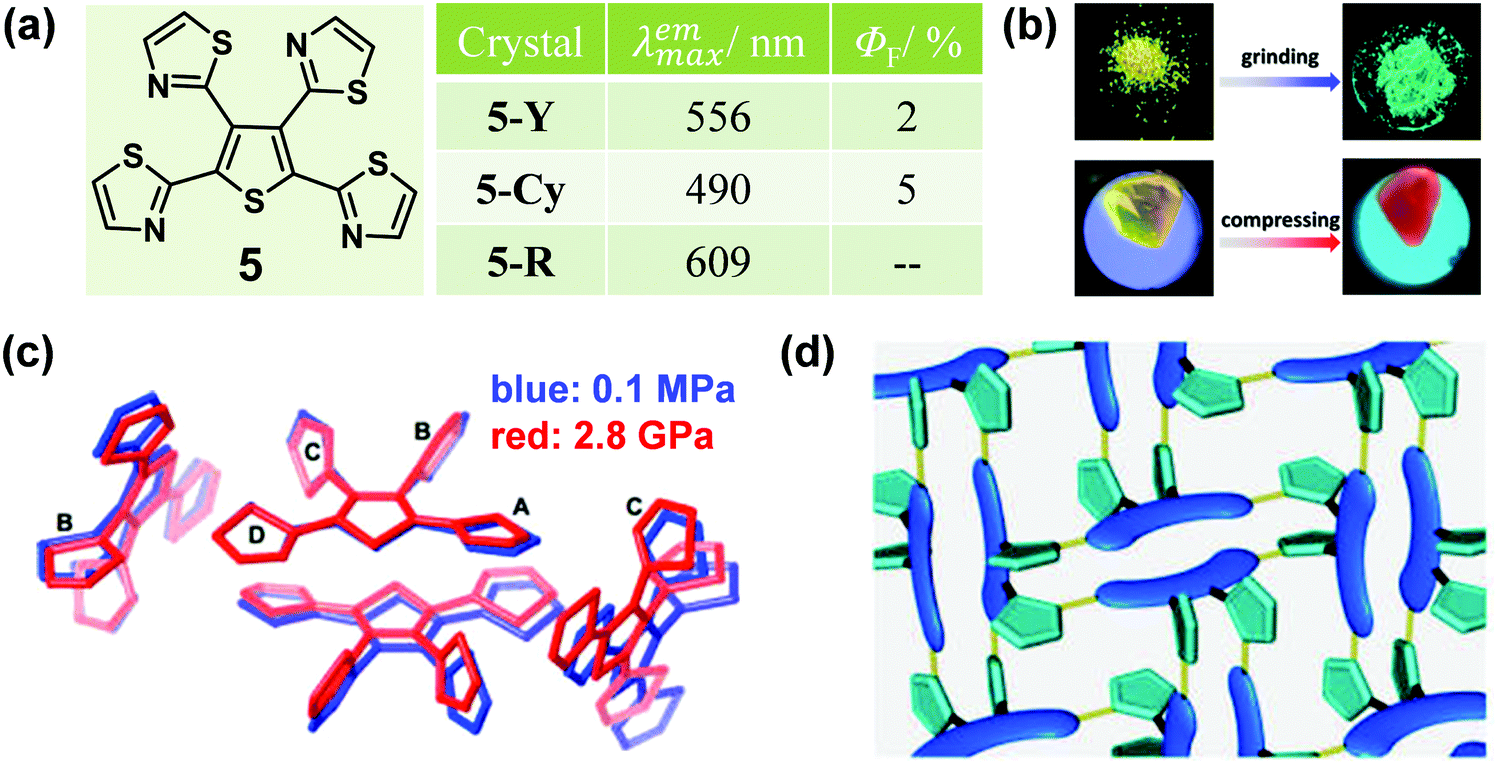 | ||
| Fig. 5 (a) Chemical structures and optical properties of 5. (b) The distinct luminescent response of 5Y upon grinding and compressing. (c) Crystal packing structures of 5Y (blue) and 5-R (red). (d) Representation of crystal packing structure of 5-Y. Blue sausages, green pentagons, and yellow cylinders demonstrate fluorophores, twisted thiazole rings B and C, and C–H⋯N hydrogen bonds between the thiazole rings, respectively. | ||
In 2015, Zhang et al. also reported a boron diketonate crystal (6-O) exhibiting distinct luminescent responses to different mechanical stimuli.45 As shown in Fig. 6b, 6-O exhibited a blue-shifted emission from 585 nm to 565 nm upon smashing and a significantly red-shifted emission to 660 nm upon compressing, and the smashing-generated 6-G could be red-shifted to 605 nm upon grinding. Two crystals with orange (6-O) and red (6-R) emission were obtained. As shown in Fig. 6c, two molecules approached in antiparallel mode and formed a dimer with a distance of 3.5 Å for 6-O and 3.6 Å for 6-R in the crystal, and the packing patterns of the dimers were distinct. In 6-O, the adjacent dimers packed in a vertical mode, giving rise to a 2D layer structure with weak non-covalent interactions. In sharp contrast, the dimers in 6-R were linearly stacked in a parallel mode, and the contact distance between the dimers was 3.3 Å, which indicated strongly continuous π–π stacking interactions in the molecular layer, giving rise to its red emission. The blue-shifted emission of 6-O upon smashing was likely attributed to the increased distance between the molecules in the dimer, and the molecular perturbations were supposed to take place only on the crack surface, as indicated by the powder XRD analysis of 6-G. The significantly red-shifted emission of 6-O upon compression was mainly ascribed to the shortened distance between the molecules in the dimers. By grinding 6-G, the emission could be changed to orange-red (λmax = 605 nm), which was possibly ascribed to the formation of π-stacked dimers, similar to that of 6-R.
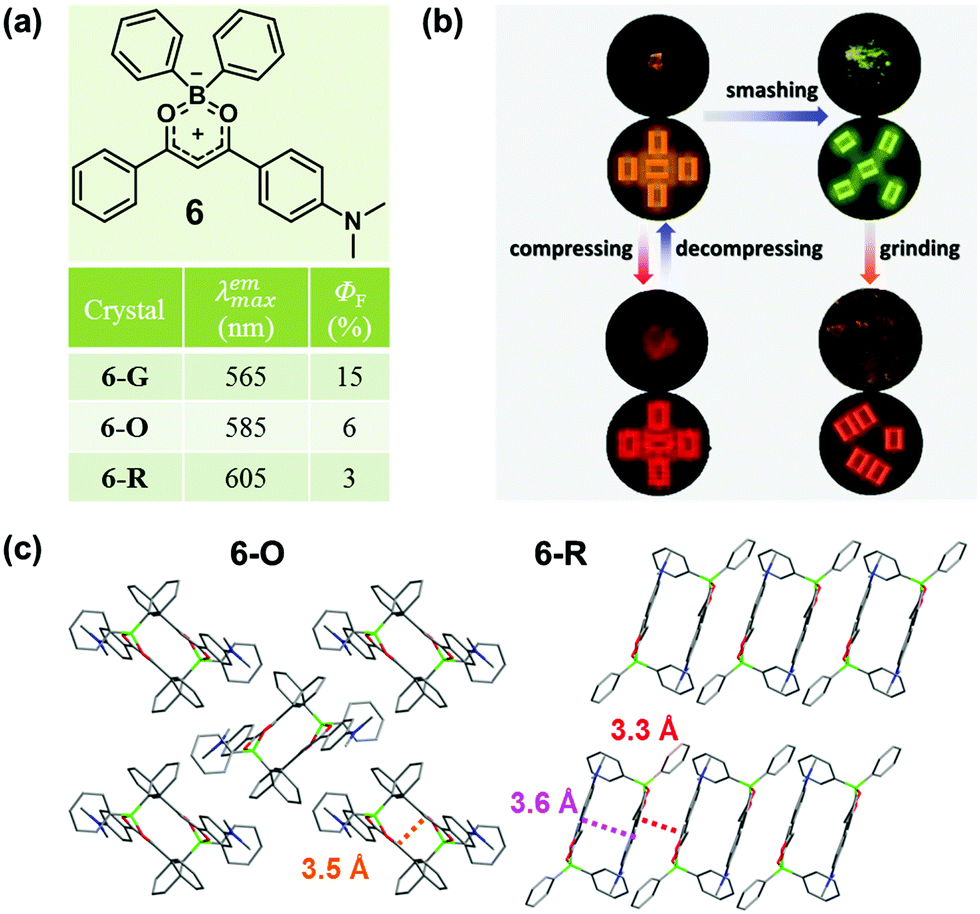 | ||
| Fig. 6 (a) Chemical structures and optical properties of 6. (b) Photographic images of 6O crystal before and after smashing, 6G before and after grinding, and 6O crystal upon compression and decompression. (c) Crystal packing structures of 6-Y and 6-R. | ||
As discussed above, the molecular packing originates from the subtle force balance of π–π interactions and van der Waals interactions.46 In 2014, Wang et al. reported a new class of 2,7-diphenylfluorenone derivatives that exhibited an AIE property with high fluorescence quantum yields in the solid state.47 Among these, two compounds (7 and 8), displayed reversible stimuli-responsive luminescence switching behavior. Compound 7 could transform between red (7-R, λmax = 601 nm) and yellow (7-Y, λmax = 551 nm) crystals, under the stimuli of temperature, pressure, or solvent vapor. In 7-R, as shown in Fig. 7c, two molecules adopted a strong π–π stacking mode with a contact distance of 3.47 Å, inducing its red-shifted emission. However, for 7-Y, no π–π interactions were observed, and the molecules were linked by hydrogen bonds with a length of 2.50 Å, accompanied by a yellow emission. These results demonstrated that the stimuli-responsive reversible phase transformations of 7 involved a structural transition with an alteration in the π–π stacking interactions. Similarly, compound 8 also exhibited luminescence response to stimuli, with the emission switching between orange (8-O, 571 nm) and yellow (8-Y, 557 nm). In 8-O, molecules were packed in pairs with strong π–π stacking interactions, in which, the adjacent molecular planes overlapped by about 40%, and the distance between them was ∼3.61 Å. This π–π stacking-bound excimer (dimer) directly generated the orange emission of 8-O upon excitation. As to 8-Y, molecules also packed in pairs, but the aromatic frameworks of the two adjacent molecules barely overlapped; thus, the π–π interactions were very weak. Therefore, the yellow luminescence was attributed to the fluorescence from the single molecule, like that in diluted solutions.
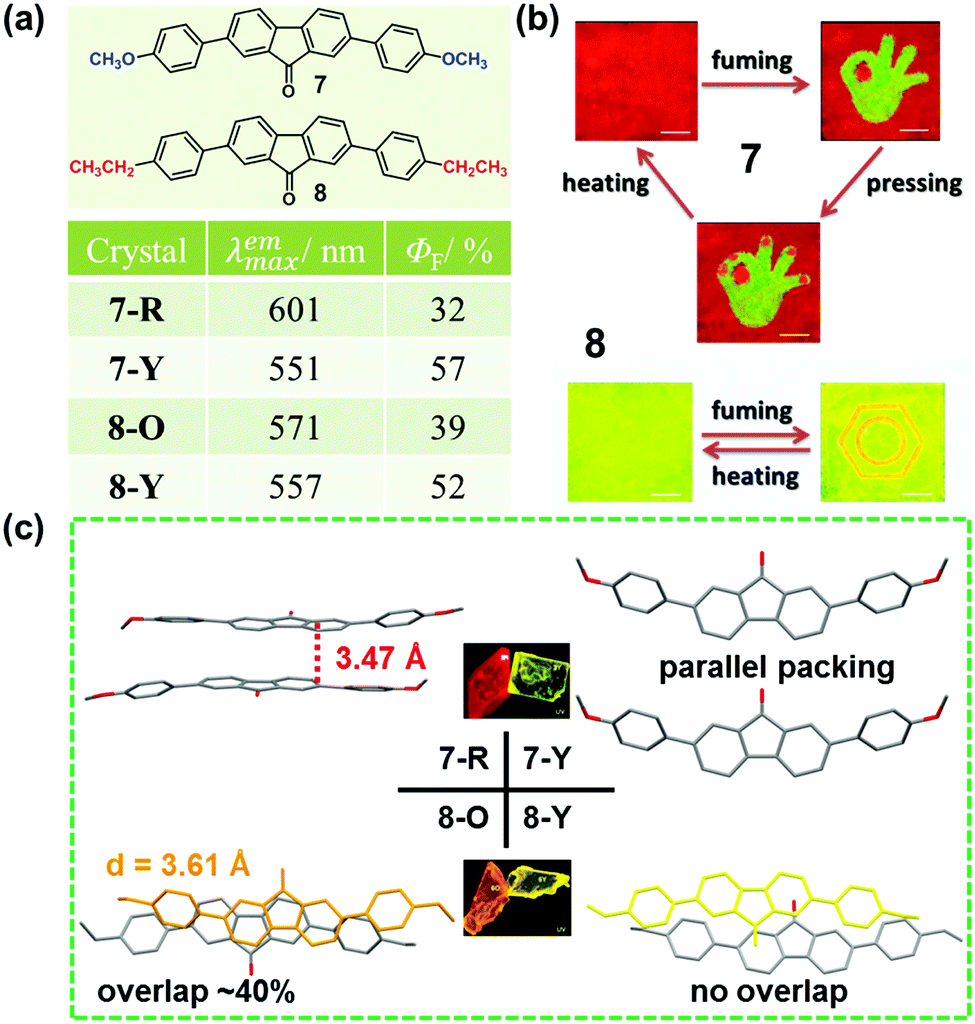 | ||
| Fig. 7 (a) Chemical structures and optical properties of 7 and 8. (b) Fluorescent images of compounds 7 and 8 upon fuming, pressing and heating. (c) Stacking modes of adjacent molecules in a single crystal. The inset depicts the fluorescence images of crystals under a fluorescence microscope. | ||
Some organic chiral π molecules exhibited solvent-dependent molecular conformation or packing, endowing the possibility of crystalline polymorphs and their corresponding dynamic structural interconversion, upon mechanical grinding or/and solvent vapor-fuming. In 2015, Enomoto et al. reported the reversible mechanofluorochromism of aminobenzopyranoxanthene (9).48 Its cis-form (9-NR) showed switchable near-IR (λmax = 758 nm) emission from the blue emission of its trans-form (9-B) (λmax = 450 nm). As shown in Fig. 8c, in 9-NR, the molecules adopted a distinct slip-stacked dimer structure with a considerable offset. The vertical distance between the xanthene planes was measured to be 3.33 Å, and the strong π–π interactions between adjacent molecules thus induced the red-shifted near-IR luminescence (λmax = 758 nm). In 9-B, the dihedral angle of the two xanthene rings of the R,R and S,S forms was 66.0°, without the presence of π–π interactions. Therefore, 9-B exhibited blue luminescence upon excitation at 365 nm. These results demonstrated that the near-IR fluorescence should be derived from the dimer configuration with close packing of the two xanthene rings, while the blue emission was from the monomer like that in solution (λmax = 430 nm).
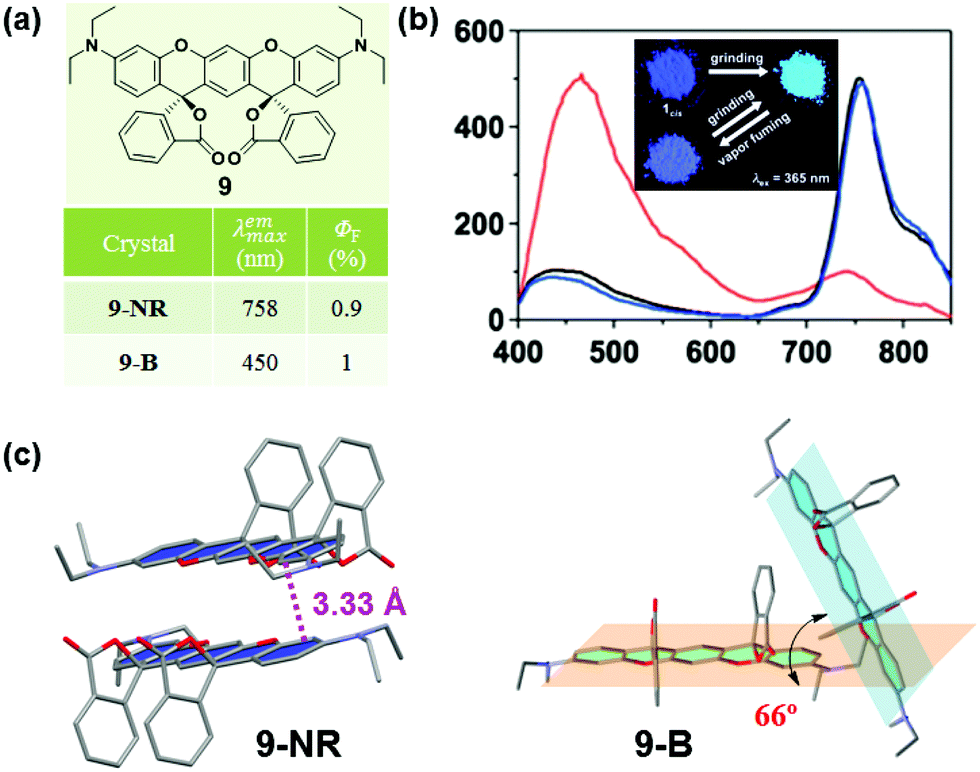 | ||
| Fig. 8 (a) Chemical structures and optical properties of 9. (b) Solid-state fluorescence spectra of 9-NR crystal (black line), ground powder of 9-NR (red line) and powder after fuming with CH2Cl2 (blue line). The inset depicts the fluorescent images of 9-NR upon grinding and vapour fuming. (c) Stacking modes of adjacent molecules in the single crystal. | ||
An effective method of adjusting the molecular interactions in the crystal state is the introduction of certain substituted groups to π molecules, such as halogen atoms and cyano groups.49–53 In 2014, Xue and Lu et al. synthesized two phenothiazine-based benzoxazole derivatives (10 and 11, Fig. 9a), and investigated the effect of the bromine atom on the molecular packing and the related MCF property.54 Upon grinding, 10 showed a moderately red-shifted emission (from yellow to orange), while 11 exhibited a significantly red-shifted emission (from green to orange). In the crystal of 10, the molecules adopted a typical herringbone packing mode with 1D π–π stacking interactions, with a distance between two adjacent molecules of 3.53 Å and a sliding angle of 34.4°. This result clearly demonstrated the formation of a J-aggregation, leading to the yellow emission of the crystal. The emission color was close to the orange emission in the ground sample, in which molecules were stacked in a smaller slippage packing mode. Whereas, in the crystal of 11, the molecules adopted a parallel arrangement pattern with a larger distance of 3.65 Å, and a larger sliding angle (60.48°) than those of 10, which induced a relative blue-shifted emission (green) compared to that of 10 (yellow) in the crystalline state. Upon grinding, the resultant amorphous sample exhibited orange emission.
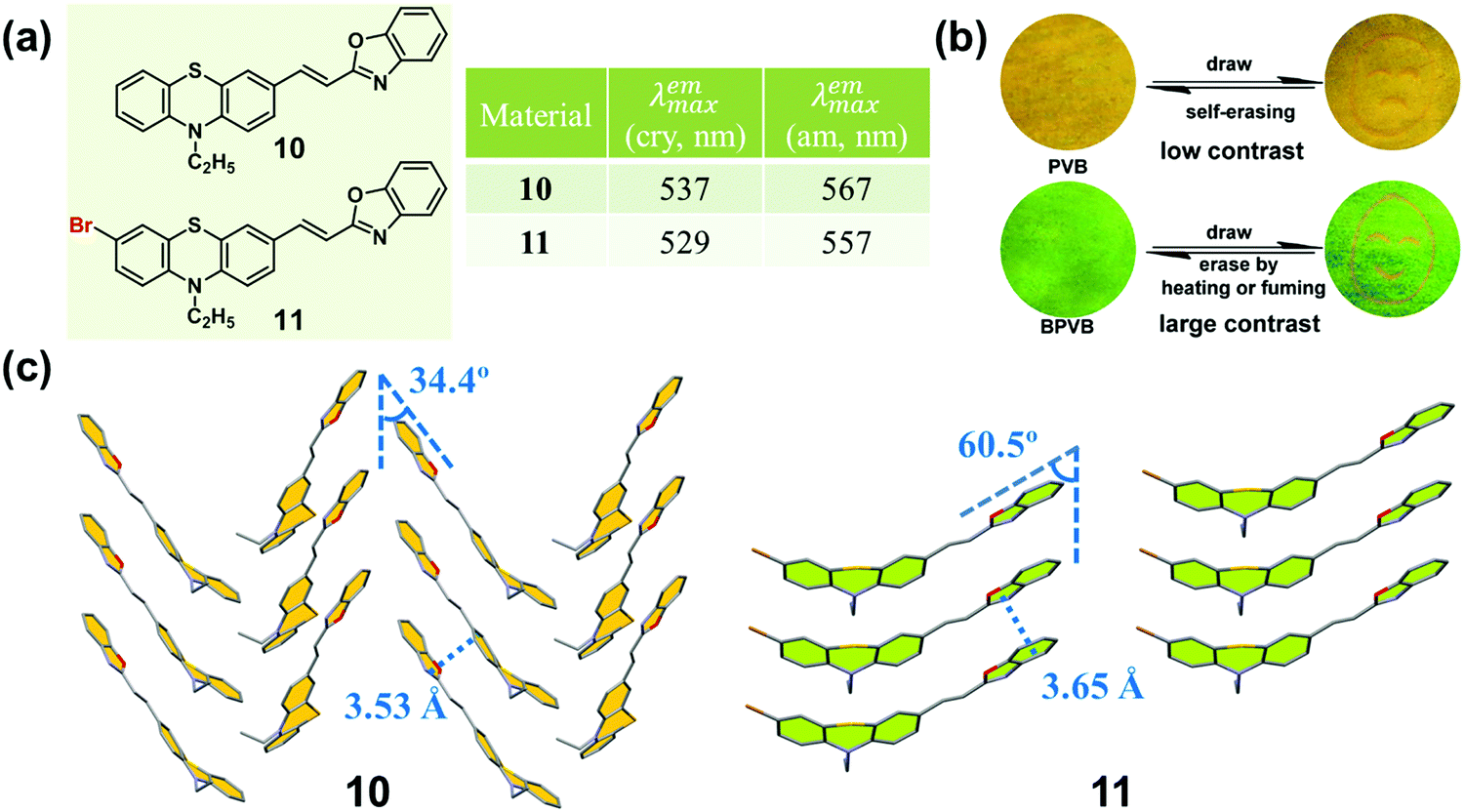 | ||
| Fig. 9 (a) Chemical structures and optical properties of 10 and 11. (b) Photographic images of 10 and 11 films in response to grinding, fuming and heating under UV light (365 nm). (c) Molecular stacking modes in single crystals. | ||
In 2010, Park et al. reported a highly luminescent cyanostilbene-based compound (12) with slip-stacking of molecular sheets, which exhibited green-blue fluorescence switching in response to pressure, temperature and solvent vapor.55 Single-crystal 12-G emitted green light (λmax = 533 nm), while a blue emissive sample (12-B, 458 nm) could be obtained by heating. As shown in Fig. 10c, in 12-G, molecules formed planar “molecular sheets”, with the aid of multiple hydrogen bonds (C–H⋯N, C–H⋯O) combined with the appropriate length of alkyl substituents. The molecular sheets were arranged in slip-stacks along the long molecular axis with a pitch angle of 26.6° and a vertical distance of 3.7 Å between the adjacent molecular sheets. This result indicated the formation of J-aggregation in 12-G. Although the single-crystal structure of 12-B could not be obtained, the information about the specific stacking changes could be monitored by small-angle X-ray scattering (SAXS), from which a different slip-stack along the shorter axis was inferred with a pitch angle of 62.8°. These molecular packing modes provided different exciton and excimer coupling in crystals. The 12-G crystal showed rather weak excitonic coupling; however, excimer formation was favored by the pronounced overlap of the π-systems. Whereas in 12-B, excimer formation was diminished, but the excitonic interaction substantially increased. The driving force for the phase transition was provided by the local dipoles, as introduced through the cyano group.
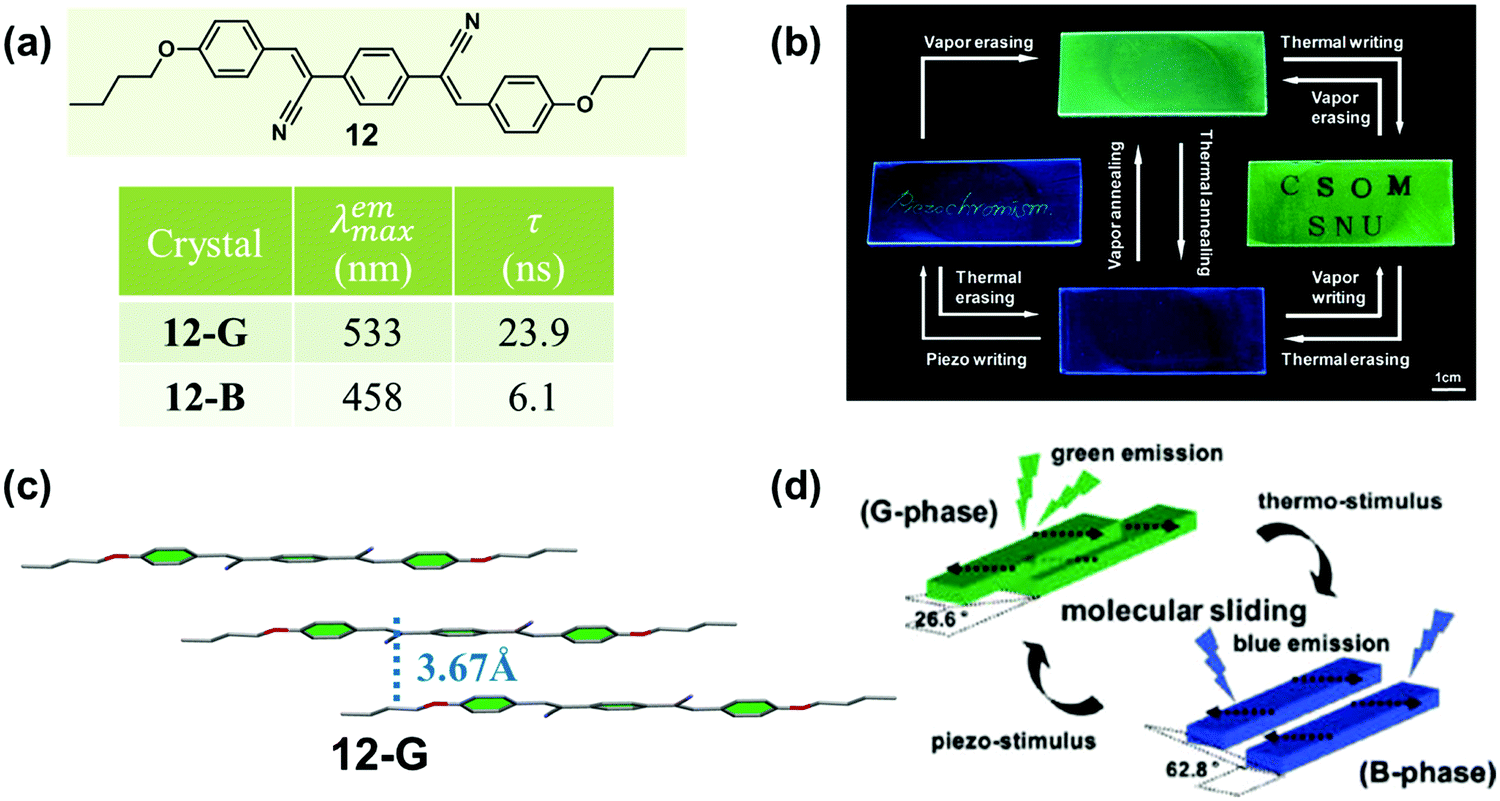 | ||
| Fig. 10 (a) Chemical structures and optical properties of 12. (b) Photos of the luminescence writing/erasing cycle in 12/PMMA film. (c) Molecular stacking modes in a 12-G crystal. (d) Illustration of two different modes of slip-stacking in 12-G and 12-B molecular sheets. | ||
According to Kitaigorodskii's close packing principle56 and Etter's hydrogen-bond rule,28 π–π interactions and hydrogen bonding interactions are the two critical factors in designing tunable-state luminescent materials. In some cases, the intermolecular hydrogen bonds not only exhibited a great influence on the molecular packing mode, but also promoted the intramolecular charge transfer (ICT) effect of the molecules, thus resulting in a luminescence bathochromic shift.57 In 2016, Wang et al. designed a coumarin hydrazine compound (13), which could form intermolecular hydrogen bonding in the solid state with concentration-dependent emission spectra and color change.58 Upon grinding, 13 displayed a red-shifted luminescence in the solid state (Fig. 11b). Crystal structures showed that two molecules formed a head-to-tail dimer through two intermolecular N–H⋯O hydrogen bonds, and the dimers constructed a herringbone arrangement. This stable hydrogen bond-directed structure promoted ICT from the electron-donating group (N,N-diethylamino) to the hydrazine moiety, leading to bathochromic shifted and enhanced luminescence in the condensed state. Upon grinding, the orderly packing dimers were converted to a slightly disordered state with a probably more intensive ICT effect, thus inducing a red-shifted emission from bright yellow (λmax = 545 nm) to orange-red (λmax = 615 nm), accompanied by an enhanced quantum yield (from 0.14 to 0.47).
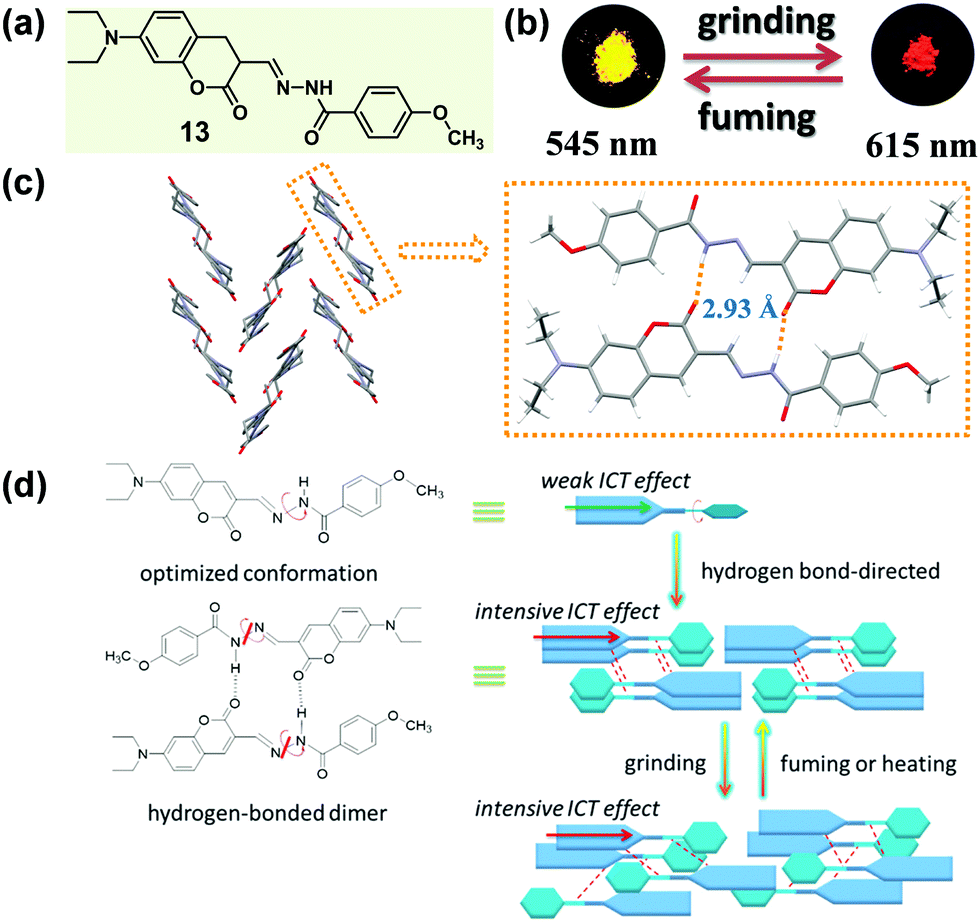 | ||
| Fig. 11 (a) Chemical structure of 13. (b) Photographic images of 13 in response to grinding and fuming under UV light (365 nm). (c) Molecular packing pattern of hydrogen-bonded dimers in the crystal. (d) Proposed mechanisms of MCF properties of 13. | ||
All the above examples are luminogens with a planar π-structure, in which the multiple-color luminescence is almost always derived from an alteration of the π–π stacking interactions in different aggregation states. The stimuli of mechanical force or hydrostatic pressure usually make the molecules pack in a more condensed phase, thus resulting in strong π–π stacking interactions, which generally leads to a red-shifted and weakened emission. However, recently, a kind of MCF material constructed with a twisted non-planar π-extended moiety has attracted increasing attention, most of which also simultaneously possessed the AIE feature.34,35 Reasonably, the inherent mechanism for their MCF property should be very different from that of the planar luminogens discussed above.
2.2 MCF materials with a twisted non-planar π-extended moiety
Chi and Xu et al. have successfully obtained a variety of MCF molecules constructed with twisted triphenylethylene or tetraphenylethylene moieties, and proposed a possible mechanism for the MCF phenomenon.33,59–63 In 2011, they reported a molecule constructed with triphenylethylene and tetraphenylethylene moieties (14), which exhibited a reversible change in emission upon grinding and annealing (or fuming).64 As shown in Fig. 12b, under illumination at 365 nm, the as-prepared sample of 14 emitted a blue fluorescence (λmax = 454 nm), whereas the ground sample gave out a strong green emission (λmax = 482 nm) with a longer fluorescence lift time (from 1.24 to 1.90 ns). The crystal structure of 14 (Fig. 12c) exhibited a highly twisted molecular conformation, in comparison with the ideal isolated free molecules, so there should be a high twist stress existing for the molecules in the crystalline state. Such a twist stress was supposed to be released upon the external force stimuli, which probably induced the disintegration of the long-range ordered crystal structure. Owing to the twisted conformation, the coplanar π–π stacking interaction became impossible. The molecules were packed via weak C–H⋯π interactions in a crystal cell, disclosing a low lattice energy. The twisted conformation and weak intermolecular interaction generated loose molecular packing and cavities in the crystalline structure, making the crystal readily destroyable by external pressure. The planarization of the twisted molecular conformation was accompanied by the destruction of the crystalline structure due to the release of the twist stress, which led to an increase in degree of molecular conjugation, thus resulting in a red-shifted fluorescence color change from 454 to 482 nm. According to this proposed mechanism, the MCF property might be a general characteristic of crystalline AIE materials.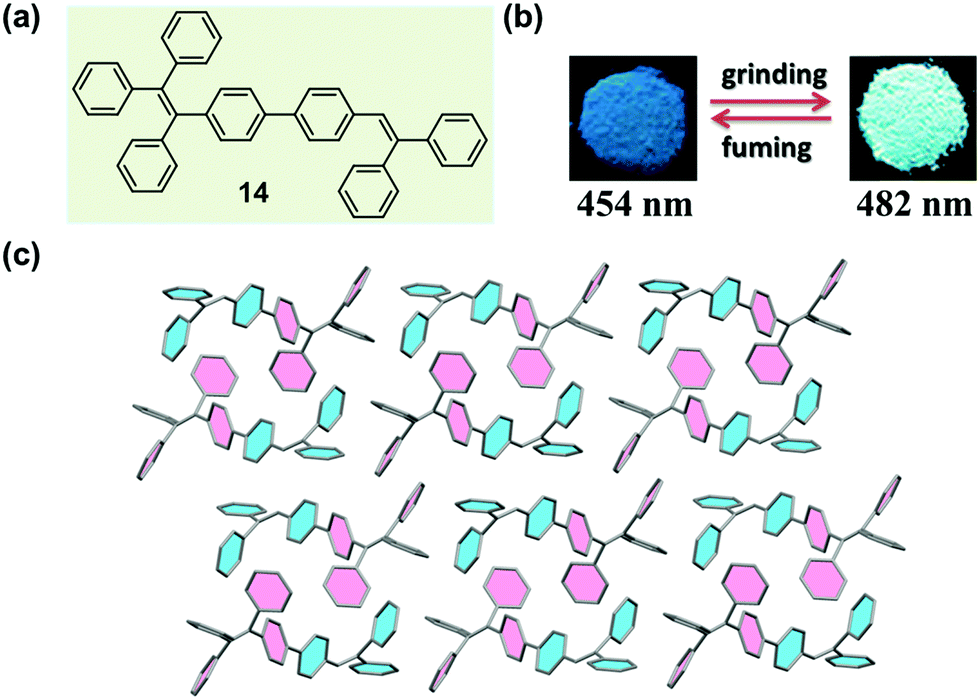 | ||
| Fig. 12 (a) Chemical structure of 14. (b) Photographic images of 14 in response to grinding and fuming under UV light (365 nm). (c) Molecular packing pattern in the crystal. | ||
Usually, the introduction of a twisted unit (tetraphenylethylene or triphenylethylene, typical AIE units) to a planar structural unit could endow the resultant molecules with an AIE characteristic, and significantly tune the molecular packing nature in the solid state. In 2015, Misra et al. reported a molecule (15) constructed with pyrenoimidazole and tetraphenylethylene units (Fig. 13a).65 Apart from the AIE characteristic, 15 also exhibited a distinct mechanochromic property in the solid state, which showed blue light emission (λmax = 461 nm) in the pristine form and yellowish green emission (λmax = 499 nm) after being ground. Generally, fluorophores containing a pyrene unit were known for their planar conformation and extensive π–π stacking interactions, but in the crystal of 15, the molecules adopted a twisted conformation due to the propeller-shaped TPE unit. This twisted conformation suppressed the π–π stacking effect between pyrene units, and, alternatively, a dimeric framework in a head-to-tail mode was formed via weak C–H⋯π interactions. Furthermore, such dimers were connected to each other in a 2D staircase-shaped framework via weak C–H⋯π interactions. Similar to the case of 14 discussed above, a loose molecular packing was generated, and the crystal was easily destroyed under external pressure. The solid-state absorption spectrum demonstrated a bathochromic shift from 417 to 446 nm upon grinding, indicating the presence of enhanced conjugation and the planarization of molecular conformation during the grinding process.
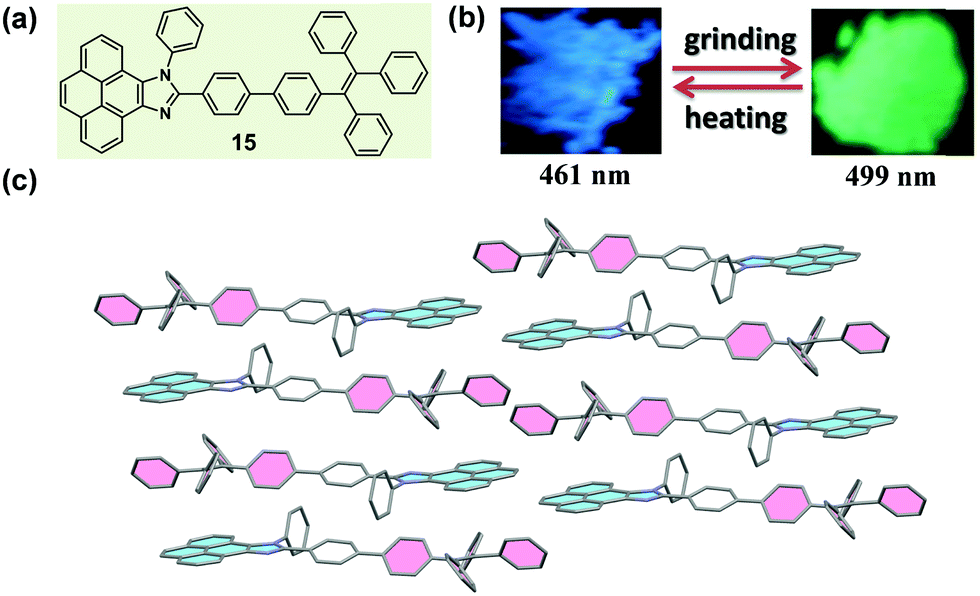 | ||
| Fig. 13 (a) Chemical structure of 15. (b) Photographic images of 15 in response to grinding and heating under UV light (365 nm). (c) Molecular packing pattern in the crystal. | ||
In some cases of MCF materials, the mechano-stimuli (like a grinding force) could not only destroy the crystallinity of crystalline materials but also have a significant effect on the amorphous-to-crystalline transition of amorphous materials. In 2014, Tao et al. reported a sort of tetrasubstituted ethylene (16), which exhibited a typically mechanochrismatic response to mechanical force.66 More interestingly, an abnormal phenomenon was observed, that the grinding process could accelerate the amorphous-to-crystalline transition upon thermal annealing.67 That is, only when an amorphous material underwent the mechano-stimulus could it crystallize by thermal annealing. As shown in the top image of Fig. 14b, when the crystalline sample of 16 was ground with a pestle or sheared with a spatula, its fluorescence switched simultaneously from an entirely blue emission (λmax = 459 nm) to a green emission (λmax = 517 nm). By directly quenching the melted compound, a pristine amorphous sample was also prepared with a green emission (λmax = 530 nm). In the crystal, the molecules adopted a highly twisted conformation similar to those reported for TPE derivatives. The large torsion shortened the effective conjugation length of the molecules, leading to its shorter-wavelength emission. Moreover, this highly twisted conformation hampered the close intermolecular π–π stacking and caused the loose molecular packing. As discussed above, such kinds of crystal had a low lattice energy, allowing them to be easily destroyed by external force. The contrast test of the grinding effect on the reversibility is depicted in the bottom image of Fig. 14b. After half of the sample was ground, the whole sample was heated to 130 °C for 1 h, but only the ground part was changed into blue-emissive crystallites. This result demonstrated that the thermal annealing-induced crystallization could not be realized without mechanical treatment. The solid-state absorption spectra revealed that after the grinding process, the absorption edge of the pristine amorphous sample was hypochromatically shifted from 490 to 475 nm; meanwhile, the emission peak was also hypochromatically shifted from 530 to 517 nm. The results indicated that the effective conjugation of the molecules in the pristine sample was even larger than that in the ground samples. The grinding process might alter their conformation to become more twisted, becoming similar to the conformation in crystals, although they were still in an amorphous state. This reduction in conformational difference would facilitate the rearrangement of the molecules into a crystalline form, when treated with thermal annealing. These results confirmed that for a rotatable intramolecular structure, the molecular conformation could be easily adjusted by mechano-grinding. In detail, the grinding of the crystalline sample (top image of Fig. 14b) could produce a relatively planar conformation from a highly twisted one, while the grinding of a pristine amorphous sample, followed by the annealing process (bottom image of Fig. 14b), might yield a relatively twisted conformation from a planar one.
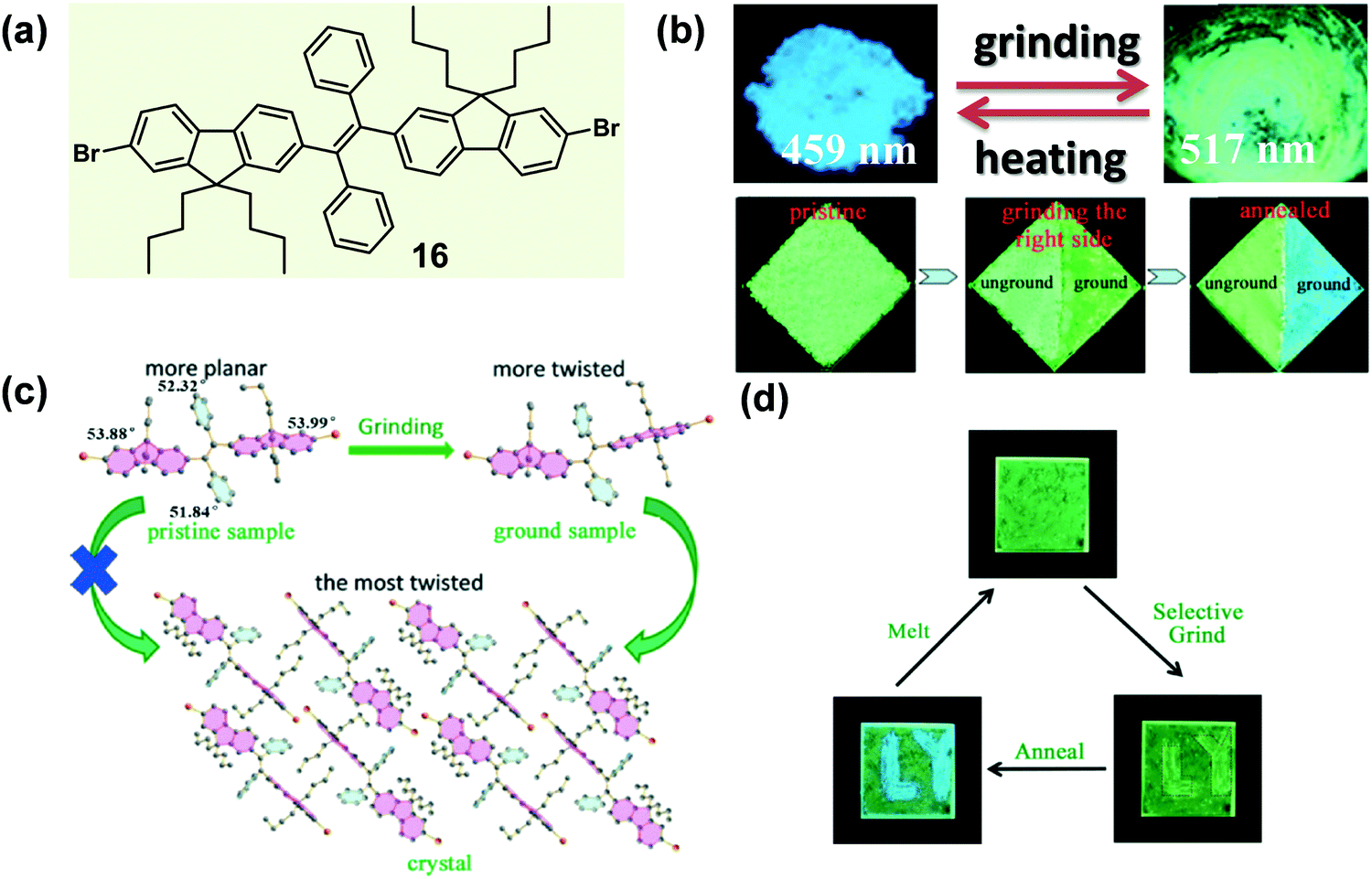 | ||
| Fig. 14 (a) Chemical structure of 16. (b) Photographic images of 16 in response to grinding and heating (top), and the comparison of the grinding effect on the thermal annealing crystallization (bottom) under UV light (365 nm). (c) Schematic illustration of conformation transition of a 16 molecule in different states (pristine amorphous sample, ground sample and crystal). (d) Procedures of writing and erasing letters on a quenched amorphous 16 film. | ||
It is well known that cyano groups play an important role in the molecular arrangement of the crystalline state through the intermolecular C–H⋯N hydrogen bonds. Recently, cyano-containing triphenylacrylonitrile (TPAN) was found to exhibit unique CIE (crystallization-induced emission) characteristics.68 In 2013, Tang et al. reported a group of TPAN-containing luminogens (17, 18 and 19) with typical AIE characteristics, which exhibited a remarkable change in emission color from blue to yellow with a shift in the maximum emissive wavelength up to 78 nm, in addition to a noticeable decrease in emission efficiency by 10.5–24% (Fig. 15a and b).69 The crystalline structure showed that all the molecules adopted a highly twisted propeller-like conformation in their crystalline state, which could shorten their effective conjugation length and avert the formation of excimers or exciplexes, making them emit bluer light like single molecules in solution. Once the crystalline lattice had been collapsed by external force, the molecules would relax to a more planar conformation with an enhanced degree of conjugation and redder fluorescence.
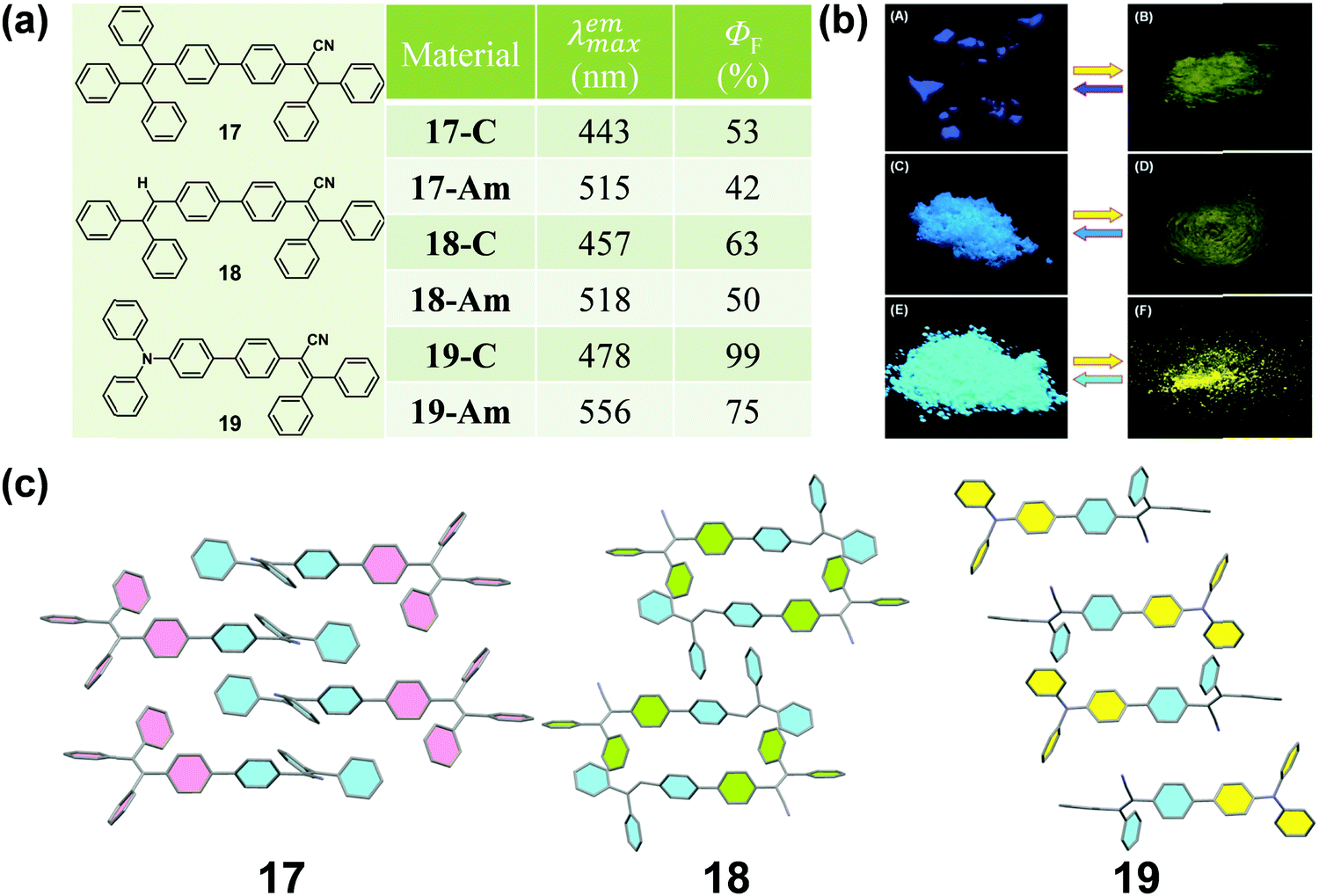 | ||
| Fig. 15 (a) Chemical structures and optical properties of 17, 18 and 19. (b) Photographic images of 17, 18 and 19 in response to grinding and fuming under UV light (365 nm). (c) Molecular packing patterns in these three crystals. | ||
Apart from the effect of molecular conformation and arrangement, the cyano group is also an ideal candidate for an excellent acceptor in fluorescent materials, due to its strong electron-withdrawing ability. In 2016, Zhao et al. reported a symmetric p-bis(2,2-dicyanovinyl)benzene-based fluorophore (20) by decorating the single benzene ring with a dicyanovinyl unit as the acceptor and p-methoxybenzene as the donor, which exhibited polymorphism-dependent emission as well as reversible mechanochromic luminescence upon external stimuli (Fig. 16).70 The single crystal of 20-G gave out a green emission (λmax = 545 nm) with a high fluorescent quantum yield of 49%, while 20-O showed a bathochromic-shifted orange emission (λmax = 560 nm) with a lower emission efficiency of 7.9%. Their crystalline structures confirmed that the molecules in both of the 20-G and 20-O crystals adopted highly twisted conformations without π–π stacking interactions. However, the dihedral angles between the dicyanovinyl unit and the 4-methoxyl phenyl moiety were a little different, being 46.27° for 20-G and 43.06° for 20-O. Thus, the relatively planar conformation of molecules in 20-O contributed to the better ICT effect and large π conjugation, resulting in a red-shifted emission.
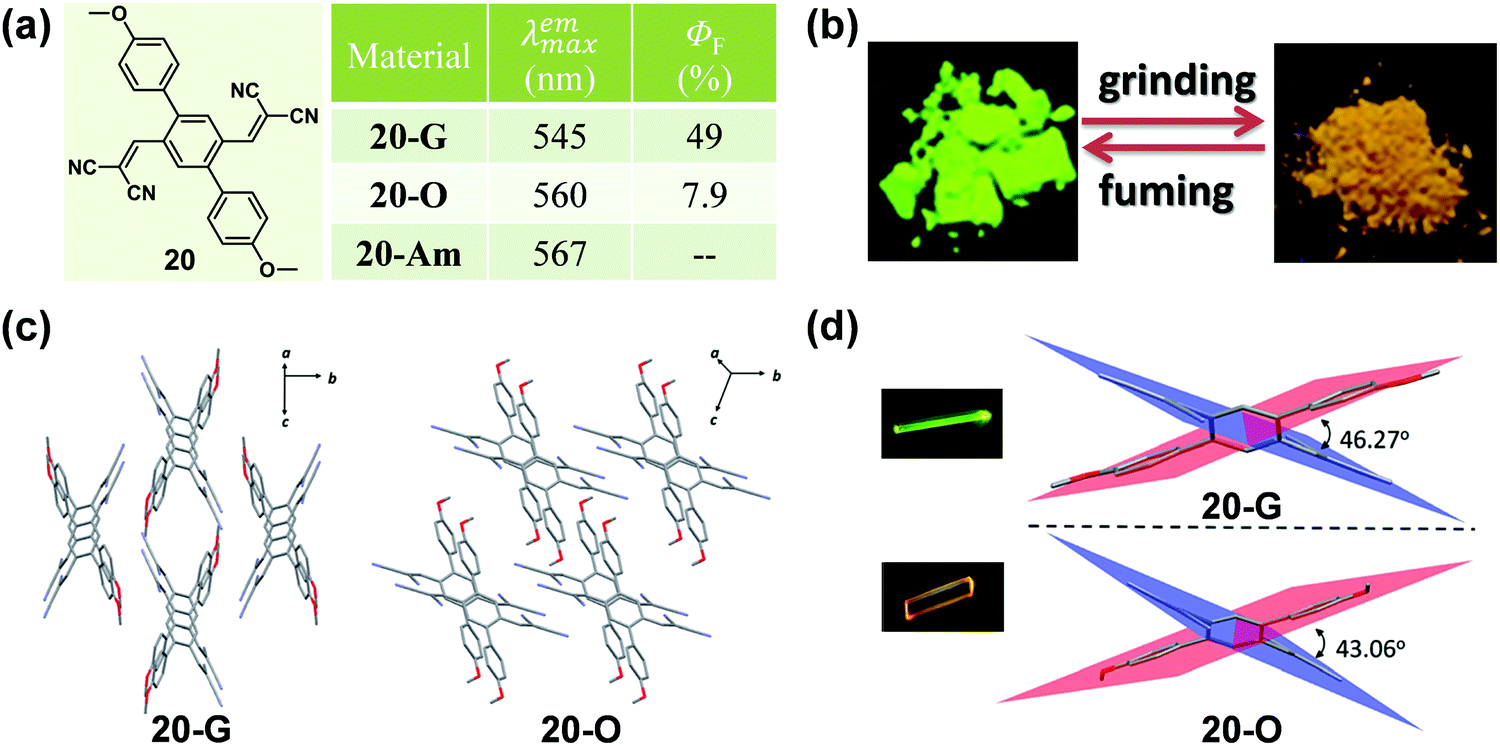 | ||
| Fig. 16 (a) Chemical structures and optical properties of 20. (b) Photographic images of 20 in response to grinding and fuming under UV light (365 nm). (c) Molecular packing in 20-G and 20-O crystals. (d) A view of the dihedral angles for 20-G and 20-O. The inset depicts the fluorescence images of crystals under a fluorescence microscope. | ||
It is well known that the ICT effect derived from the molecular electronic structure of donor–acceptor (D–A) or donor–π–acceptor (D–π–A) type fluorophores could be tuned effectively with an alteration in the twisted angles between the constructing units, which could thereafter affect the emissive behavior.71–73 Recently, many successful MCF compounds have been reported by introducing an electron-withdrawing acceptor to a twisted AIE structure. In 2015, Tian et al. reported a molecular crystal of acridonyl-tetraphenylethene with a well-defined D–A structure (21), which exhibited an intriguing turn-on and color-tuned luminescence in response to mechanical grinding and hydrostatic compression.74 The pristine powder demonstrated weak emission under UV irradiation; however, after grinding, the fluorescent quantum yield (ΦF) could be largely enhanced by a 43-fold increase from 0.01 to 0.43. Furthermore, the highly emissive ground powder could recover to the initial low emissive case upon heating at 250 °C for 1 min or fuming with organic solvents, accompanied by an alteration in the molecular conformation and reconstruction of the crystalline structure. As shown in Fig. 17c, by increasing the hydrostatic pressure to 2.98 GPa, the pristine colorless crystal gradually changed to light-yellow, accompanied by an enhanced and red-shifted emission spectrum. In the crystal, the molecules of 21 adopted a highly twisted conformation with a dihedral angle of 75° between the AD (acridone) and TPE units, without the face-to-face π–π interactions between the adjacent molecules. The steric hindrance introduced by the twisted TPE unit afforded a relatively loose packing, making the amorphization and the accompanying emissive change easy under the mechanical stimulus. Theoretical calculation disclosed that the almost orthogonal conformation between the TPE and AD units fully separated the electronic distribution and inhibited the ICT process, leading to emission from a locally excited state in the crystalline state. When the molecule was under the mechanical stimulus, the twisted conformation could be changed to a more planar one by the force perturbation, which would result in an overlap of the frontier orbitals between donor and acceptor and the formation of an ICT state, hence inducing the change in luminescence from a nearly non-emissive crystalline state to a bright cyan-emissive amorphous state. This finding provided an effective approach to designing luminescent “turn-on” MCF materials.
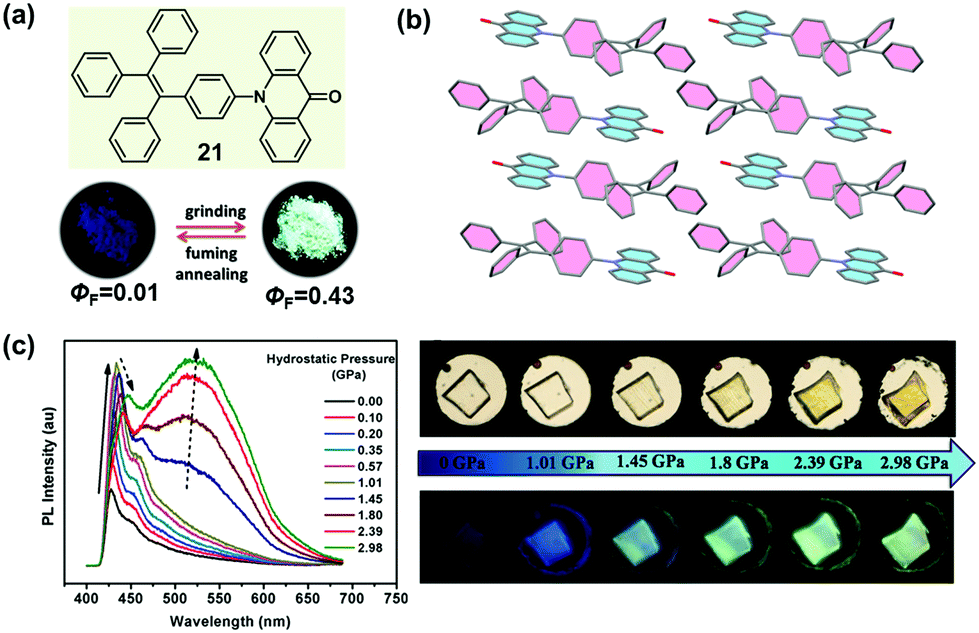 | ||
| Fig. 17 (a) Chemical structure of 21 and its photographic images in response to grinding and fuming under UV light (365 nm). (b) Molecular packing in the crystal. (c) Visible and fluorescence images of a single crystal of 21 under different hydrostatic pressures and corresponding fluorescence spectra. | ||
In 2015, Ling et al. reported a Λ-shaped D–π–A–π–D molecule 22, which was constructed with an electronic-withdrawing maleimide unit, an electronic-donating carbazole unit and a π-bridging benzene ring.7522 exhibited typical AEE, CIEE and polymorphism-dependent fluorescence properties. Three kinds of crystals, 22-Y1, 22-Y2 and 22-R, were cultured, providing a unique opportunity to investigate their morphology-dependent luminescence and the inherent mechanism. 22-Y1 and 22-Y2 emitted similar yellow-green emissions (λmax = 553 and 557 nm) with ultrahigh fluorescent quantum yields of 72% and 80%, respectively, while 22-R exhibited a red emission (λmax = 636 nm) with a much lower efficiency of 23%. The yellow emission (close to that of 22-Y1 and 22-Y2) of the pristine powder could be reversibly changed into a weak orange emission (λmax = 602 nm, ΦF = 8%) upon grinding and heating or fuming. As shown in Fig. 18c, molecules in crystals of 22-C1 and 22-C2 adopted a highly twisted and unsymmetrical conformation, and the dihedral angles of the maleimide ring with the carbazole ring (φD–A) linked with the same benzene ring were 1.0° and 77.5° in 22-Y1, and 1.2° and 77.9° in 22-Y2. However, the molecules in 22-R adopted a much more planar and symmetrical conformation with φD–A = 16.0°, indicating a larger intramolecular conjugation, in accordance with its redder emission. The molecular packing modes were also totally different in these three crystals. In the case of 22-R, two molecules were packed together in a dimer-like structure with an opposite direction, through weak C–H⋯π and π–π interactions with a contact distance of 3.34 Å between two carbazole rings. This further explained its relatively redder emission wavelength (636 nm vs. 602 nm) in comparison with that of the amorphous form obtained by grinding. As to 22-Y2, two adjacent molecules formed an intertwined structure with a crossing arrangement of carbazole moieties, which inhibited the internal rotations and blocked the non-radioactive relaxation to a larger degree, resulting in its higher quantum yield than that of 22-Y1.
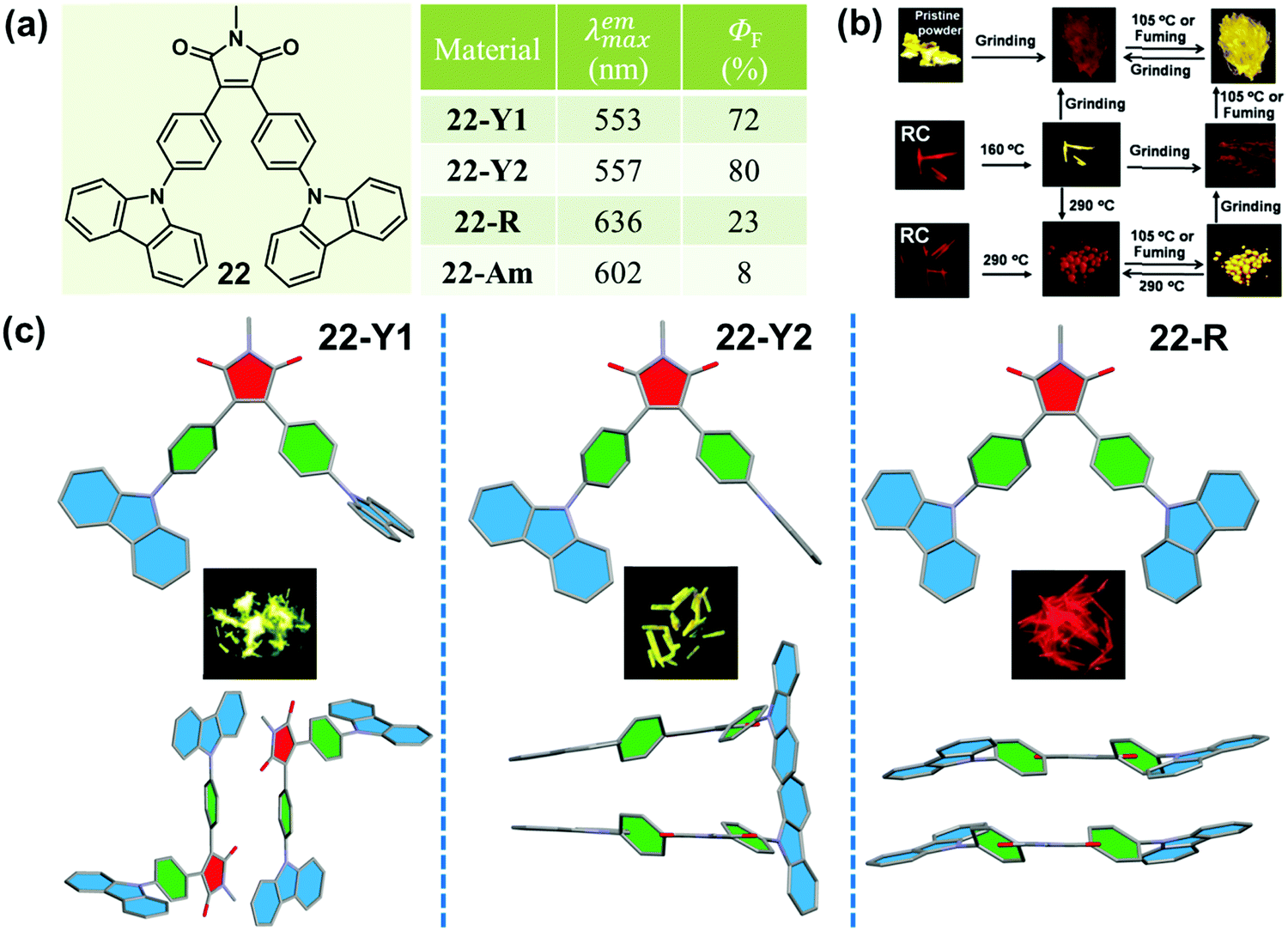 | ||
| Fig. 18 (a) Chemical structures and optical properties of 22. (b) Photographic images of 22 in response to different treatments under UV light (365 nm). (c) Molecular conformations and packing pattern of adjacent molecules in the crystal. The inset depicts the fluorescence images of crystals under a fluorescence microscope. | ||
Crystallization-induced (enhanced) emission (CIE or CIEE) is an interesting phenomenon present in plenty of AIEgen systems, and the related deformation and reconstruction of the crystalline structure upon external stimulus might induce distinct changes in emission efficiency and/or color.76–79 In previous works, Tang et al. found that a group of diphenyldibenzofulvene (DPDBF) molecules exhibited a typical AIE characteristic and a distinct CIEE effect.80,81 Among them, compound 23 displayed polymorphism-dependent fluorescence properties, and two single crystals were cultured. 23-G had a green emission (λmax = 500 nm), while there was yellow emission for 23-Y (λmax = 545 nm).82 In both crystals, the molecules adopted a highly twisted propeller-like conformation, which ruled out any specific strong intermolecular interactions, such as π–π stacking or H/J-aggregates. Thus, the various emission colors should be ascribed to the different molecular conformations, as partially confirmed by the different torsion angles of the two phenyl rings. As shown in Fig. 19a, the torsion angles (θ1 = 62.5°, 57.0° and θ2 = 51.1°, 51.4°) in 23-G were larger than those in 23-Y (θ1 = 52.9°, θ2 = 47.8°), indicating a more twisted conformation and a lower degree of intramolecular conjugation in 23-G. This well explained its bluer emission. Actually, the inherent reason for the different conformations was derived from their different molecular packing modes. In 23-G, all the molecules packed in an approximately parallel mode, whereas in 23-Y, adjacent molecules adopted a vertical arrangement. Considering the steric effect, undoubtedly, the space occupied by a molecule in 23-Y was much smaller than that occupied by a molecule in 23-G. As viewed from the cone packing model (Fig. 19c and d), molecules in 23-Y really packed more tightly than those in 23-G. For the propeller-like molecules, in theory, molecules should adopt a very twisted (probably close to orthogonal) conformation to minimize the molecular potential energy. Accordingly, the more twisted molecular conformation in 23-G was reasonable, accounting for its bluer emission. Thus, an understanding of the molecular packing mode in the crystalline state could also provide meaningful guidance for the design of MCF molecules, despite an insufficient understanding of the molecular conformations and packing styles in the amorphous state without distinct D–A structures. However, the weak and red-shifted emission of 23 in the amorphous state could only be ascribed to the formation of excimers and the further planarization of the molecular conformation upon external force, because of the lack of single crystals.
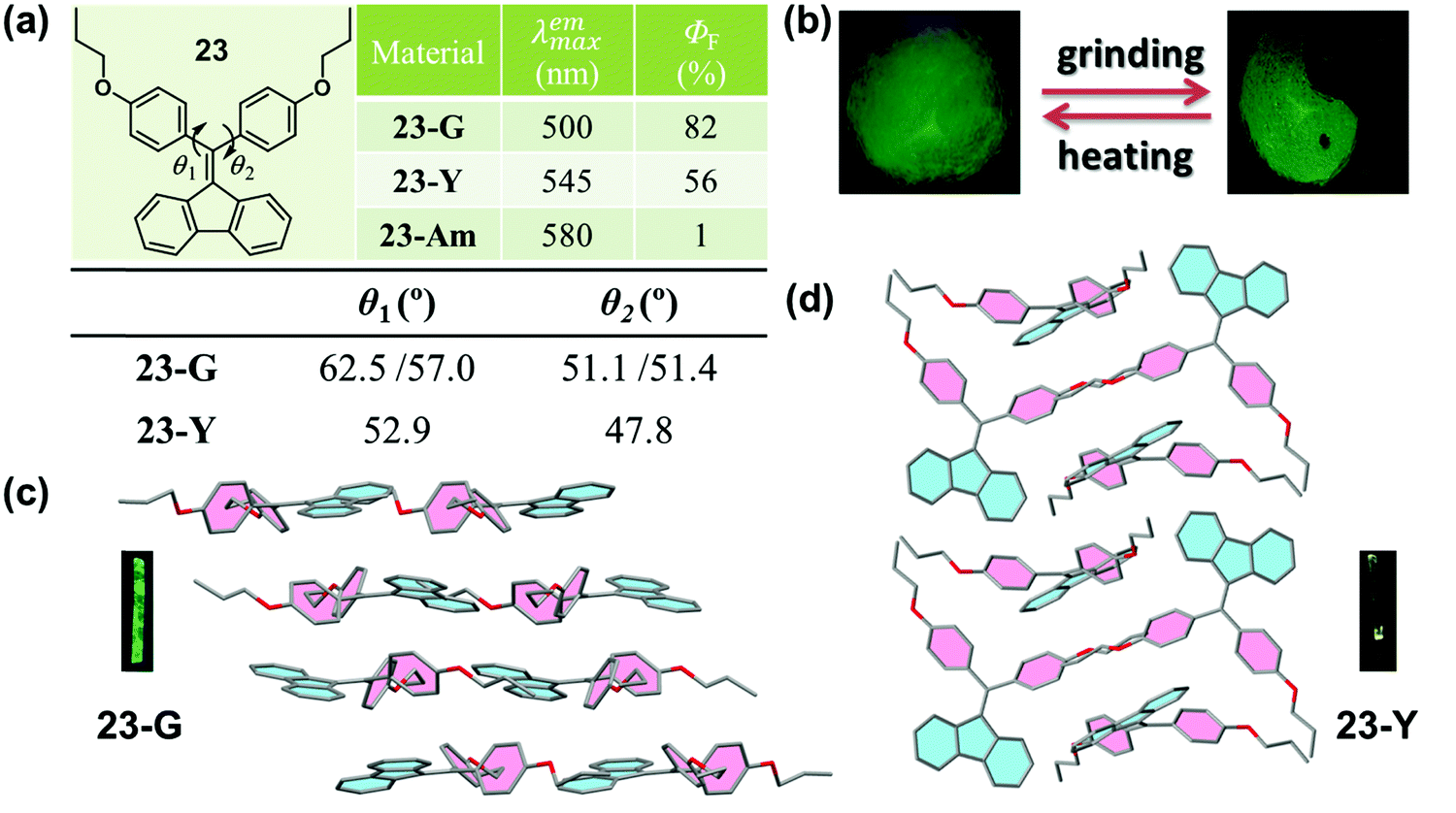 | ||
| Fig. 19 (a) Chemical structure and optical properties of 23, and torsion angles θ1 and θ2 in two single crystals of 23. (b) Photographic images of 23 in response to grinding and heating under UV light (365 nm). (c) Molecular packing in a crystal of 23-G. (d) Molecular packing in a crystal of 23-Y. The inset depicts the fluorescence images of crystals under a fluorescence microscope. | ||
The modulation of the molecular structure by altering substituent positions can induce a specific arrangement of dyes, which can tune the emission properties in the crystalline state, thus affecting the MCF property. A typical example, reported by Chujo et al. in 2016, could well describe the modulation of the sensitivity to mechanical force in diiodo boron diiminates by altering the substitution position of the iodine groups.83 Crystals 24 and 25 exhibited a cyan emission (λmax = 474 nm) and a green emission (λmax = 495 nm) with different substituent positions of the iodine atom. The emission would be weakened and red-shifted to 534 nm or 538 nm upon grinding. The dihedral angles between the phenyl rings linked to the imine carbons and the boron-containing six-membered ring, were 49° and 55° for 24, but 42° and 48° for 25, showing the planarity of the molecular conformation (Fig. 20b). The dihedral angles confirmed the greater planar structure of the molecules in 25, which induced the better π conjugation system and coincided well with its relatively red emission in the crystalline state in comparison to that of 24. The molecular conformations could also be explained by the molecular packing mode. As shown in Fig. 20c, the adjacent molecules in 25 adopted a vertical arrangement, while there was an approximately parallel packing mode with a slipping angle (∼30°) in contiguous molecular chains for 24. From this comparison, molecules in crystal 25 packed more tightly with a more planar structure, thus inducing its redder emission in the crystalline state and less significant MCF property.
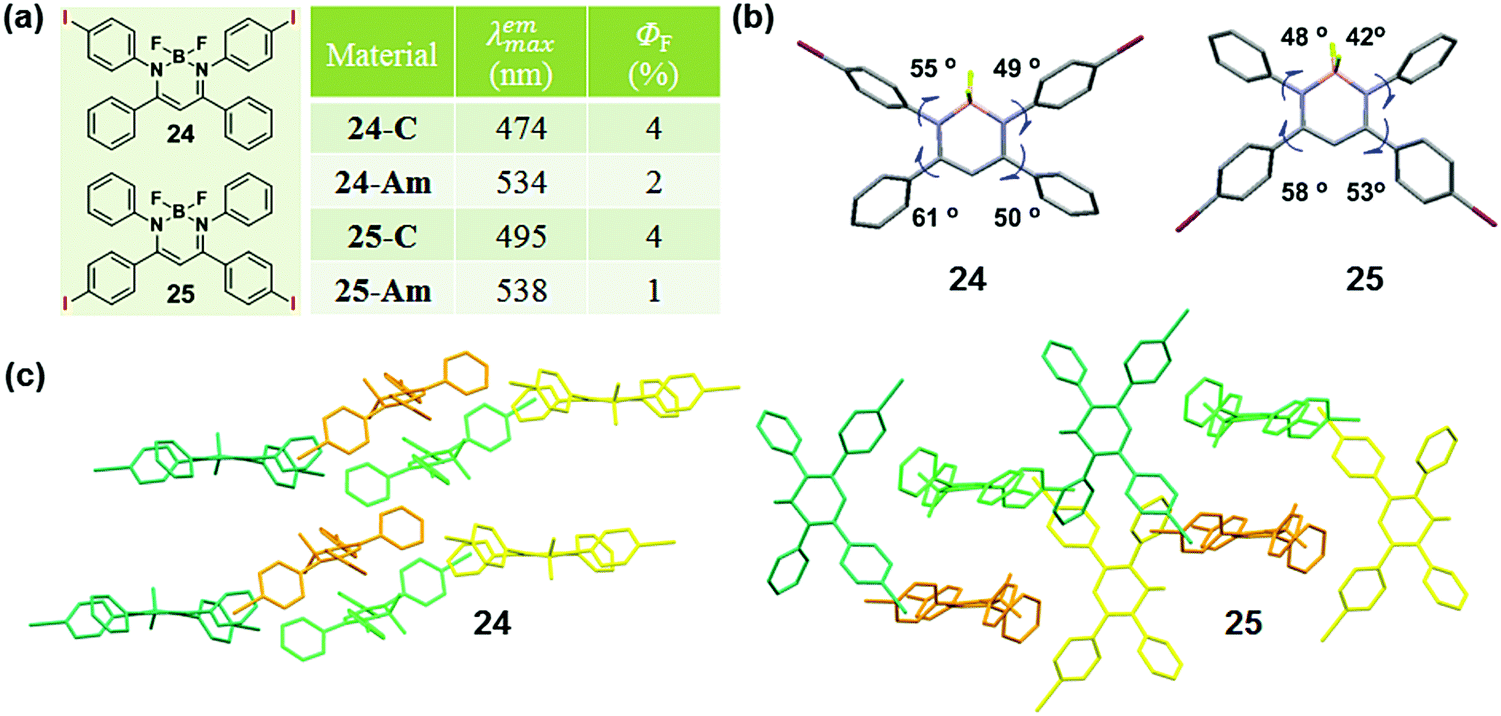 | ||
| Fig. 20 (a) Chemical structure and optical properties of 24 and 25. (b) Torsion angles of molecules for 24 and 25 in the crystal. (c) Molecular packing pattern of 24 and 25 in the crystal. | ||
This phenomenon of a packing-dependent emission could be also observed in a highly twisted butterfly-like bis(diarylmethylene)dihydroanthracenes (26) system, as reported by Tang et al. in 2015.84 As shown in Fig. 21, 26 exhibited typical AIE characteristic with different emission colors in different phases: blue light emission for 26-B (λmax = 425 nm), cyan light emission for 26-Cy (λmax = 464 nm), and weak yellow light emission (λmax = 520 nm) for the amorphous powder (26-Am). The crystalline structures demonstrated that the average volume occupied by a single molecule in the crystal lattice was 805.6 Å3 for 26-B, while it was a little smaller for 26-G (778.3 Å3). The higher packing density of 26-G led to its relatively smaller dihedral angles; hence inducing better electronic communication and redder light emission. Moreover, the adjacent molecules exhibited stronger intermolecular interactions with each other, which could help to further rigidify the molecular conformation and block the nonradiative pathways, thus resulting in a much higher emission efficiency (76.8% vs. 27.7%). The much lower fluorescent quantum yield (3.3%) of the amorphous powder should be ascribed to the poor molecular arrangement, which provided a lot of room for the intramolecular rotation and vibration. In addition, the relatively redder emission should be ascribed to the more planar conformation in the amorphous state.
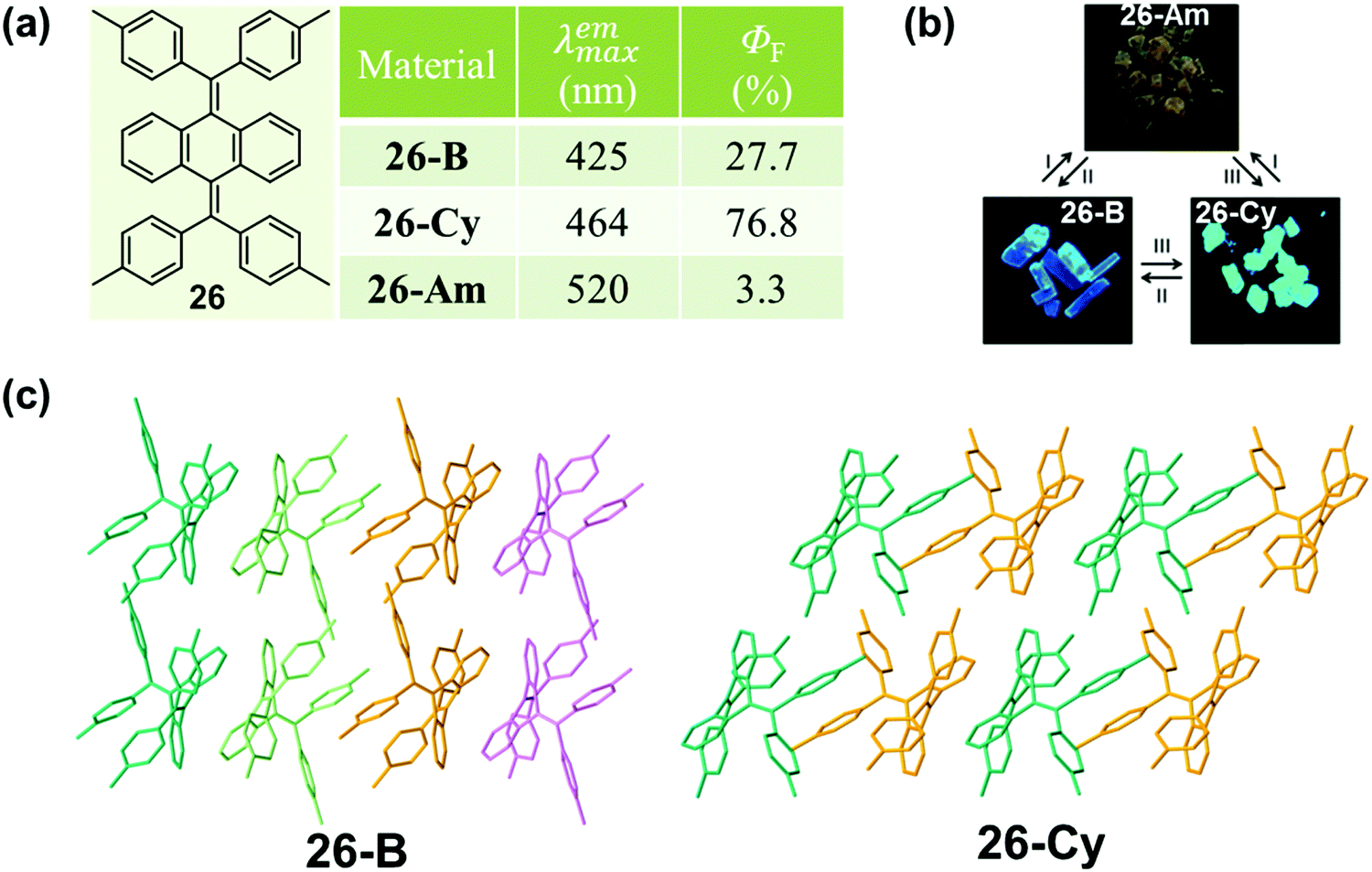 | ||
| Fig. 21 (a) Chemical structure and optical properties of 26. (b) Photographic images of 26 in response to different treatments under UV light (365 nm). (Conditions: heating to melt and cooling by liquid nitrogen (I), heating at 170 °C for more than 1 h or fuming with CH2Cl2 or CHCl3 vapor for 3 h (II), and fuming with acetone vapor for 3 h (III).) (c) Molecular packing pattern in the crystal. | ||
However, despite this deep insight into conformation-dependent emission in the crystalline state, the relationship between the photo-physical properties, the molecular conformation and packing styles was still difficult to determine for the twisted MCF molecules. In 2014, Zou et al. investigated the emissive behavior of TPE (27), a star AIE molecule, under high pressure by using a diamond anvil cell technique with associated spectroscopic measurements, such as steady-state photoluminescence, time-resolved emission decay measurements, high-pressure synchrotron powder X-ray diffraction and high-pressure infrared measurements.85 As shown in Fig. 22a, upon increasing the pressure up to 10 GPa, the emission of 27 could gradually be changed from deep-blue (λmax = 448 nm) to spring green (488 nm). The changing trend in λmax and emission efficiency could be divided into three stages: 0.1–1.5 GPa, 1.5–5.3 GPa, and 5.3–10 GPa. In the range of 0.1 to 1.5 GPa, the emission color exhibited a faint red shift with decreased efficiency, which should be attributed to the additional nonradiative decay channels originating from the closer intermolecular interactions due to the unit-cell contraction of TPE under pressure, without conformational planarization. In the pressure range of 1.5–5.3 GPa, the emission efficiency was dramatically enhanced due to the further strengthening of the intermolecular interactions, which could significantly restrict the movement of the aromatic rings, thus more efficiently suppressing the energy loss through intramolecular rotation. On the other hand, there was a larger red shift in the PL spectra when the pressure was over 4 GPa, as a result of the conformational planarization associated with the deformation of the intermolecular interaction (C–H⋯π and C–H⋯C) network. When the pressure was above 5.3 GPa, the crystalline sample transformed to an amorphous phase with pronounced conformational planarization, and the λmax of the PL spectra was persistently red-shifted, accompanied by decreased efficiency. On one hand, the decrease in emission efficiency was ascribed to the insufficient restriction of intramolecular rotation (RIR) in the amorphous state; on the other hand, further excessive compression would lead to close interactions of the panel parts of the molecules, thus quenching the excited states, resulting in decreased emission efficiency. This work provided important information on the transformation of molecular conformations and packing styles under high pressure.
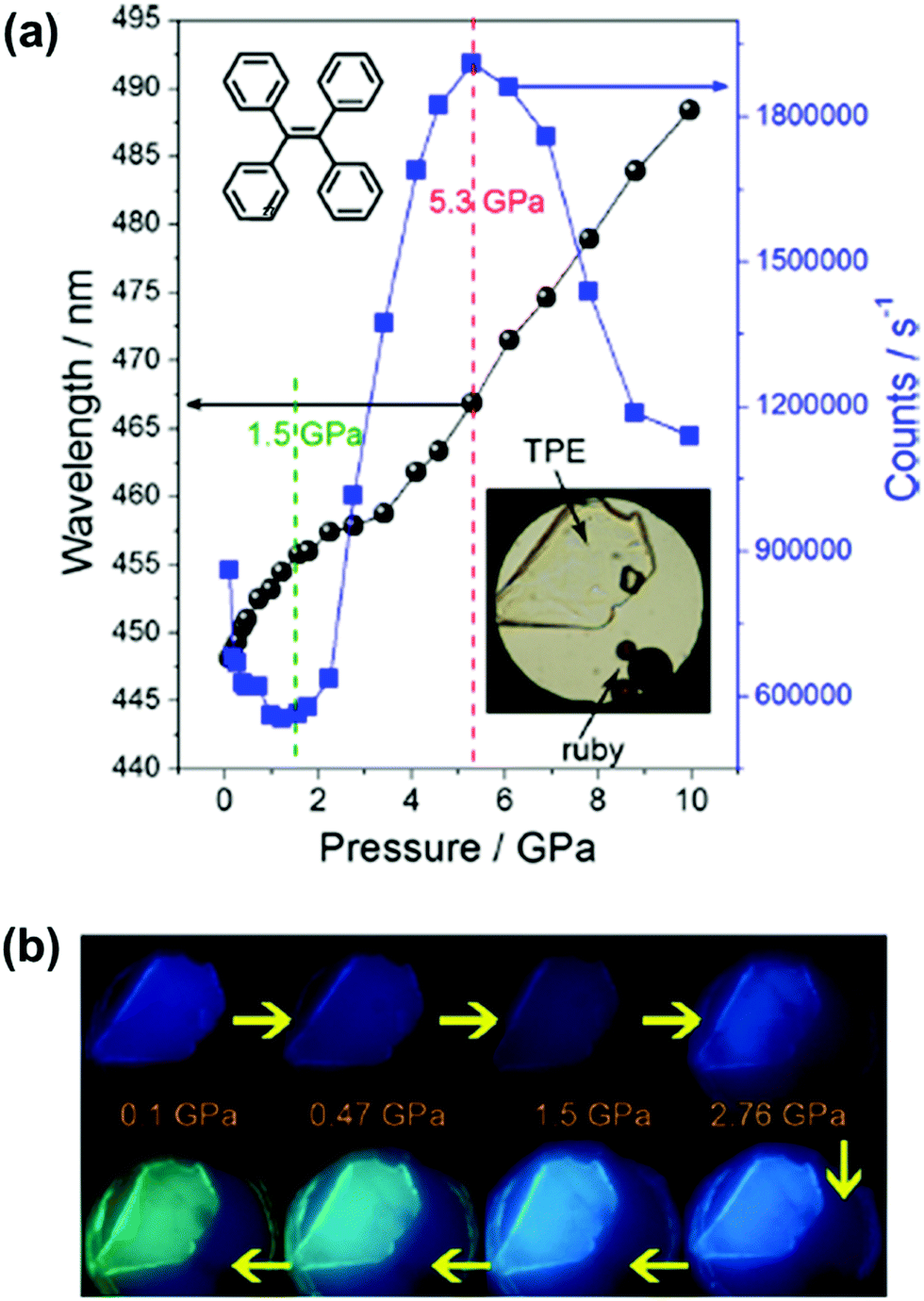 | ||
| Fig. 22 (a) Pressure-dependent PL maximum (left axis) and intensity (right axis) of TPE crystals. The inset panel includes the molecular structure of TPE and an optical photograph of TPE crystals in DAC. (b) Corresponding typical photographs under UV irradiation (λex = 375 nm). | ||
Recently, Walsh et al. reported an absorbate-induced piezochromism phenomenon in a porous molecular crystal (28), which showed potential applications in the direct detection of gases (Fig. 23a).86 They presented quantum chemical calculations of the electronic response to pressure based on the crystalline structure (Fig. 23b), which could provide some useful information for understanding pressure-induced modulations in the electronic structure of MCF materials. The valence band maximum (VBM) and conduction band minimum (CBM) of both molecule and crystal are shown in Fig. 23c. In the perfect solid, the electronic levels of both the VBM and the CBM were dependent on the intermolecular distance, and the band gap (Eg) was sensitive to the cell pressure. In the equilibrium configuration, solid-state calculations predicted Eg = 3.54 eV. The origin of the electronic response to stress could be understood by examining the electron density of the frontier bands. Upon compression, the valence band was destabilized as the out-of-phase, destructive, intermolecular π-overlap increased, resulting in a decreased ionization potential. However, the intermolecular interactions of the conduction band had an overlap with the constructive wave function to some extent, as a result of the spatially helical wave function (the product of the molecular propeller-like geometry). Actually, this overlap could be stabilized under pressure. It was therefore expected that a decrease in sheet separation should cause a redshift in optical absorption under compressing pressure. As shown in Fig. 23d, the Eg could decrease to 3.36 eV under 71 MPa pressure. These results coincided well with the generally red-shifted luminescence behavior upon pressure observed in MCF materials.
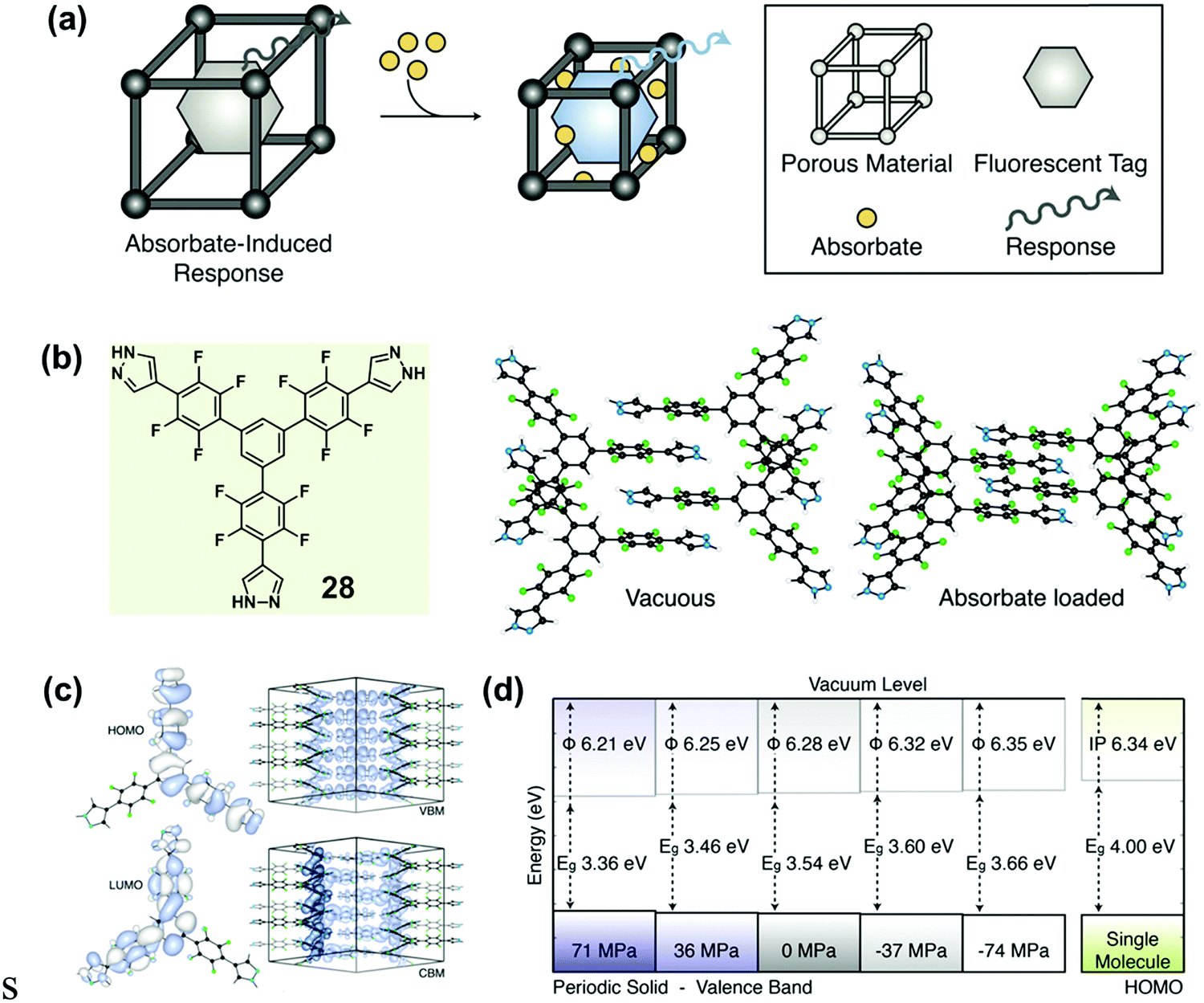 | ||
| Fig. 23 (a) Schematic illustration of porous materials with an absorbate-induced response property. (b) Chemical structure of 28 and the molecular packing in vacuous state and absorbate-loaded state. (c) HOMO/LUMO (left) of a single molecule of 28, and the valence band maximum (VBM) and conduction band minimum (CBM) of 28 in the solid state. (d) The changes in work function and electronic band gap of the solid under different pressures. | ||
3. Other mechanical force responsive materials
The numerous examples discussed above illustrated how the molecular conformation and packing affect the luminescent behavior of organic dyes in the solid state and the related mechanism of the force–packing–luminescence process. Besides the fast-growing mechanical-responsive fluorescent materials, there are some kinds of abnormal stimuli-responsive phase transitions, accompanied by specific luminescence-switching cases, which could also be attributed to the inherent structural changes in the single molecule (conformation) or a collective of molecules (packing) levels.3.1 Shear-triggered crystallization materials
As is well known, molten organic molecules would be driven to the crystalline state at temperatures below their melting point. Although supercooled liquids could be formed by a rapid cooling, stable ones are rare, because the crystalline organic materials readily undergo a phase transformation to an energetically favorable crystalline phase upon subsequent treatment by heating. Recently, a thermally stable supercooled liquid of diketopyrrolopyrrole (DPP) derivatives (29) was reported by Kim et al., with a unique shear-triggered crystallization and light emission property.8729 could induce a 25-fold enhancement in PL quantum efficiency, accompanied by a color change from red to greenish yellow. This phenomenon was quite similar to the MCF property discussed above, but in contrast to the order-to-disorder transformation under force stimuli, the shear-triggered crystallization underwent a distinctive disorder-to-order transition.As shown in Fig. 24b, no strong hydrogen bonding, π–π interactions, or halogen bonding were observed in the crystal structure of 29. The origin of the thermal stability was the subtle force balance between aromatic interactions among the DPP core units and van der Waals interactions among the aliphatic side chains, which induced an unattainable yet required large critical radius (r*, the minimum size of nucleus which can continue to grow spontaneously) originating from a small ΔG between the supercooled liquid and the crystalline solid states.
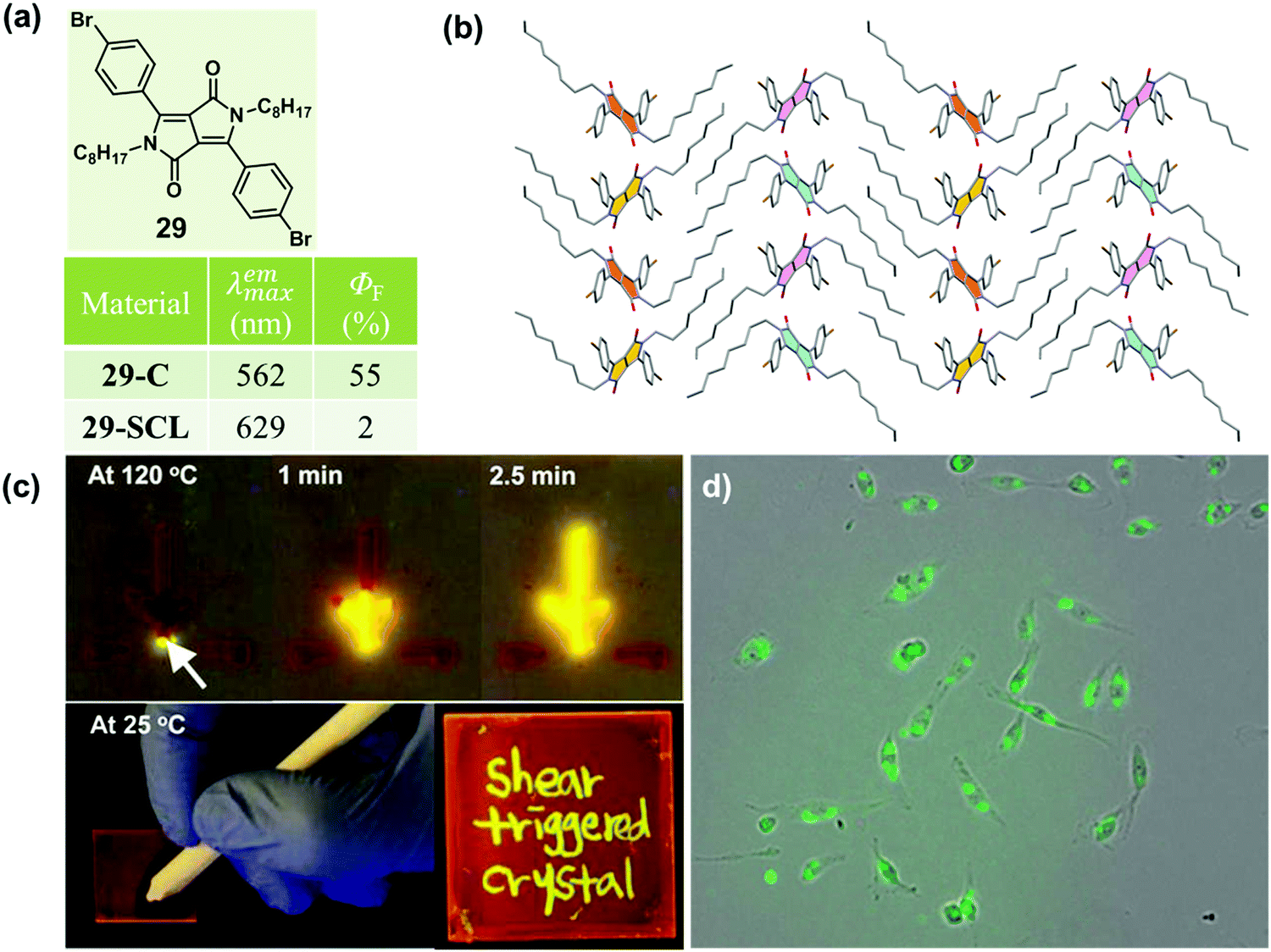 | ||
| Fig. 24 (a) Chemical structure and optical properties of 29. (b) Molecular packing pattern in the crystal. (c) Different shear-triggered lighting-up crystallization of 29 at 120 °C (up images) and 25 °C (bottom images). (d) Shear-triggered crystallization by living cell attachment. | ||
Interestingly, upon applying a shear force, the dim orange-red fluorescent supercooled liquid transformed to bright greenish yellow fluorescent crystals: the shear force acted as a trigger for crystallization via molecular agitation in this system. Interestingly, the supercooled liquid of 29 exhibited distinct shear-triggered lighting-up crystallization propagated at different temperatures. As shown in the top images of Fig. 24c, the entire supercooled liquid could crystallize spontaneously when the force stimulus was applied at the tip of the liquid film at 120 °C. Whereas at 25 °C, crystallization could only take place where the force was applied. Thus fluorescent patterning by direct writing on the supercooled liquid film could be accomplished at 25 °C. Furthermore, the threshold shear stress value was as small as 0.9 kPa, which was comparable to the fibroblast cell's traction force per unit area (1–5 kPa),88 providing an application in the detection of cell attachment and cell contraction, as shown in Fig. 24d.
3.2 Mechanoluminescent materials
Mechanoluminescence (ML) or triboluminescence (TL) is the emission of light caused by an application of mechanical stress to a solid. Despite its long history since the first report in 1605, the inherent mechanism of the ML property has remained uncertain until now.89 However, many reported cases demonstrated that the ML property was concerned with crystalline polymorphs, indicating the underlying connection between the ML property and molecular packing patterns.90–92 Recently, several bright ML molecules have been reported, which also possessed the typical AIE characteristic.59,93–95 Among them, two crystalline polymorph systems with a distinct ML property provided some important information for a deep understanding of the underlying structure–mechanoluminescence relationship.In 2016, our group reported a methoxy-substituted TPE derivative (30), which possessed two crystalline polymorphs, 30-C1 and 30-C2. Excitingly, 30-C1 displayed a strong mechanoluminescence, while 30-C2 was ML-inactive. The pristine powder of 30 exhibited a high-contrast MCF behavior upon grinding, accompanied by the piezoelectric effect.95 As illustrated in the crystalline structure (Fig. 25c and d), the most notable difference between the two polymorphs was the dihedral angle (θ) between the ethylenic bonds of adjacent molecules. In the case of 30-C1, two adjacent molecules adopted a vertical stacking mode with θ = 89.3°. However, in 30-C2, two adjacent molecules were packed in a fully parallel packing mode with θ = 0°. This difference in the molecular arrangement induced totally different intermolecular interactions in the crystalline state. The quantity of the C–H⋯π and C–H⋯O hydrogen bonds was almost 4-fold and 2-fold in 30-C1 than those in 30-C2, respectively. The much stronger intermolecular interactions resulted in the better compression-resistance property of 30-C1. Thus, upon a mechanical force stimulus like grinding, the larger crystal would fracture and change into smaller crystals rather than the crystalline structure collapsing entirely. As proposed in previous work, the fracture of the crystals and the concomitant creation of new surfaces were responsible for the ML excitation.96–98 In contrast, as confirmed by powder XRD, 30-C2 was easy to collapse and transform to the amorphous state even upon a light grinding force, giving rise to its ML-inactive property. Based on these results, a possible mechanism for the ML process was promoted: the more rigid molecular stacking mode and stronger interactions between molecules could block the nonradiative pathway by decreasing the molecule slippage upon mechanical force, and the large crystal would fracture and change into smaller crystals accompanied by the creation of new surfaces, inducing the energy brought from the mechanical force to be released in a radiative channel with the ML phenomenon.
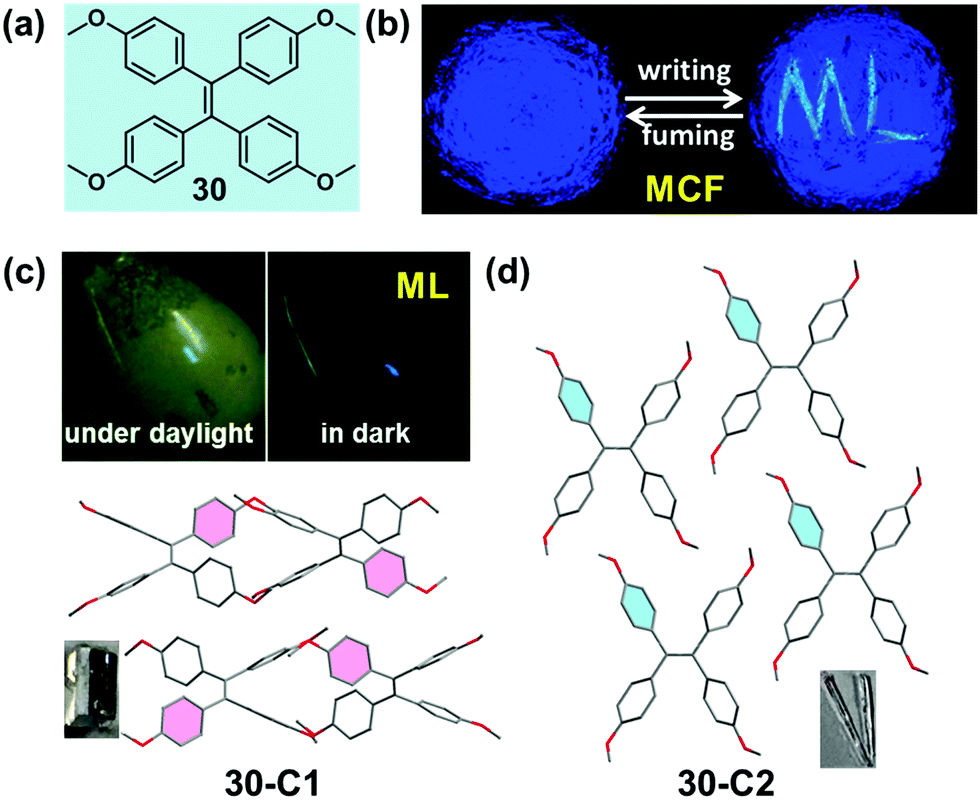 | ||
| Fig. 25 (a) Chemical structure of 30. (b) MCF property: photographic images of 30 in response to grinding and fuming under UV light (365 nm). (c) ML property and molecular packing pattern of the 30-C1 crystal. (d) Molecular packing pattern in the 30-C2 crystal. The inset depicts the images of the crystals. | ||
A similar molecular packing-dependent ML property was also illustrated by Chi and Xu et al. recently (Fig. 26).59 Compound 31, a thiophene-2-carbaldehyde substituted TPE derivative, had two crystalline polymorphs. While 31-C1 had the ML property, 31-C2 was ML-inactive. By grinding the 31-C1 crystal with a pestle or shearing it with a spatula, a highly bright green light emission peaking at 517 nm was observed, coinciding well with its PL emission spectra. The distinct ML property of crystalline polymorphs should be attributed to their different molecular packing, similar to the case of 30. The asymmetric units in both polymorphs were composed of two crystallographically independent molecules, and the big difference was the packing mode of the two thiophene rings. In the case of 31-C2, they were packed in an anti-parallel arrangement with a dihedral angle of 3.7°, whereas in 31-C1, they were almost vertical to each other (θ = 85.8°). The vertical packing pattern of adjacent molecules induced stronger intermolecular interactions in the crystalline lattice of 31-C1, thus giving rise to the ML property according to the possible mechanism proposed by us and discussed above.
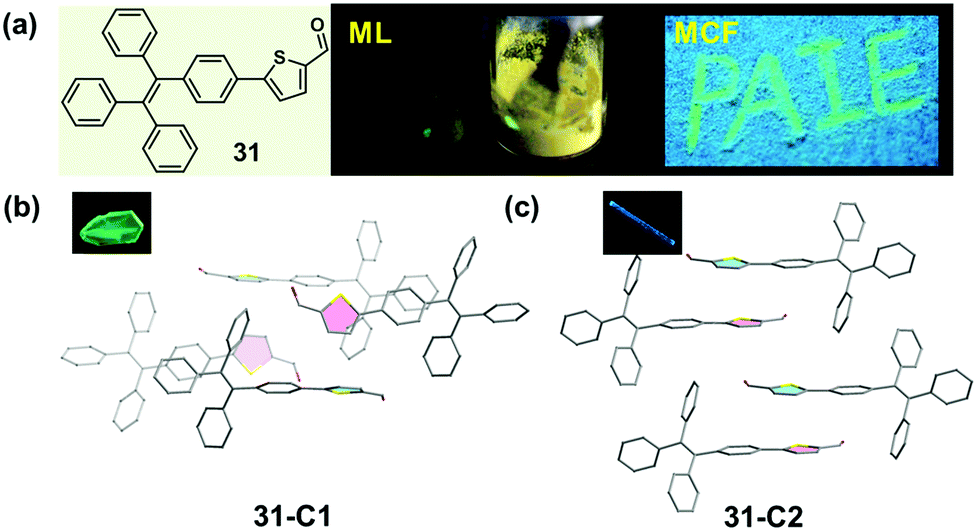 | ||
| Fig. 26 (a) Chemical structure and MCF property of 31, and ML property of 31-C1. (b) Molecular packing pattern in the 31-C1 crystal. (c) Molecular packing pattern in the 31-C2 crystal. The inset depicts the fluorescence images of the crystals under a fluorescence microscope. | ||
So far, bright organic ML compounds have shown a rapid increase in the past several years, but the correlations between molecular packing and the ML property were still uncertain and independent in different cases.99–102 However, based on the present results, there is no doubt that the molecular packing mode plays an important role in the ML property, and a deeper insight into the ML phenomenon would be accomplished with the development of this research field.
4. Outlook
In this paper, based on recent research on mechanochromic fluorescent materials with accurate crystalline structures, we have tried hard to build a relationship between the molecular packing mode in the aggregated state and the emissive performance, with an attempt to understand the hidden thoughts of the authors. Thanks to the enthusiasm of scientists, crystalline polymorphs have provided lots of important information, especially on the molecular packing status, frequently accompanied by a change in the molecular conformation. In some cases, the formed excimers (e.g. compound 5) and dimers (e.g. compound 9) are present as the emissive moieties. Thus, upon aggregation, many different parameters would affect the emissive behavior, and the stimulations could change the molecular packing and give rise to the accompanying changes, such as the conformation, π–π interactions, and the order–disorder alignment. From this point of view, the energy levels of a single molecule will not be adequate to explain the emissive behavior of luminogens in the aggregated states or upon stimuli. Therefore, the heavily emphasized electronic properties of the single molecule, as presented in the proposed design principles for luminogens, should be seriously reconsidered, because the changed molecular packing status, intermolecular interactions, and conformation mentioned above, could surely affect the ICT effect and energy levels, possibly resulting in some new-born energy levels. Thus, to achieve a distinct mechanochromic luminescence performance, a D–A structure constructed with both planar and twisted moieties (AIE unit) would be an ideal model molecule. The D–A structure could induce changes in the ICT effect, accompanied by an alteration in the molecular conformation upon the application of mechanical force, which would significantly affect the electronic structures of the molecules. The existence of a twisted moiety, like an AIE unit, could contribute to a loose packing mode in the crystalline state, thus providing amorphization upon exposure to the external mechanical stimulus and endowing it with high emission efficiency in the solid state. The planar moiety could provide the possibility of intermolecular π–π stacking interactions, hence possibly inducing the formation of excimers during the disintegration of the crystalline structure. On the other hand, some substituted groups that could promote the construction of intermolecular interactions in the aggregated state are also important parameters, such as alkyl chains, halogen atoms, cyano groups, and elements like boron, oxygen, nitrogen etc.From the above selected examples, it could be proposed that under different statuses, no matter whether in normal aggregated states or subject to high pressure or solvent fuming or grinding, the molecules are liable to adopt the most stable existing form under those conditions. Just for relative stability, the packing status and the conformation could be changed accordingly, sometimes accompanied by the formed dimers or excimers derived from the strong intermolecular interactions. Actually, many scientists have strengthened their research on how packing status affects performance. Thanks to the high sensitivity of fluorescence, MCF materials, presented in this review paper, have provided lots of useful and important information. However, for the further and rapid development of MCF materials, perhaps, at least three issues should be considered seriously: the quantification of the stimulation and the corresponding property changes including emission, related theory, and the accurate structure–packing–performance relationship.
As partially demonstrated in the cases of 21 and 27, the experimental results under accurate pressures could provide much more detailed information for an understanding of the inherent mechanism. If other stimulations could be quantified, we could know MCF materials much better and more deeply. To meet this point, perhaps, scientists from other areas, for example, mechanics, electronics, and tribology, should be involved. Also, some targeted equipment might need to be constructed for the required experiments.
Undoubtedly, the theories extracted from the experiments provide important guidance for further development. In the case of compound 28, the quantum chemical calculations of electronic response to pressure based on the crystalline structure, could provide useful information for understanding pressure-induced modulations in the electronic structure of MCF materials. However, the present theoretical calculations could not accurately predict the possible molecular packing status and the related conformation under different conditions in the aggregated states. Surely, much more detailed information should be collected for the optimization of present theories, especially the quantification of the stimulation and the corresponding property changes.
As demonstrated in all the cases discussed in this review paper and in some other multi-component MCF material cases utilizing a host–guest molecular assembly strategy,103–106 the intermolecular interactions (π–π interactions and van der Waals interactions) play a key role in molecular conformation and packing. The phase transformations are always accompanied by the destruction and reconstruction of intermolecular interactions. Additionally, we could find out the determined effect of molecular conformation or packing in many “abnormal” emission phenomena, like the vibration-induced emission reported recently.107,108 Inherently, from the viewpoint of relatively stable molecular existence under different conditions, the molecular status should surely be derived from its electronic structure, or be considered as an explicit behavior of the inherent molecular structure. Since we know much less about how the electronic properties of the single molecule with different bulk and length affect the molecular packing and conformation under different conditions, we could not design a molecular structure exactly according to a desired performance. That is to say, there is a lack of an accurate structure–packing–performance relationship. Regardless of the seemingly many examples of MCF materials, lots of additional examples, related information, rational analysis, and deep thinking, are still badly needed, especially those elaborately designed cases with specific thoughts. Not easy, but so important! Actually, the importance of the accurate structure–packing–performance relationship is not only limited to MCF materials, but all kinds of organic solid materials, including OFET, OPV, and OLED. For example, the observed abnormal room temperature phosphorescence of cyanoacetic acid by us recently, could further confirm the key role of molecular packing in solid state, since there are no aromatic rings in cyanoacetic acid.109 Science is the systematic study of natural phenomena and how they affect us and our environment. In this viewpoint, continuous investigation and deeper understanding into molecular packing and how it affects material performance, might promote the development of materials science and subsequent practical applications.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 21325416 and 51573140), and the National Fundamental Key Research Program (2013CB834701).Notes and references
- H. Kang, W. Lee, J. Oh, T. Kim, C. Lee and B. J. Kim, Acc. Chem. Res., 2016, 49, 2424–2434 CrossRef CAS PubMed.
- L. Yuan, W. Y. Lin, K. B. Zheng and S. S. Zhu, Acc. Chem. Res., 2013, 46, 1462–1473 CrossRef CAS PubMed.
- P. L. Burn, S. C. Lo and I. D. W. Samuel, Adv. Mater., 2007, 19, 1675–1688 CrossRef CAS.
- J. E. Kwon and S. Y. Park, Adv. Mater., 2011, 23, 3615–3642 CrossRef CAS PubMed.
- J. Mei, N. L. C. Leung, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, Chem. Rev., 2015, 115, 11718–11940 CrossRef CAS PubMed.
- J. Mei, Y. N. Hong, J. W. Y. Lam, A. J. Qin, Y. H. Tang and B. Z. Tang, Adv. Mater., 2014, 26, 5429–5479 CrossRef CAS PubMed.
- B. K. An, J. Gierschner and S. Y. Park, Acc. Chem. Res., 2012, 45, 544–554 CrossRef CAS PubMed.
- A. P. de Silva, H. Q. N. Gunaratne, T. Gunnlaugsson, A. J. M. Huxley, C. P. McCoy, J. T. Rademacher and T. E. Rice, Chem. Rev., 1997, 97, 1515–1566 CrossRef CAS PubMed.
- A. Ajayaghosh and V. K. Praveen, Acc. Chem. Res., 2007, 40, 644–656 CrossRef CAS PubMed.
- R. S. Clegg and J. E. Hutchison, J. Am. Chem. Soc., 1999, 121, 5319–5327 CrossRef CAS.
- C. Kuemin, L. Nowack, L. Bozano, N. D. Spencer and H. Wolf, Adv. Funct. Mater., 2012, 22, 702–708 CrossRef CAS.
- R. J. Li, W. P. Hu, Y. Q. Liu and D. B. Zhu, Acc. Chem. Res., 2010, 43, 529–540 CrossRef CAS PubMed.
- C. Reese and Z. N. Bao, Mater. Today, 2007, 10, 20–27 CrossRef CAS.
- S. Casalini, C. A. Bortolotti, F. Leonardi and F. Biscarini, Chem. Soc. Rev., 2017, 46, 40–71 RSC.
- M. Campoy-Quiles, T. Ferenczi, T. Agostinelli, P. G. Etchegoin, Y. Kim, T. D. Anthopoulos, P. N. Stavrinou, D. D. C. Bradley and J. Nelson, Nat. Mater., 2008, 7, 158–164 CrossRef CAS PubMed.
- J.-H. Im, I.-H. Jang, N. Pellet, M. Grätzel and N.-G. Park, Nat. Nanotechnol., 2014, 9, 927–932 CrossRef CAS PubMed.
- W. Y. Nie, H. H. Tsai, R. Asadpour, J. C. Blancon, A. J. Neukirch, G. Gupta, J. J. Crochet, M. Chhowalla, S. Tretiak, M. A. Alam, H. L. Wang and A. D. Mohite, Science, 2015, 347, 522–525 CrossRef CAS PubMed.
- Y. Dang, Y. Zhou, X. Liu, D. Ju, S. Xia, H. Xia and X. Tao, Angew. Chem., Int. Ed., 2016, 55, 3447–3450 CrossRef CAS PubMed.
- Y. Dang, D. Ju, L. Wang and X. Tao, CrystEngComm, 2016, 18, 4476–4484 RSC.
- Y. Dang, C. Zhong, G. Zhang, D. Ju, L. Wang, S. Xia, H. Xia and X. Tao, Chem. Mater., 2016, 28, 6968–6974 CrossRef CAS.
- T. Seki, Y. Takamatsu and H. Ito, J. Am. Chem. Soc., 2016, 138, 6252–6260 CrossRef CAS PubMed.
- J. Wu, Y. Cheng, J. Lan, D. Wu, S. Qian, L. Yan, Z. He, X. Li, K. Wang, B. Zou and J. You, J. Am. Chem. Soc., 2016, 138, 12803–12812 CrossRef CAS PubMed.
- Y. Hong, J. W. Y. Lam and B. Z. Tang, Chem. Commun., 2009, 4332–4353 RSC.
- Y. Hong, J. W. Y. Lam and B. Z. Tang, Chem. Soc. Rev., 2011, 40, 5361–5388 RSC.
- Q. Li and Z. Li, Adv. Sci., 2017, 1600484 CrossRef.
- K. Araki and T. Mutai, Photochemistry, 2015, 43, 191–225 Search PubMed.
- H. Yamada, C. Xu, A. Fukazawa, A. Wakamiya and S. Yamaguchi, Macromol. Chem. Phys., 2009, 210, 904–916 CrossRef CAS.
- M. C. Etter, Acc. Chem. Res., 1990, 23, 120–126 CrossRef CAS.
- F. Chen, T. Tian, B. Bai, J. Wang, H. Wang and M. Li, J. Mater. Chem. C, 2016, 4, 4451–4458 RSC.
- K. Ariga, T. Mori and J. P. Hill, Adv. Mater., 2012, 24, 158–176 CrossRef CAS PubMed.
- Y. Sagara, S. Yamane, M. Mitani, C. Weder and T. Kato, Adv. Mater., 2016, 28, 1073–1095 CrossRef CAS PubMed.
- Z. Ma, Z. Wang, M. Teng, Z. Xu and X. Jia, ChemPhysChem, 2015, 16, 1811–1828 CrossRef CAS PubMed.
- Z. G. Chi, X. Q. Zhang, B. J. Xu, X. Zhou, C. P. Ma, Y. Zhang, S. W. Liu and J. R. Xu, Chem. Soc. Rev., 2012, 41, 3878–3896 RSC.
- L. Wang, K. Q. Ye and H. Y. Zhang, Chin. Chem. Lett., 2016, 27, 1367–1375 CrossRef CAS.
- Y. Q. Dong, J. W. Lam and B. Z. Tang, J. Phys. Chem. Lett., 2015, 3429–3436 CrossRef CAS PubMed.
- P. C. Xue, J. P. Ding, P. P. Wang and R. Lu, J. Mater. Chem. C, 2016, 4, 6688–6706 RSC.
- S. Varughese, J. Mater. Chem. C, 2014, 2, 3499–3516 RSC.
- R. Gawinecki, G. Viscardi, E. Barni and M. A. Hanna, Dyes Pigm., 1993, 23, 73–78 CrossRef CAS.
- Y. Sagara and T. Kato, Nat. Chem., 2009, 1, 605–610 CrossRef CAS PubMed.
- H. Nam, B. Boury and S. Y. Park, Chem. Mater., 2006, 18, 5716–5721 CrossRef CAS.
- Y. Dong, B. Xu, J. Zhang, X. Tan, L. Wang, J. Chen, H. Lv, S. Wen, B. Li, L. Ye, B. Zou and W. Tian, Angew. Chem., Int. Ed., 2012, 51, 10782–10785 CrossRef CAS PubMed.
- G. Zhang, J. Lu, M. Sabat and C. L. Fraser, J. Am. Chem. Soc., 2010, 132, 2160–2162 CrossRef CAS PubMed.
- G. R. Krishna, M. S. R. N. Kiran, C. L. Fraser, U. Ramamurty and C. M. Reddy, Adv. Funct. Mater., 2013, 23, 1422–1430 CrossRef CAS.
- K. Nagura, S. Saito, H. Yusa, H. Yamawaki, H. Fujihisa, H. Sato, Y. Shimoikeda and S. Yamaguchi, J. Am. Chem. Soc., 2013, 135, 10322–10325 CrossRef CAS PubMed.
- L. Wang, K. Wang, B. Zou, K. Ye, H. Zhang and Y. Wang, Adv. Mater., 2015, 27, 2918–2922 CrossRef CAS PubMed.
- X. C. Du, F. Xu, M. S. Yuan, P. C. Xue, L. Zhao, D. E. Wang, W. J. Wang, Q. Tu, S. W. Chen and J. Y. Wang, J. Mater. Chem. C, 2016, 4, 8724–8730 RSC.
- M.-S. Yuan, D.-E. Wang, P. Xue, W. Wang, J.-C. Wang, Q. Tu, Z. Liu, Y. Liu, Y. Zhang and J. Wang, Chem. Mater., 2014, 26, 2467–2477 CrossRef CAS.
- M. Tanioka, S. Kamino, A. Muranaka, Y. Ooyama, H. Ota, Y. Shirasaki, J. Horigome, M. Ueda, M. Uchiyama, D. Sawada and S. Enomoto, J. Am. Chem. Soc., 2015, 137, 6436–6439 CrossRef CAS PubMed.
- H. Moon, R. Zeis, E.-J. Borkent, C. Besnard, A. J. Lovinger, T. Siegrist, C. Kloc and Z. Bao, J. Am. Chem. Soc., 2004, 126, 15322–15323 CrossRef CAS PubMed.
- J. W. Chung, B.-K. An and S. Y. Park, Chem. Mater., 2008, 20, 6750–6755 CrossRef CAS.
- S. K. Park, J. H. Kim, S.-J. Yoon, O. K. Kwon, B.-K. An and S. Y. Park, Chem. Mater., 2012, 24, 3263–3268 CrossRef CAS.
- S. Shin, S. H. Gihm, C. R. Park, S. Kim and S. Y. Park, Chem. Mater., 2013, 25, 3288–3295 CrossRef CAS.
- P. Rajamalli, P. Gandeepan, M. J. Huang and C. H. Cheng, J. Mater. Chem. C, 2015, 3, 3329–3335 RSC.
- P. Xue, B. Yao, J. Sun, Q. Xu, P. Chen, Z. Zhang and R. Lu, J. Mater. Chem. C, 2014, 2, 3942–3950 RSC.
- S.-J. Yoon, J. W. Chung, J. Gierschner, K. S. Kim, M.-G. Choi, D. Kim and S. Y. Park, J. Am. Chem. Soc., 2010, 132, 13675–13683 CrossRef CAS PubMed.
- A. I. Kitaigorodskii, Organic Chemical Crystallography, Consultants Bureau, New York, 1961 Search PubMed.
- K. Wang, S. Huang, Y. Zhang, S. Zhao, H. Zhang and Y. Wang, Chem. Sci., 2013, 4, 3288–3293 RSC.
- H. Y. Yu, W. Ren, H. G. Lu, Y. A. Liang and Q. S. Wang, Chem. Commun., 2016, 52, 7387–7389 RSC.
- B. J. Xu, J. J. He, Y. X. Mu, Q. Z. Zhu, S. K. Wu, Y. F. Wang, Y. Zhang, C. J. Jin, C. C. Lo, Z. G. Chi, A. Lien, S. W. Liua and J. R. Xu, Chem. Sci., 2015, 6, 3236–3241 RSC.
- B. J. Xu, M. Y. Xie, J. J. He, B. Xu, Z. G. Chi, W. J. Tian, L. Jiang, F. L. Zhao, S. W. Liu, Y. Zhang, Z. Z. Xu and J. R. Xu, Chem. Commun., 2013, 49, 273–275 RSC.
- D. P. Ou, T. Yu, Z. Y. Yang, T. G. Luan, Z. Mao, Y. Zhang, S. W. Liu, J. R. Xu, Z. G. Chi and M. R. Bryce, Chem. Sci., 2016, 7, 5302–5306 RSC.
- C. J. Chen, J. Y. Liao, Z. G. Chi, B. J. Xu, X. Q. Zhang, D. B. Kuang, Y. Zhang, S. W. Liu and J. R. Xu, J. Mater. Chem., 2012, 22, 8994–9005 RSC.
- X. Q. Zhang, Z. G. Chi, B. J. Xu, C. J. Chen, X. Zhou, Y. Zhang, S. W. Liu and J. R. Xu, J. Mater. Chem., 2012, 22, 18505–18513 RSC.
- B. J. Xu, Z. G. Chi, J. Y. Zhang, X. Q. Zhang, H. Y. Li, X. F. Li, S. W. Liu, Y. Zhang and J. R. Xu, Chem. – Asian J., 2011, 6, 1470–1478 CrossRef CAS PubMed.
- T. Jadhav, B. Dhokale, S. M. Mobin and R. Misra, J. Mater. Chem. C, 2015, 3, 9981–9988 RSC.
- Y. Lv, Y. Liu, D. Guo, X. Ye, G. Liu and X. Tao, Chem. – Asian J., 2014, 9, 2885–2890 CrossRef CAS PubMed.
- Y. Lv, Y. Liu, X. Ye, G. F. Liu and X. T. Tao, CrystEngComm, 2015, 17, 526–531 RSC.
- W. Z. Yuan, Y. Gong, S. Chen, X. Y. Shen, J. W. Y. Lam, P. Lu, Y. Lu, Z. Wang, R. Hu, N. Xie, H. S. Kwok, Y. Zhang, J. Z. Sun and B. Z. Tang, Chem. Mater., 2012, 24, 1518–1528 CrossRef CAS.
- W. Z. Yuan, Y. Tan, Y. Gong, P. Lu, J. W. Y. Lam, X. Y. Shen, C. Feng, H. H. Y. Sung, Y. Lu, I. D. Williams, J. Z. Sun, Y. Zhang and B. Z. Tang, Adv. Mater., 2013, 25, 2837–2843 CrossRef CAS PubMed.
- N. L. Jing, N. Zhang, H. Kang, S. Zhou, H. Sun, S. Yin, N. Zhao and B. Z. Tang, Chem. Sci., 2017, 8, 577–582 RSC.
- Y. Li, F. Shen, H. Wang, F. He, Z. Xie, H. Zhang, Z. Wang, L. Liu, F. Li, M. Hanif, L. Ye and Y. Ma, Chem. Mater., 2008, 20, 7312–7318 CrossRef CAS.
- R. Hu, E. Lager, A. Aguilar-Aguilar, J. Liu, J. W. Y. Lam, H. H. Y. Sung, I. D. Williams, Y. Zhong, K. S. Wong, E. Peña-Cabrera and B. Z. Tang, J. Phys. Chem. C, 2009, 113, 15845–15853 CAS.
- Z. R. Grabowski, K. Rotkiewicz and W. Rettig, Chem. Rev., 2003, 103, 3899–4032 CrossRef PubMed.
- Q. K. Qi, J. Y. Qian, X. Tan, J. B. Zhang, L. J. Wang, B. Xu, B. Zou and W. J. Tian, Adv. Funct. Mater., 2015, 25, 4005–4010 CrossRef CAS.
- X. F. Mei, G. X. Wen, J. W. Wang, H. M. Yao, Y. Zhao, Z. H. Lin and Q. D. Ling, J. Mater. Chem. C, 2015, 3, 7267–7271 RSC.
- Y. Q. Dong, J. W. Y. Lam, A. J. Qin, Z. Li, J. Z. Sun, H. H. Y. Sung, I. D. Williams and B. Z. Tang, Chem. Commun., 2007, 40–42 RSC.
- W. Z. Yuan, X. Y. Shen, H. Zhao, J. W. Y. Lam, L. Tang, P. Lu, C. L. Wang, Y. Liu, Z. M. Wang, Q. Zheng, J. Z. Sun, Y. G. Ma and B. Z. Tang, J. Phys. Chem. C, 2010, 114, 6090–6099 CAS.
- P. Galer, R. C. Korosec, M. Vidmar and B. Sket, J. Am. Chem. Soc., 2014, 136, 7383–7394 CrossRef CAS PubMed.
- R. Yoshii, A. Hirose, K. Tanaka and Y. Chujo, J. Am. Chem. Soc., 2014, 136, 18131–18139 CrossRef CAS PubMed.
- Y. Dong, J. W. Lam, A. Qin, Z. Li, J. Sun, H. H. Sung, I. D. Williams and B. Z. Tang, Chem. Commun., 2007, 40–42 RSC.
- H. Tong, Y. Dong, Y. Hong, M. Häussler, J. W. Y. Lam, H. H. Y. Sung, X. Yu, J. Sun, I. D. Williams, H. S. Kwok and B. Z. Tang, J. Phys. Chem. C, 2007, 111, 2287–2294 CAS.
- X. Luo, J. Li, C. Li, L. Heng, Y. Q. Dong, Z. Liu, Z. Bo and B. Z. Tang, Adv. Mater., 2011, 23, 3261–3265 CrossRef CAS PubMed.
- M. Yamaguchi, S. Ito, A. Hirose, K. Tanaka and Y. Chujo, J. Mater. Chem. C, 2016, 4, 5314–5319 RSC.
- Z. He, L. Zhang, J. Mei, T. Zhang, J. W. Y. Lam, Z. Shuai, Y. Q. Dong and B. Z. Tang, Chem. Mater., 2015, 27, 6601–6607 CrossRef CAS.
- H. Yuan, K. Wang, K. Yang, B. Liu and B. Zou, J. Phys. Chem. Lett., 2014, 5, 2968–2973 CrossRef CAS PubMed.
- C. H. Hendon, K. E. Wittering, T. H. Chen, W. Kaveevivitchai, I. Popov, K. T. Butler, C. C. Wilson, D. L. Cruickshank, O. S. Miljanic and A. Walsh, Nano Lett., 2015, 15, 2149–2154 CrossRef CAS PubMed.
- K. Chung, M. S. Kwon, B. M. Leung, A. G. Wong-Foy, M. S. Kim, J. Kim, S. Takayama, J. Gierschner, A. J. Matzger and J. Kim, ACS Cent. Sci., 2015, 1, 94–102 CrossRef CAS PubMed.
- N. Q. Balaban, U. S. Schwarz, D. Riveline, P. Goichberg, G. Tzur, I. Sabanay, D. Mahalu, S. Safran, A. Bershadsky, L. Addadi and B. Geiger, Nat. Cell Biol., 2001, 3, 466–472 CrossRef CAS PubMed.
- J. I. Zink, Acc. Chem. Res., 1978, 11, 289–295 CrossRef CAS.
- G. E. Hardy, W. C. Kaska, B. P. Chandra and J. I. Zink, J. Am. Chem. Soc., 1981, 103, 1074–1079 CrossRef CAS.
- G. E. Hardy, J. I. Zink, W. C. Kaska and J. C. Baldwin, J. Am. Chem. Soc., 1978, 100, 8001–8002 CrossRef CAS.
- L. M. Sweeting and A. L. Rheingold, J. Am. Chem. Soc., 1987, 109, 2652–2658 CrossRef CAS.
- J. Yang, Z. Ren, Z. Xie, Y. Liu, C. Wang, Y. Xie, Q. Peng, B. Xu, W. Tian, F. Zhang, Z. Chi, Q. Li and Z. Li, Angew. Chem., Int. Ed., 2017, 56, 880–884 CrossRef CAS PubMed.
- B. J. Xu, W. L. Li, J. J. He, S. K. Wu, Q. Z. Zhu, Z. Y. Yang, Y. C. Wu, Y. Zhang, C. J. Jin, P. Y. Lu, Z. G. Chi, S. W. Liu, J. R. Xu and M. R. Bryce, Chem. Sci., 2016, 7, 5307–5312 RSC.
- C. Wang, B. Xu, M. Li, Z. Chi, Y. Xie, Q. Li and Z. Li, Mater. Horiz., 2016, 3, 220–225 RSC.
- B. P. Chandra and J. I. Zink, Inorg. Chem., 1980, 19, 3098–3102 CrossRef CAS.
- B. P. Chandra and J. I. Zink, Phys. Rev. B: Condens. Matter Mater. Phys., 1980, 21, 816–826 CrossRef CAS.
- J. I. Zink, Naturwissenschaften, 1981, 68, 507–512 CrossRef CAS.
- J. Guo, X.-L. Li, H. Nie, W. Luo, S. Gan, S. Hu, R. Hu, A. Qin, Z. Zhao, S.-J. Su and B. Z. Tang, Adv. Funct. Mater., 2017, 27, 1606458 CrossRef.
- K. K. Neena, P. Sudhakar, K. Dipak and P. Thilagar, Chem. Commun., 2017, 53, 3641–3644 RSC.
- H. Nakayama, J.-i. Nishida, N. Takada, H. Sato and Y. Yamashita, Chem. Mater., 2012, 24, 671–676 CrossRef CAS.
- J. Nishida, H. Ohura, Y. Kita, H. Hasegawa, T. Kawase, N. Takada, H. Sato, Y. Sei and Y. Yamashita, J. Org. Chem., 2016, 81, 433–441 CrossRef CAS PubMed.
- D. Yan, H. Yang, Q. Meng, H. Lin and M. Wei, Adv. Funct. Mater., 2014, 24, 587–594 CrossRef CAS.
- D. Yan, J. Lu, J. Ma, S. Qin, M. Wei, D. G. Evans and X. Duan, Angew. Chem., Int. Ed., 2011, 50, 7037–7040 CrossRef CAS PubMed.
- D. Yan and D. G. Evans, Mater. Horiz., 2014, 1, 46–57 RSC.
- G. Fan and D. Yan, Sci. Rep., 2014, 4, 4933–4940 CrossRef CAS PubMed.
- Z. Zhang, Y. S. Wu, K. C. Tang, C. L. Chen, J. W. Ho, J. Su, H. Tian and P. T. Chou, J. Am. Chem. Soc., 2015, 137, 8509–8520 CrossRef CAS PubMed.
- W. Chen, C. L. Chen, Z. Zhang, Y. A. Chen, W. C. Chao, J. Su, H. Tian and P. T. Chou, J. Am. Chem. Soc., 2017, 139, 1636–1644 CrossRef CAS PubMed.
- Z. Li, M. Fang and Q. Li, CN Pat., 201710214462.7, 2017 Search PubMed.
| This journal is © the Partner Organisations 2017 |