
MOFs-Derived Co@CN bi-functional catalysts for selective transfer hydrogenation of α,β-unsaturated aldehydes without use of base additives†
Xiaomei
Liu
a,
Shujuan
Cheng
a,
Jilan
Long
*a,
Wei
Zhang
*b,
Xiaohong
Liu
c and
Dapeng
Wei
b
aChemical Synthesis and Pollution Control Key Laboratory of Sichuan Province, College of Chemistry and Chemical Engineering, China West Normal University, Nanchong 637000, P. R. China. E-mail: xlong612@126.com
bChongqing Institute of Green and Intelligent Technology, Chinese Academy of Sciences, Chongqing 400714, P. R. China. E-mail: andyzhangwei@163.com
cDepartment of Laboratory Medicine, Southwest Hospital, Third Military Medical University, Chongqing 400038, P. R. China
First published on 5th June 2017
Abstract
Transfer hydrogenation protocols involve versatile and environmentally benign methods capable of providing alternatives to traditional hydrogenation processes. However, most of the non-noble metal catalysts used for transfer hydrogenation processes proceed with the aid of base additives or phosphorus-containing ligands to obtain good yields. We report a green and sustainable catalytic system by fabrication of a Co@CN bi-functional nanocatalyst with a new N-containing [Co(TPA)(ted)0.5] as a sacrificial template. This catalytic system was employed effectively for selective transfer hydrogenation of an α,β-unsaturated aldehyde (cinnamaldehyde, CAL) to obtain a high yield of an α,β-unsaturated alcohol (cinnamyl alcohol, COL) under a mild reaction condition without use of additives. During thermolysis, the retained N atoms were distributed uniformly in catalysts and endowed the catalysts with abundant basic sites, and Co2+ in [Co(TPA)(ted)0.5] were converted in situ into Co nanoparticles. Experimental results indicated obvious coordination interactions between Co nanoparticles and N atoms in these bi-functional catalysts. By optimizing the reaction conditions, a catalytic system with Co@CN-900 as a catalyst and n-hexanol as the proton donor exhibited the optimal activity for this transformation, achieving 99% selectivity for COL with full conversion. In addition, a reasonable reaction mechanism for the transfer hydrogenation of CAL to COL was proposed. Furthermore, this catalytic system also demonstrated good recyclability as well as wide substrates compatibility.
Introduction
Selective hydrogenation of α,β-unsaturated aldehydes (e.g. cinnamaldehyde (CAL)) to generate the corresponding unsaturated alcohols that can be used as vital intermediates for the preparation of flavors, fragrances, and pharmaceuticals has attracted extensive interest from chemical and pharmaceutical researchers.1,2 However, owing to the conjugated effect, selective hydrogenation of α,β-unsaturated aldehydes leads to the coexistence of different products (Fig. 1) with low selectivities.3 Moreover, with regard to thermodynamics, selective hydrogenation of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond over the CC bond is a greater challenge due to the higher bond energy of CO.4–6
O bond over the CC bond is a greater challenge due to the higher bond energy of CO.4–6
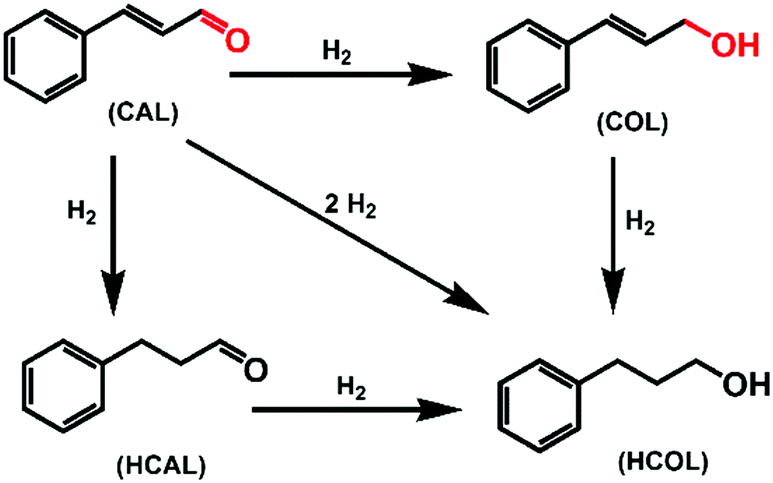 | ||
| Fig. 1 Reaction processes for the selective hydrogenation of cinnamaldehyde (CAL). | ||
Noble-metal catalysts such as Pt, Ru, Au, and Ir have been reported to be able to complete this reaction.7–10 However, the high costs and rarity of noble metal catalysts hinder their practical applications in industry. Most recently, non-noble metal catalysts have been developed as alternatives for this transformation. In particular, Co, Cu, and Fe nanoparticles and their alloy catalysts, as well as some Co and Fe complexes, have attracted considerable attention due to their high selectivity for unsaturated alcohols.5,11–14 However, the high H2 pressures or temperatures eclipse these systems to some extent (some of the relevant studies are shown in Table S1, ESI†).14,15 Compared with traditional H2-based hydrogenation, transition metal nanoparticles catalyse transfer hydrogenation reactions with alcohols as proton donors, therefore avoiding high H2 pressures and autoclaving. This represents a safe, green, sustainable and atom-economic hydrogenation pathway.16,17
Until now, the transfer hydrogenation method has been used in various types of reactions,18,19 and many non-noble metal catalysts have been used for transfer hydrogenation reactions, such as soluble Fe, Cu, and Co complexes,12,20,21 as well as some heterogeneous Fe, Co, and Ni nanoparticles and their alloy nanoparticles (Table S1, ESI†).9,22–24 However, most of these non-noble metal catalytic systems have proceeded with the aid of base additives (such as Et3N, NaOH, KOt-Bu) or P-containing ligands to obtain a relatively high selectivity.21,23–25 From the perspective of environmental protection, addition of base additives and phosphorus ligands does not meet the requirements of sustainable chemistry. Therefore, more green and highly efficient catalytic systems are needed to be developed as alternatives for selective transfer hydrogenation of cinnamaldehyde (CAL) to obtain a high yield of cinnamyl alcohol (COL).
We report a new metal–organic framework (MOF)-derived N-doped carbon layer26,27 supported Co bi-functional nanocatalyst (Co@CN) that can be effectively employed for selective transfer hydrogenation of an α,β-unsaturated aldehyde (CAL) to obtain a high yield of an α,β-unsaturated alcohol (COL) under a mild reaction condition without the use of additives. [Co(TPA)(ted)0.5] (Co-MOF) was adopted as a self-template due to its N-containing character and well-defined framework structure.28 After thermolysis, the N atoms could be retained in catalysts to endow the catalysts with abundant basic sites. The latter have been shown to have a similar role as base additives,29,30 and thereby ensure that the transfer hydrogenation reaction could proceed smoothly in the absence of base additives. Subsequently, all of the Co@CN materials were employed as the catalysts for the selective transfer hydrogenation of CAL to synthesize COL. The preparation conditions of catalysts and the different proton donors were investigated as well. The optimal Co@CN-900 catalysts showed 99% selectivity for COL with quantitative conversion under a mild condition with n-hexanol as the proton donor. Moreover, this catalytic system also exhibited excellent recyclability and substrates compatibility.
Experimental
Synthesis of [Co(TPA)(ted)0.5] (Co-MOF)
[Co(TPA)(ted)0.5] was prepared according to a previous report.31 Typically, 3.36 mmol Co(NO3)·6H2O, 3.37 mmol terephthalic acid (TPA) and 1.76 mmol 1,4-diazabicyclooctane (TED) were suspended in 40 mL N,N-dimethylformamide (DMF) and heated to 120 °C in a autoclave for 2 days. Then, the solids were collected by filtration and dried in an oven at 80 °C. The final green product was [Co(TPA)(ted)0.5] ([Co(C8H4O4)(C6H12N2)0.5]).Synthesis of Co@CN catalysts
The Co@CN catalysts were synthesized by calcining the Co-MOF ([Co(TPA)(ted)0.5]) in Ar flow. The Co-MOF was heated in a tubular furnace at a heating rate of 2 °C min−1 from room temperature to 200 °C and maintained at this temperature for 2 h to remove the solvent. Then, the calcining temperature was increased to the target temperature, such as 600 °C, 700 °C, 800 °C, and 900 °C, respectively, and kept at the target temperature for 8 h. After cooling to ambient temperature, black catalysts were obtained and labelled as Co@CN-600, Co@CN-700, Co@CN-800 and Co@CN-900, respectively.Catalyst tests
Selective transfer hydrogenation of CAL proceeded in 25 mL Schlenk tubes with alcohols as proton donors and solvent at 80 °C. Before reactions, the catalysts were treated by H2 flow for 1 h in a tubular furnace to stop them being oxidized by Co nanoparticles in air. Then, the H2 was degassed and purged by N2 flow for 1 h. After cooling to ambient temperature, the catalysts were transferred to a Schlenk tube, which was degassed and purged several times with pure N2 to remove air, followed by injection of CAL and isopropanol. Then, the mixture was heated in an oil bath at 80 °C. Then, 48 h after the reaction, the catalysts were isolated from the mixture by magnetic separation (Fig. S1, ESI†). The final products were analysed by gas chromatography-mass spectrometry (GC-MS).Recycling experiments of catalysts were conducted at optimal conditions. Typically, the isolated catalysts were washed with the solvent and dried in an oven at 80 °C. Furthermore, the catalysts were reduced at 400 °C in H2 flow before reuse for the next run.
Results and discussion
The Co-MOF and catalysts were first evaluated by powder X-ray diffraction (XRD) to confirm their structure. The XRD pattern of Co-MOF (Fig. 2a) matched exactly the reported XRD pattern in the literature,32–34 thereby confirming the structure of Co-MOF in the present work. The N-doped carbon layers-supported Co nanocatalysts (Co@CN) were synthesized by the thermolysis of Co-MOF in Ar flow. The XRD patterns of Co@CN are shown in Fig. 2b. The XRD diffraction peaks at 44.4°, 51.6° and 76.1° corresponded to the (1 1 1), (2 0 0) and (2 2 0) lattice planes of metallic Co, respectively, demonstrating that the Co2+ in Co-MOF are reduced in situ to zero-valent Co nanoparticles by the carbon layers at high temperature. In addition, a distinct peak of the Co@CN-900 catalysts appeared at ≈25.8°, which could be assigned to the formation of graphitic carbon layers,35 and this speculation was confirmed by transmission electron microscopy (TEM). The graphitic carbon layers were derived from calcining of the organic ligands of MOFs at high temperature.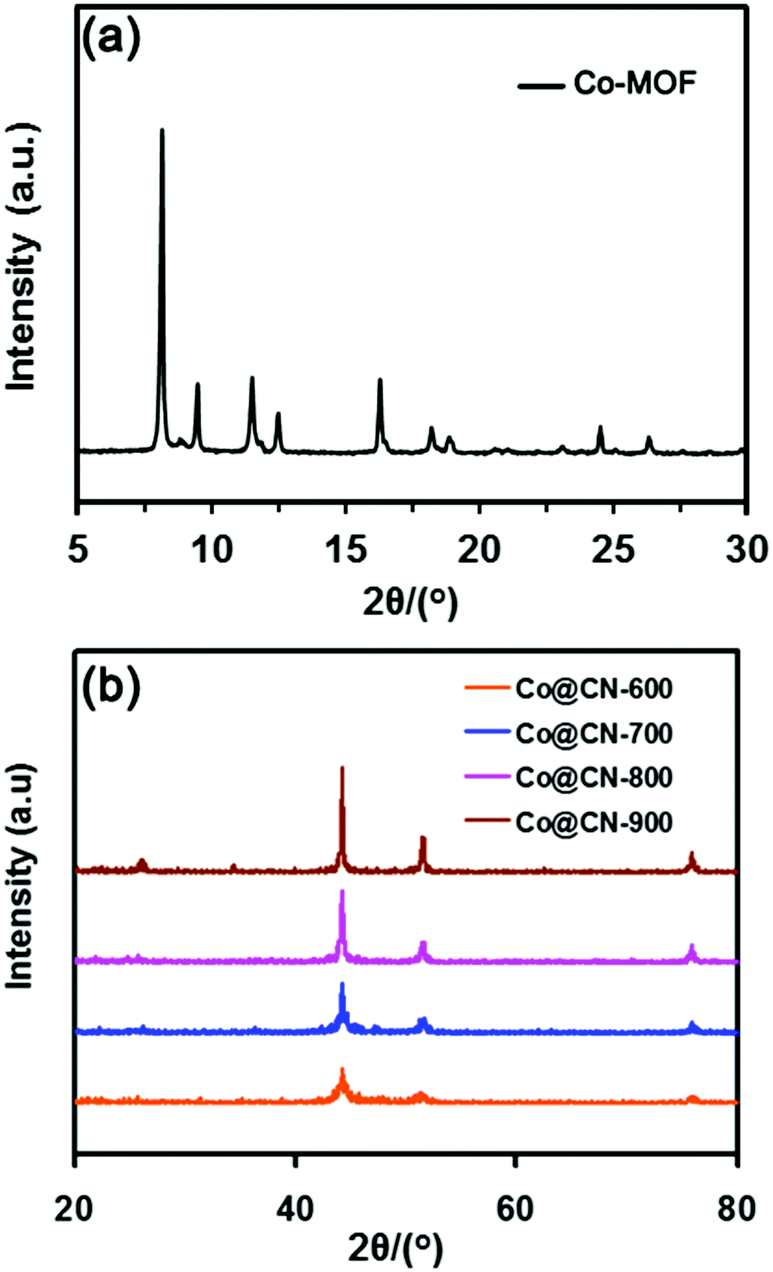 | ||
| Fig. 2 XRD patterns of Co-MOF (a) and Co@CN (b) catalysts. | ||
Subsequently, to further confirm that Co2+ was converted into metallic Co during thermolysis, X-ray photoelectron spectroscopy (XPS) measurements were conducted. The Co 2p peaks at 781.98 eV and 797.98 eV with the shakeup peaks could be assigned to the inherent characteristics of high-spinning Co2+ in Co-MOF (Fig. 3a)36 whereas, after thermolysis, the peaks were transferred into lower bonding energies of 3.48 eV and 3.78 eV, further indicating that Co2+ were converted to Co nanoparticles during thermolysis at high temperature.37
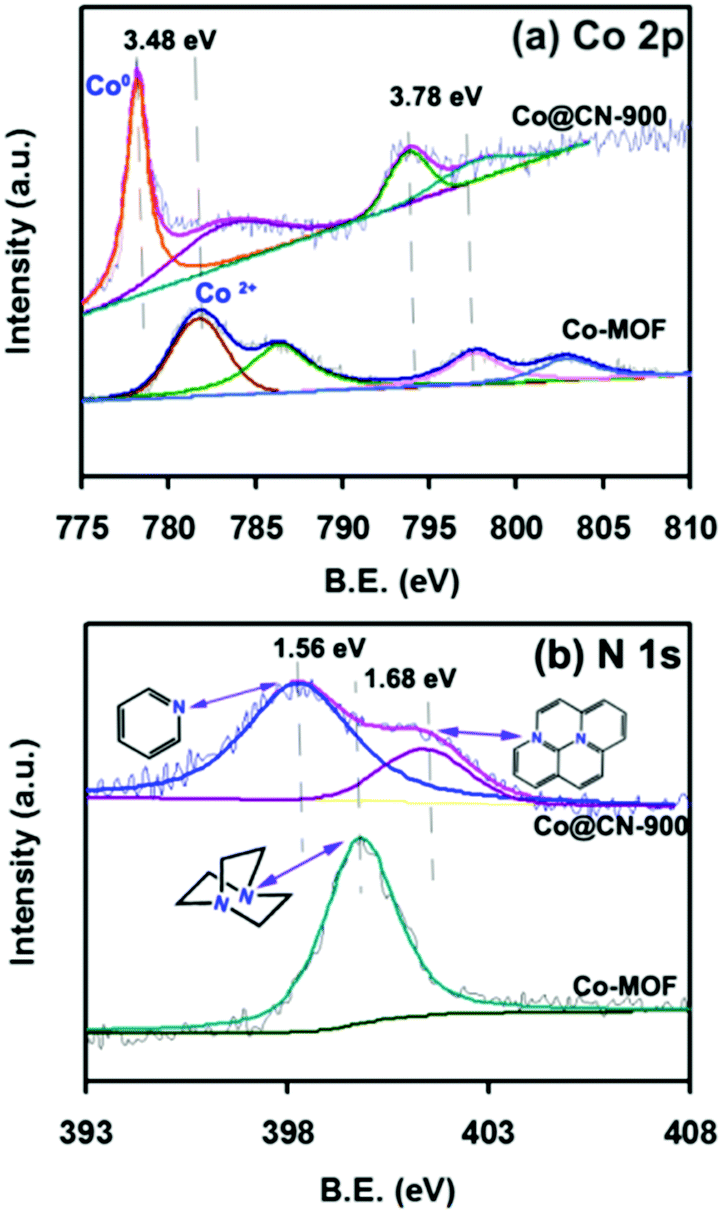 | ||
| Fig. 3 XPS patterns of Co-MOF and Co@CN-900, (a) Co 2p and (b) N 1s. | ||
The N 1s peaks at 399.80 eV could be assigned to the N atom of 1,4-diazabicyclooctane (ted) coordinated to a Co2+ (Fig. 3b).38,39 After calcining, two new peaks appeared at 398.24 eV and 401.48 eV, which could be assigned to the pyridinic nitrogen and graphitic nitrogen,40–42 demonstrating that the N atoms were successfully doped into the catalysts. It is worth noting that the N sites can endow the catalysts with abundant basic sites,37 which have crucial roles in facilitating electron/proton transfer.
To confirm the existence of basic sites, temperature-programmed desorption (TPD) of CO2 (CO2-TPD) was conducted (Fig. S2, ESI†). In general, the strength of basic sites is determined by the desorption temperatures of CO2: the higher the desorption temperature, the stronger is the basicity.43 As shown in Fig. S2 (ESI†), Co@CN-800 and Co@CN-900 showed two obvious CO2 desorption peaks, strongly demonstrating that the Co@CN catalysts contained abundant basic sites.
The precise surface appearances of catalysts were observed by scanning electron microscopy (SEM). As shown in Fig. 4, the catalysts maintained a basic framework configuration with the parental Co-MOF (the coordinated structure between metal ions and organic ligands is shown in Fig. S3a (ESI†), a typical SEM image of Co-MOF is shown in Fig. S3b, ESI†), except that the surface of the catalysts became rougher with an increase in thermolysis temperatures. Furthermore, large numbers of mesoporous structures appeared at the surface of catalysts when the thermolysis temperatures were increased to ≤800 °C and ≤900 °C. Such mesoporous structures in catalysts are thought to be beneficial for mass transfer during reaction processes.
 | ||
| Fig. 4 SEM images of Co@CN. (a) Co@CN-600, (b) Co@CN-700, (c) Co@CN-800 and (d) Co@CN-900. | ||
More structural morphologies were revealed by TEM. As shown in Fig. 5, with an increase in calcining temperatures from 600 °C to 900 °C, the mean particle size of Co nanoparticles increased gradually from 8.18 nm to 15.48 nm (Fig. 5a–d). Furthermore, high-resolution transmission electron microscopy (HR-TEM) showed that each Co nanoparticle was surrounded by carbon layers with an interplanar spacing of 0.34 nm, which is in accordance with the lattice plane of graphitic structures (Fig. 5e).34,40,44 The surrounding graphitic carbon layers have important roles in “fixing” the Co nanoparticles into catalysts to improve the stability of catalysts.37 Moreover, energy-dispersive X-ray spectroscopy (EDS) results further identified the coexistence of Co, C and N elements (Fig. 5f).
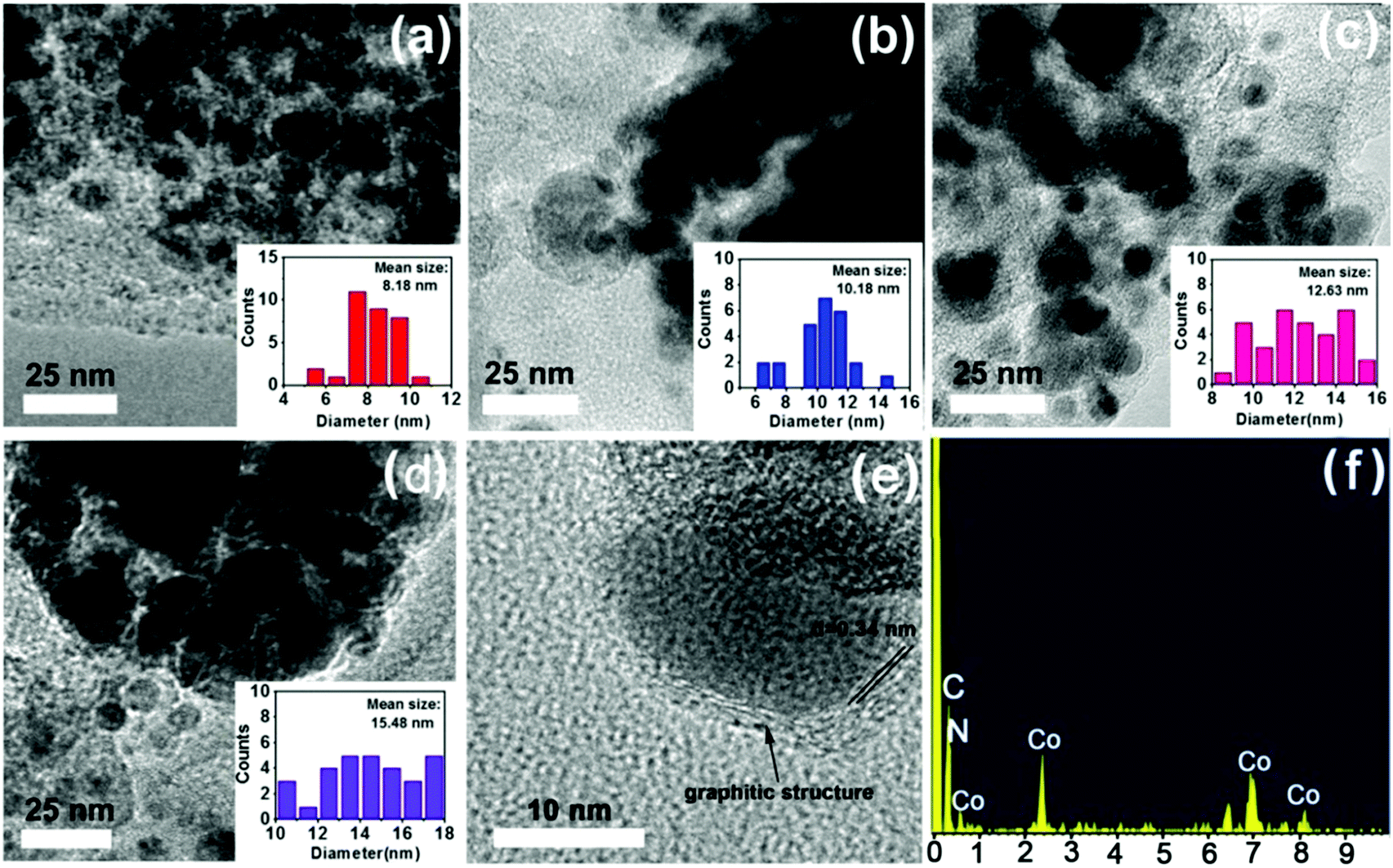 | ||
| Fig. 5 TEM images with corresponding particle distribution. (a) Co@CN-600, (b) Co@CN-700, (c) Co@CN-800, (d) Co@CN-900, and (e) HR-TEM for an individual nanoparticle and the EDS result of Co@CN-900 (f). | ||
Fig. 6 shows the elemental maps of a typical Co nanoparticle wrapped by N-doped carbon layers. They reveal the N and C atoms to be distributed homogeneously in the catalysts, further indicating that the N atoms were doped successfully into the catalysts. Moreover, the superimposed elemental mapping of Co, C and N indicate the Co nanoparticles to be surrounded by the N-doped carbon layer, which has a significant role in confining Co nanoparticles into the catalyst, thereby improving the stability of metal catalysts.
 | ||
| Fig. 6 Elemental mapping for each Co nanoparticle. (a) Co, (b) C, (c) N and (d) superimposed elemental mapping of Co, C and N. | ||
The specific surface area of Co-MOF and catalysts was determined by N2 absorption/desorption measurements (BET). As shown in Fig. S4a (ESI†), the N2 adsorption curve and desorption curve can overlap with each other, indicating that there were only microporous structures in Co-MOF, and the results of pore-distribution studies further supported this appraisal (Fig. S4b, ESI†). After calcining, an obvious hysteresis loop was observed at each BET curve (Fig. S4c, ESI†), suggesting formation of the mesoporous structures that had been observed from SEM. The pore sizes of catalysts were mainly 6–7 nm (Fig. S4d, ESI†). Co content was determined by atomic absorption spectrophotometry (AAS). The proportions of C and N were detected by elemental analyses. As shown in Table S2 (ESI†), the content of Co, C and N in catalysts was around 37 wt%, 58 wt% and 2 wt%, respectively.
Selective transfer hydrogenation of CAL to COL over Co@C catalysts at 80 °C are shown in Table 1. A blank run without catalysts showed no conversion (Table 1, entry 1), whereas the conversions of CAL increased when the Co@CN catalysts were added to the mixture (Table 1, entries 2–5). In general, the conversion of CAL improved dramatically with an increase in the calcining temperature of catalysts (Table 1, entries 2–5). Co@CN-900 proved to be the best catalysts for this transformation, achieving 80% conversion for CAL with 90% selectivity for COL (Table 1, entry 5). This series of catalysts showed good selectivity toward COL to further optimize the reaction conditions.
|
|
||||
|---|---|---|---|---|
| Entry | Cat. | Sol. | Con. | Sel. |
| (%) | (%) | |||
| a 1 mmol cinnamaldehyde, Co 10 mmol% catalyst, 2 mL i-PrOH, 48 h, 80 °C. b 100 °C. c 70 °C. d 60 h. e 15 mmol% catalyst. f 1 mL CH3OH. g 35 h. h CN-900 was prepared by using nitric acid to remove Co from Co@CN-900. | ||||
| 1 | — | i-PrOH | — | — |
| 2 | Co@CN-600 | i-PrOH | 16 | 81 |
| 3 | Co@CN-700 | i-PrOH | 37 | 89 |
| 4 | Co@CN-800 | i-PrOH | 55 | 90 |
| 5 | Co@CN-900 | i-PrOH | 80 | 90 |
| 6b | Co@CN-900 | i-PrOH | 90 | 55 |
| 7c,d | Co@CN-900 | i-PrOH | 88 | 91 |
| 8e | Co@CN-900 | i-PrOH | 97 | 90 |
| 9e | Co@CN-900 | 2-Butanol | 92 | 82 |
| 10 | Co@CN-900 | n-Hexanol | >99 | 99 |
| 11e | Co@CN-900 | n-Octylalcohol | Trace | Trace |
| 12e | Co@CN-900 | CH3OH | — | — |
| 13e,f,g | Co@CN-900 | Ethanediol | >99 | 88 |
| 14e,f | Co@CN-900 | Glycerol | >99 | 77 |
| 15e | Co@CN-900 | HCOOH | — | — |
| 16h | CN-900 | n-Hexanol | Trace | Trace |
| 17e | Co@CN-900 | Toluene | — | — |
To optimize the reaction conditions, the reaction temperatures were investigated. Increasing the reaction temperature to 100 °C improved the conversion of CAL, whereas the selectivity of COL fell from 90% to 55% (Table 1, entry 6). Lowering the reaction temperature and prolonging the reaction time could achieve 88% conversion, but the selectivity of COL showed no significant improvement (Table 1, entry 7). Increasing the ratio of catalysts could improve the conversion dramatically, achieving 97% conversion for CAL, nevertheless, the selectivity of COL showed almost no change (Table 1, entry 8).
To optimize the reaction system further, different alcohols were employed as proton donors. First, 2-butanol showed relatively high activity, providing 92% conversion for CAL with 82% selectivity for COL (Table 1, entry 9). Subsequently, n-hexanol and n-octylalcohol were also studied in our system. Surprisingly, we could achieve 99% selectivity for COL with full conversion by using n-hexanol as a proton donor (Table 1, entry 10). Conversely, almost no product was obtained when n-octylalcohol was used for this transformation (Table 1, entry 11), which could be attributed to the high steric hindrance of the long carbon chain. Subsequently, CH3OH was used as a solvent and proton-donor for this transformation. As shown in Table 1, almost no conversion was observed (Table 1, entry 12). Ethanediol and glycerol were also investigated. Owing to the viscous property of ethanediol and glycerol, 1 mL CH3OH was added to ethanediol and glycerol to improve the performance of mass transfer. However, the selectivities of COL were very low even though both of them exhibited >99% conversions (Table 1, entries 13 and 14).
Formic acid is one of most common proton donors in transfer hydrogenation reactions. Therefore, formic acid was also considered in this work. Nevertheless, essentially no reaction occurred (Table 1, entry 15). The reason, as we have mentioned above, is that the N atoms in catalysts can endow the catalysts with many basic sites, which have remarkable roles in facilitating proton transfer.37 However, the basic sites may lose efficiency through acid–base reactions when they come into contact with formic acid, so no product is detected in this system. This hypothesis also proves that the basic sites (N atoms) had significant roles in our catalytic system. Subsequently, to confirm the role of metal nanoparticles, CN-900, which was prepared by removing the Co nanoparticles from Co@CN-900 catalysts, was also tested at the optimal condition (Table 1, entry 16). However, no reaction was observed in this system, proving the crucial roles of Co nanoparticles, and indicating the strong coordination interactions between Co nanoparticles and N atoms in this bi-functional catalyst.
Notably, to prove that alcohols were the only source of H atoms, we employed toluene as a solvent for this transformation under a N2 atmosphere (Table 1, entry 17). As expected, no conversion was detected in this system, firmly indicating that the alcohols were the only source of protons. Moreover, the transfer hydrogenation reaction essentially stopped after removal of the catalyst from the mixture at 16 h, strongly suggesting that the reaction proceeded mostly on the surface of catalysts (Fig. S5, ESI†).
For recyclability, the catalysts were separated by magnetic separation and washed with n-hexanol and methanol several times. After drying in an oven at 80 °C, the catalysts were reduced by H2 at 400 °C for 2 h, and then purged by N2 flow for 1 h before reuse in the next run. The catalysts showed good recyclability, and almost no apparent loss in conversion or selectivity were observed in the transfer hydrogenation of CAL even after four runs (Fig. S6, ESI†). Moreover, after the reaction, the catalysts were removed with a magnet and then the liquid mixture was measured using AAS to ascertain if the Co nanoparticles had leached from the catalysts. The AAS results showed that only trace amounts of Co (<0.03%) leached into the liquid phase during the reaction. This indicated that the Co nanoparticles were highly stabilized against leaching, and also showed the significant role of graphitic carbon layers in fixing the Co nanoparticles into the catalysts.
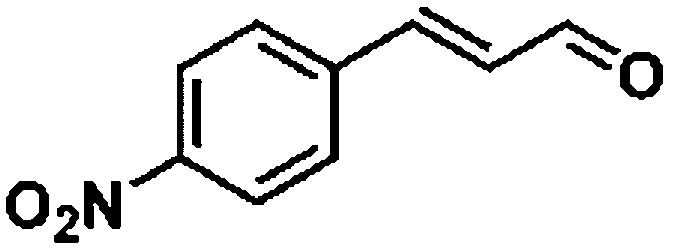
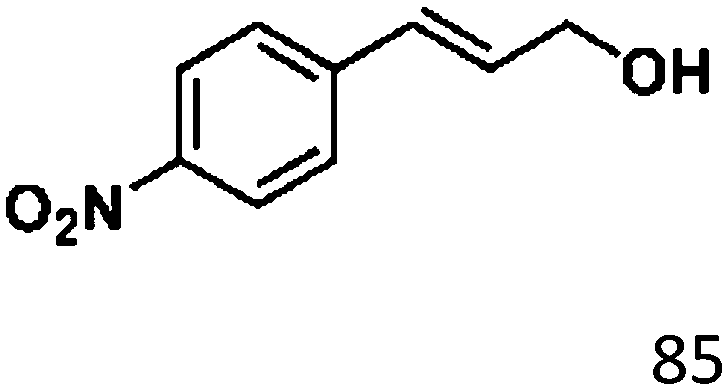
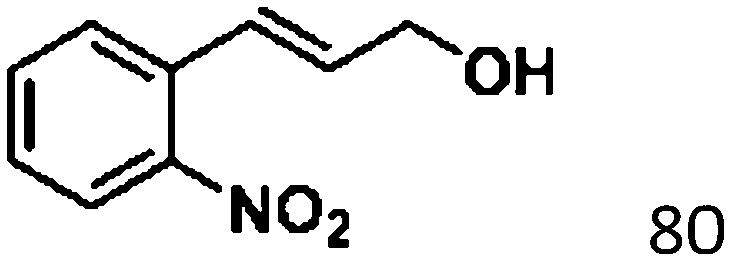
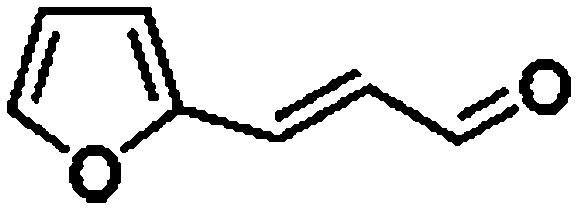
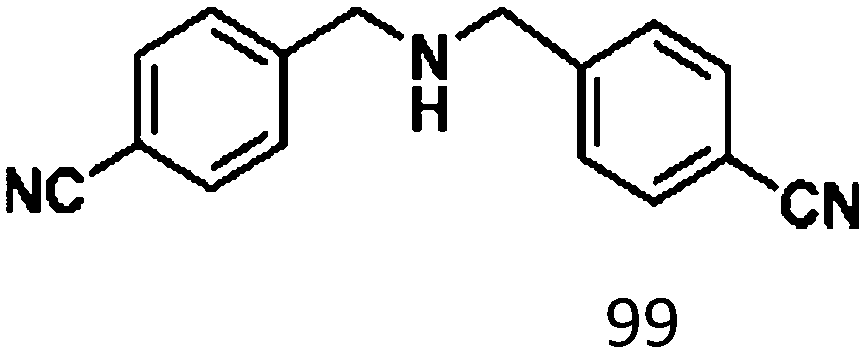
For a promising heterogeneous catalyst, compatibility is considered to be a significant factor. Subsequently, the scope of application of this system was investigated under optimal conditions. Typically, substrates with electron-donating groups, such as 4-methyl-cinnamaldehyde, 4-methoxy-cinnamaldehyde and 3-methoxy-cinnamaldehyde, were investigated first, after 40–44 h reactions, and achieved >90% yields for the corresponding products (Table 2, entries 1–3). Remarkably, 2-methoxy-cinnamaldehyde helped to make transfer hydrogenation proceed smoothly despite an ortho substituent resulting in high steric hindrance, providing a 90% yield for the desired product (Table 2, entry 4). Electron-withdrawing groups-substituted CALs were also able to conduct selective transfer hydrogenation by improving the reaction temperature slightly, achieving >80% yields for the corresponding products (Table 2, entries 5–8). A heteroatom-containing substrate could also be hydrogenated after prolonging the reaction time, affording ≈80% yield for the corresponding unsaturated alcohol (Table 2, entry 9).

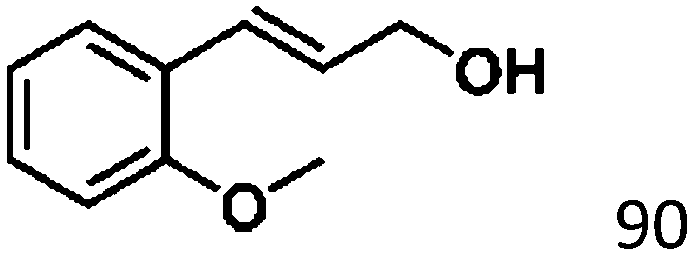

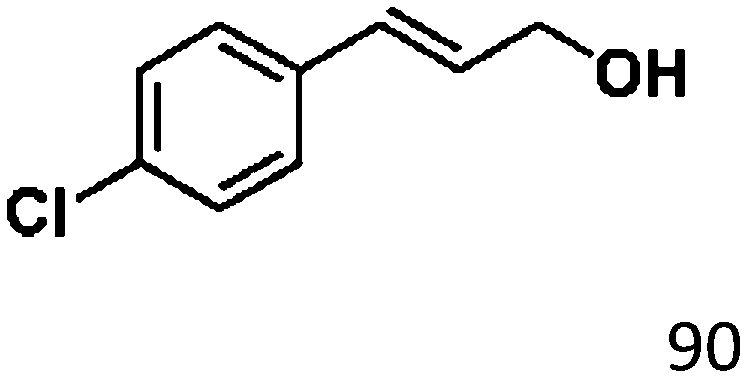
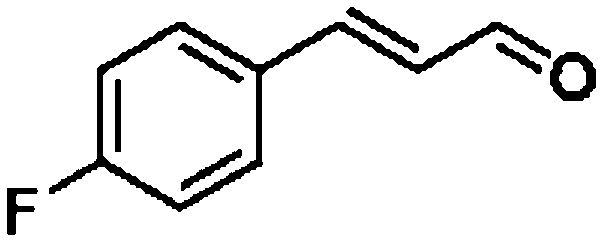
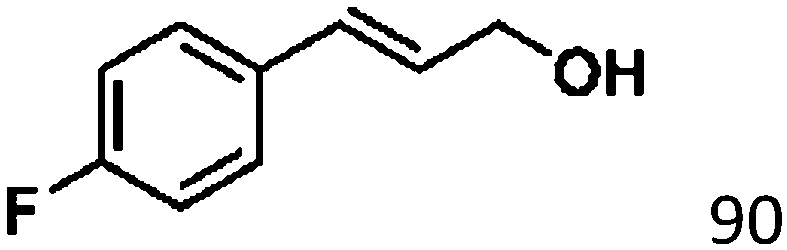

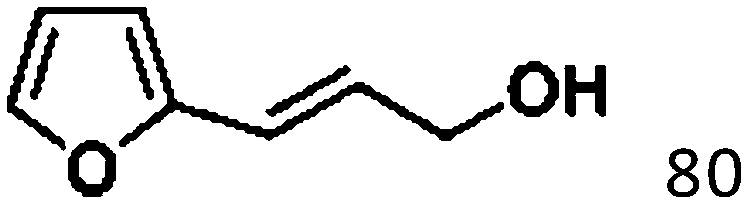
To further investigate the scope of application of this system, different unsaturated groups were tested.
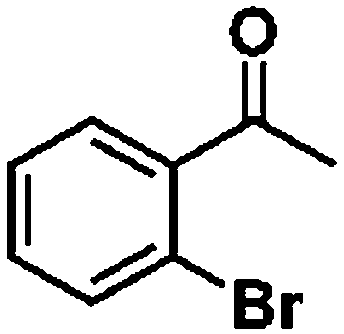
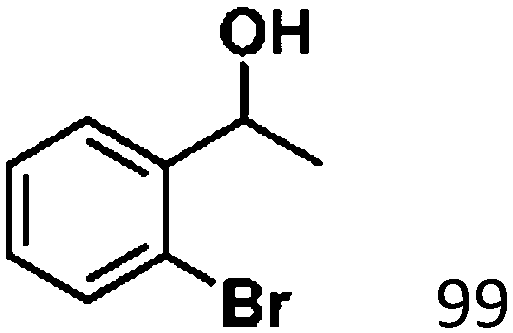
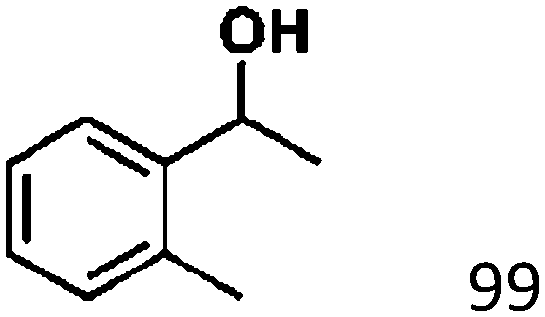
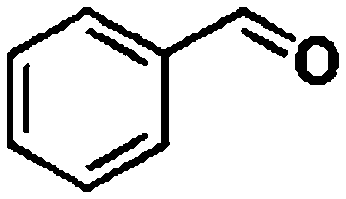
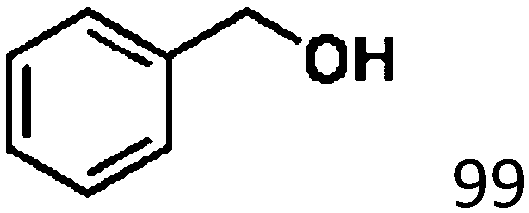
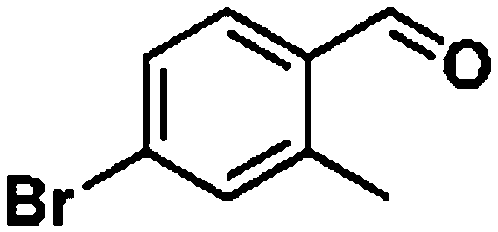
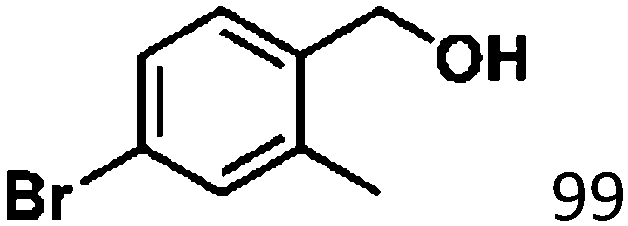
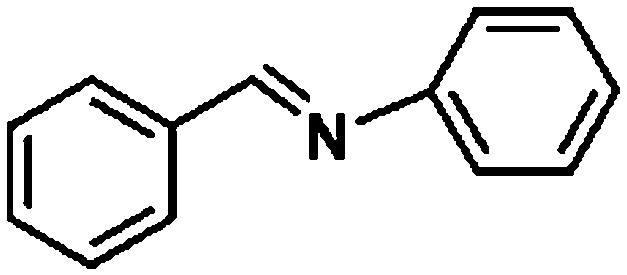
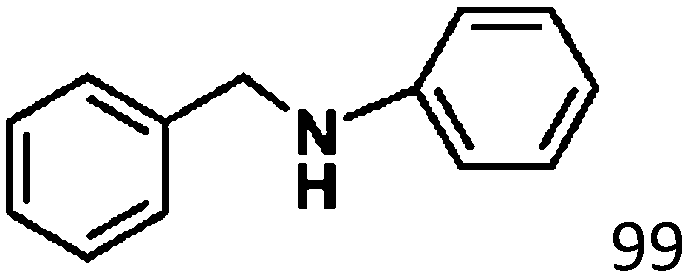
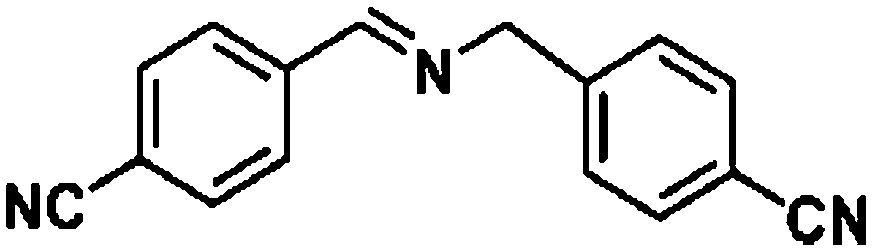
Ketones such as 2-bromoacetophenone and 2-methylacetophenone could help to make transfer hydrogenation proceed smoothly, affording 99% yields for the corresponding products (Table 3, entries 1 and 2). Benzaldehyde and 4-bromo-2-methylbenzaldehyde were hydrogenated in our system, giving ≈99% yields for the desired products (Table 3, entries 3 and 4). Notably, it was more difficult to achieve selective transfer hydrogenation of imines owing to cleavage of the CN bond. However, in this system, imines could undergo transfer hydrogenation successfully, thereby providing the corresponding amines with 99% yields (Table 3, entries 5 and 6). Selective transfer hydrogenation of nitrobenzene was a greater challenge than that for other unsaturated bonds owing to the p–π conjugative effect of the nitro group.45 However, in this system, the nitro group could also be hydrogenated to afford a quantitative yield of the corresponding product by improving the reaction temperature and prolonging the reaction time (Table 3, entry 7). In general, hydrogenation of the NO2 group was more difficult than that of the C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N bond. However, in this system, the NO2 group was hydrogenated rather than the CN bond, indicating that the oxygen atom was preferentially adsorbed on Co catalysts to promote hydrogenation of the NO2 group. This phenomenon is in accordance with studies reporting experimental conditions.46,47
N bond. However, in this system, the NO2 group was hydrogenated rather than the CN bond, indicating that the oxygen atom was preferentially adsorbed on Co catalysts to promote hydrogenation of the NO2 group. This phenomenon is in accordance with studies reporting experimental conditions.46,47


Finally, a reasonable reaction mechanism was proposed (Fig. 7). First, the CO bond at the end of the molecule can be absorbed on the metal nanoparticle more easily than the CC bond in the middle of the molecule because of the steric effect. Meanwhile, owing to the low d-electron density of Co, the CO bond was absorbed strongly on metallic Co species,22 leading to the formation of a CAL-Co intermediate (Fig. 7, II). Meanwhile, the n-hexanol was absorbed on the surface of cobalt nanoparticle with a linear interaction through the O−H of the alcohol moiety (Fig. 7, II), which has been shown by some researchers.22,48 Subsequently, the protons were released from n-hexanol with the aid of basic sites to generate the aldehyde and metal hydride, which is hypothesized to be the catalytically active species (Fig. 7, III and IV).22,49,50 Notably, it has been reported that base additives are required during the transfer hydrogenation process to facilitate proton release, but no base additives were added in our system. Therefore, basic sites, which were shown by CO2-TPD measurements, could have the same roles as base additives. Subsequently, the 2 H attacked the activated CAL to produce the COL product (Fig. 7, V). During this process, the basic sites not only transfer protons from the metal hydride to the activated CAL, but also prevent the products from “stacking” on the surface of catalysts to maintain the consecutive activities of catalysts.51
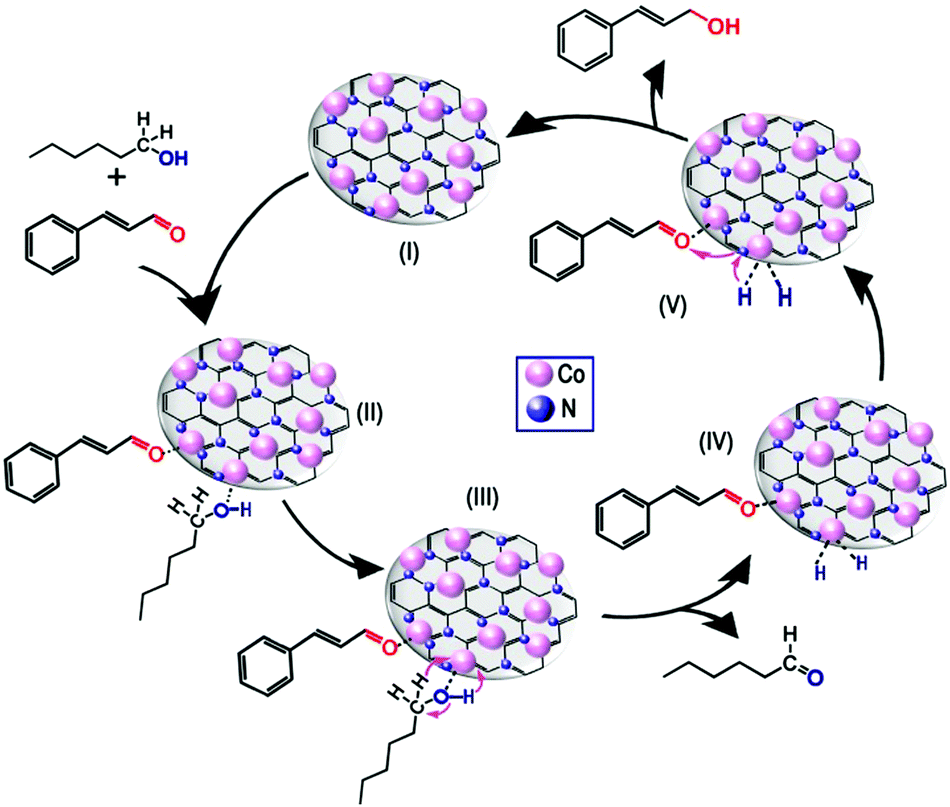 | ||
| Fig. 7 Proposed mechanism for the transfer hydrogenation of cinnamaldehyde to cinnamyl alcohol over Co@CN-900 catalysts. | ||
Conclusions
We developed a new N-doped carbon matrix-encapsulated Co bi-functional nanocatalyst (Co@CN) with a novel N-containing Co-MOF as a sacrificial template. The Co@CN bi-functional nanocatalyst could be employed effectively for selective transfer hydrogenation of an α,β-unsaturated aldehyde to obtain a high yield of an α,β-unsaturated alcohol under a mild reaction condition in the absence of additives. The experimental results indicated obvious coordination interactions between Co nanoparticles and N atoms in these bi-functional catalysts. Moreover, increases in the calcining temperature saw the catalytic activity enhance dramatically. By optimizing the reaction conditions, Co@CN-900 with n-hexanol as a proton donor exhibited optimal activity for this transformation, achieving 99% selectivity for COL with full conversion. Also, a reasonable reaction mechanism for the transfer hydrogenation of CAL to COL over catalysts was proposed. In addition, different substrates helped transfer hydrogenation to proceed smoothly in this catalytic system, indicating the wide compatibility of this system. Moreover, the catalysts showed good recyclability, and high conversion and selectivity could be maintained even up to four runs. This combination of recyclable, highly efficient and environmentally benign characteristics makes this system a highly promising candidate in many other hydrogenation reactions.Acknowledgements
This work was supported by research funding from the Scientific Research Fund of the Chemical Synthesis and Pollution Control Key Laboratory of Sichuan Province (CSPC2015-1-2), Scientific Research Fund of China West Normal University (412553), Scientific Research Fund of Sichuan Provincial Education Department (466768, 17TD0036), Scientific Research Fund of Science & Technology of Sichuan Province (2017JY0015), Youth Innovation Promotion Association of CAS (2015316) and The National High Technology Research and Development Program of China (863 Program 2015AA021107).Notes and references
- L. Saudan, A. Acc. Chem. Res., 2007, 40, 1309 CrossRef CAS PubMed.
- K. Bauer, D. Garbe and H. Surburg, Flavors and Fragrances, Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH, Weinheim, 2003 Search PubMed.
- H. L. Liu, L. Zhong and Y. W. Li, Ind. Eng. Chem. Res., 2015, 54, 1487 CrossRef CAS.
- H. N. Chen, D. A. Cullen and J. Z. Larese, J. Phys. Chem. C, 2015, 119, 28885 CAS.
- P. Maity, S. Yamazoe and T. Tsukuda, ACS Catal., 2013, 3, 182 CrossRef CAS.
- S. Jiménez, J. A. López, M. A. Ciriano, C. Tejel, A. Martíne and R. A. Sánchez-Delgado, Organometallics, 2009, 28, 3193 CrossRef.
- M. T. Zhao, K. Yuan, Y. Wang, G. D. Li, J. Guo, L. Gu, W. P. Hu, H. J. Zhao and Z. Y. Tang, Nature, 2016, 539, 76 CrossRef CAS PubMed.
- J. Zhao, J. Ni, J. H. Xu, J. T. Xu, J. Cen and X. N. Li, Catal. Commun., 2014, 54, 72 CrossRef CAS.
- G. Wienhöfer, F. A. Westerhaus, K. Junge, R. Ludwig and M. Beller, Chem. – Eur. J., 2013, 19, 7701 CrossRef PubMed.
- M. S. Ide, B. Hao, M. Neurock and R. J. Davis, ACS Catal., 2012, 2, 671 CrossRef CAS.
- S. Fleischer, S. L. Zhou, K. Junge and M. Beller, Angew. Chem., Int. Ed., 2013, 52, 5120 CrossRef CAS PubMed.
- L. J. Malobela, J. Heveling, W. G. Augustyn and L. M. Cele, Ind. Eng. Chem. Res., 2014, 53, 13910 CrossRef CAS.
- M. Manikandan, A. K. Venugopal, A. S. Nagpure, S. Chilukuri and R. Thirumalaiswamy, RSC Adv., 2016, 6, 3888 RSC.
- Z. Y. Guo, X. X. Chao, L. Zhou, T. W. Goh, X. L. Li, D. T. A. Thiel and W. Y. Huang, ACS Catal., 2014, 4, 1340 CrossRef CAS.
- N. Gorgas, B. Stöger, L. F. Veiros and K. Kirchner, ACS Catal., 2016, 6, 2664 CrossRef CAS PubMed.
- H. L. Peng, Z. Y. Mo, S. J. Liao, H. G. Liang, L. J. Yang, F. Luo, H. Y. Song, Y. L. Zhong and B. Q. Zhang, Sci. Rep., 2013, 3, 1765 CrossRef.
- L. F. Lai, J. R. Potts, D. Zhan, L. Wang, C. K. Poh, C. H. Tang, H. Gong, Z. X. Shen, J. Y. Lin and R. S. Ruoff, Energy Environ. Sci., 2012, 5, 7936 CAS.
- D. E. Karakasa, F. Durapb, A. Baysalb, Y. S. Ocakc, K. Rafikovad, E. C. Kayac, H. Temelc and C. Kayanb, J. Organomet. Chem., 2016, 824, 33 CrossRef.
- (a) D. E. Karakasa, F. Durapb, A. Baysalb, Y. S. Ocakc, K. Rafikovad, E. C. Kayac, H. Temelc and C. Kayanb, J. Organomet. Chem., 2016, 824, 104 CrossRef; (b) F. Wang, R. Shi, Z. Q. Liu, P. J. Shang, X. Pang, S. Shen, Z. Feng, C. Li and W. Shen, ACS Catal., 2013, 3, 890 CrossRef CAS; (c) J. L. Long, Y. Zhou and Y. W. Li, Chem. Commun., 2015, 51, 2331 RSC.
- G. H. Wang, X. H. Deng, D. Gu, K. Chen, H. Tüysüz, B. Spliethoff, H. J. Bongard, C. Weidenthaler, W. Schmidt and F. Schüth, Angew. Chem., Int. Ed., 2016, 55, 11101 CrossRef CAS PubMed.
- (a) N. Meyer, A. J. Lough and R. H. Morris, Chem. – Eur. J., 2009, 15, 5605 CrossRef CAS PubMed; (b) Q. Fan, Y. Liu, Y. Zheng and W. Yan, Chem. Eng. China., 2008, 2, 63 CrossRef CAS.
- M. Kennema, I. B. D. de Castro, F. Meemken and R. Rinaldi, ACS Catal., 2017, 7, 2437 CrossRef CAS.
- G. H. Lai, R. X. Zhou, X. X. Han and X. M. Zheng, Chin. Chem. Lett., 2004, 15, 1494 CAS.
- X. Han, R. Zhou, B. Yue and X. Zheng, Catal. Lett., 2006, 109, 157 CrossRef CAS.
- J. R. Cabrero-Antonino, R. Adam, K. Junge, R. Jackstell and M. Beller, Catal. Sci. Technol., 2017, 7, 1981 CAS.
- M. T. Zhao, K. Deng, L. C. He, Y. Liu, G. D. Li, H. J. Zhao and Z. Y. Tang, J. Am. Chem. Soc., 2014, 136, 1738 CrossRef CAS PubMed.
- L. He, F. Weniger, H. Neumann and M. Beller, Angew. Chem., Int. Ed., 2016, 55, 2 CrossRef.
- (a) Y. Cui, Y. Yue, G. Qian and B. Chen, Chem. Rev., 2011, 112, 1126 CrossRef PubMed; (b) M. Latroche, S. Surblé, C. Serre, C. Mellot-Draznieks, P. L. Llewellyn, J. H. Lee, J. S. Chang, S. H. Jhung and G. Férey, Angew. Chem., Int. Ed., 2006, 45, 8227 CrossRef CAS PubMed; (c) Y. K. Hwang, D. Y. Hong, J. S. Chang, S. H. Jhung, Y. K. Seo, J. Kim, A. Vimont, M. Daturi, C. Serre and G. Férey, Angew. Chem., Int. Ed., 2008, 47, 4144 CrossRef CAS PubMed.
- M. H. Zhang, J. Yang, W. Q. Kan, Y. Y. Liu and J. F. Ma, Cryst. Growth Des., 2016, 1, 265 Search PubMed.
- J. Zhao, Y. N. Wang, W. W. Dong, Y. P. Wu, D. S. Li and Q. C. Zhang, Inorg. Chem., 2016, 7, 3265 CrossRef PubMed.
- D. N. Dybtsev, H. Chun and K. Kim, Angew. Chem., Int. Ed., 2004, 38, 5033 CrossRef PubMed.
- Y. Zhao, H. Wu, T. J. Emge, Q. Gong, N. Nijem, Y. J. Chabal, L. Kong, D. C. Langreth, H. Liu, H. Zeng and J. Li, Chem. – Eur. J., 2011, 17, 5101 CrossRef CAS PubMed.
- Z. Liang, M. Marshall and A. L. Chaffee, Microporous Mesoporous Mater., 2010, 132, 305 CrossRef CAS.
- K. Tan, N. Nijem, P. Canepa, Q. Gong, J. Li, T. Thonhauser and Y. J. Chabal, Chem. Mater., 2012, 24, 3153 CrossRef CAS.
- W. Zhong, H. L. Liu, C. H. Bai, S. J. Liao and Y. W. Li, ACS Catal., 2015, 5, 1850 CrossRef CAS.
- M. C. Biesinger, B. P. Payne, A. P. Grosvenor, L. W. M. Lau, A. R. Gerson and R. C. Smart, Appl. Surf. Sci., 2011, 257, 2717 CrossRef CAS.
- J. L. Long, K. Shen, L. Chen and Y. W. Li, J. Mater. Chem. A, 2016, 4, 10254 CAS.
- M. C. Biesingera, B. P. Payne, A. P. Grosvenor, L. W. M. Lau, A. R. Gerson and R. C. Smart, Appl. Surf. Sci., 2011, 7, 2717 CrossRef.
- F. G. Souza, P. Richa, A. Siervo, G. E. Oliveira, C. H. M. Rodrigues, M. Nele and J. C. Pinto, Macromol. Mater. Eng., 2008, 8, 675 CrossRef.
- F. Jaouen, J. Herranz, M. Lefevre, J. P. Dodelet, U. I. Kramm, I. Herrmann and E. A. Ustinov, ACS Appl. Mater. Interfaces, 2009, 1, 1623 CAS.
- X. Wang and Y. W. Li, J. Mol. Catal. A. Chem., 2016, 420, 56 CrossRef CAS.
- X. L. Li, H. L. Wang, J. T. Robinson, H. Sanchez, G. Diankov and H. J. Dai, J. Am. Chem. Soc., 2009, 131, 15939 CrossRef CAS PubMed.
- M. L. Dieuzeide, M. Jobbagy and N. Amadeo, Catal. Today, 2013, 213, 50 CrossRef CAS.
- Y. Endo and J. E. Backvall, Chem. – Eur. J., 2011, 17, 12596 CrossRef CAS PubMed.
- J. L. Long, B. L. Yin, Y. W. Li and L. Zhang, J. AIChE Journal, 2014, 10, 3565 CrossRef.
- C. Y. Hsu, T. C. Chiu, M. H. Shih, W. J. Tsai, W. Y. Chen and C. H. Lin, Phys. Chem. C, 2010, 114, 4502 CrossRef CAS.
- X. Xu, Y. Li, Y. Gong, P. Zhang, H. Li and Y. Wang, J. Am. Chem. Soc., 2012, 134, 16987 CrossRef CAS PubMed.
- (a) D. Al-Mawlawi and J. M. J. Saleh, Chem. Soc., Faraday Trans., 1981, 77, 2965 RSC; (b) Y. H. Zhou, P. H. Lv and G. C. Wang, J. Mol. Catal. A: Chem., 2006, 258, 203 CrossRef CAS; (c) O. Borck, I. H. Svenum and A. Borg, Surf. Sci., 2009, 603, 2378 CrossRef CAS; (d) I. H. Svenum, O. Borck, K. Schulte, L. E. Walle and A. Borg, Surf. Sci., 2009, 603, 2370 CrossRef CAS; (e) X. Y. Pang, C. Wang, Y. H. Zhou, J. M. Zhao and G. C. Wang, THEOCHEM, 2010, 948, 1 CrossRef CAS.
- J. Long, K. Shen and Y. Li, ACS Catal., 2017, 7, 275 CrossRef CAS.
- S. M. S. Joseph, J. E. Bäckvall, G. A. Pher and P. Brandt, Chem. Soc. Rev., 2006, 35, 237 RSC.
- S. Y. Chin, F. J. Lin and A. N. Ko, Catal. Lett., 2009, 132, 389 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: TPD, BET, SEM, et al. See DOI: 10.1039/c7qm00189d |
| This journal is © the Partner Organisations 2017 |