
Morphology control enables thickness-insensitive efficient nonfullerene polymer solar cells†
Xiaoyu
Liu
ab,
Long
Ye
 *c,
Wenchao
Zhao
b,
Shaoqing
Zhang
*a,
Sunsun
Li
b,
Gregory M.
Su
d,
Cheng
Wang
d,
Harald
Ade
*c and
Jianhui
Hou
*b
*c,
Wenchao
Zhao
b,
Shaoqing
Zhang
*a,
Sunsun
Li
b,
Gregory M.
Su
d,
Cheng
Wang
d,
Harald
Ade
*c and
Jianhui
Hou
*b
aSchool of Chemistry and Biology Engineering University of Science and Technology Beijing, Beijing 100083, China. E-mail: shaoqingz@ustb.edu.cn
bState Key Laboratory of Polymer Physics and Chemistry, Beijing National Laboratory for Molecular Sciences, Institute of Chemistry, Chinese Academy of Sciences, Beijing 100190, P. R. China. E-mail: hjhzlz@iccas.ac.cn
cDepartment of Physics, North Carolina State University, Raleigh, North Carolina 27695, USA. E-mail: hwade@ncsu.edu; lye4@ncsu.edu
dAdvanced Light Source, Lawrence Berkeley National Laboratory, Berkeley, CA, USA
First published on 22nd June 2017
Abstract
Owing to the use of cost-effective materials and excellent stability, nonfullerene polymer solar cells (PSCs) have great potential for realizing large-area industrial production. In contrast to fullerene-based devices, non-fullerene PSCs have exhibited a superior photovoltaic performance with up to 12% efficiency and long-term thermal stability. Presently, one of the major factors hindering industrial production is the high sensitivity of the power conversion efficiency (PCE) to thickness variations, which can significantly affect the manufacturing yields and production costs of roll-to-roll processing. Specifically, the device fill factors and PCEs of many high-efficiency nonfullerene PSCs show a significant loss when the thickness of the active layer is over 100 nm. In order to achieve high output capabilities earlier, there is an urgent need to find a processing method to fabricate high-efficiency thick-film nonfullerene PSCs. Controlling the morphology and performance sensitivity in thick-film non-fullerene devices is a great challenge in the field. Here, we present a simple morphology optimization method via thermal annealing to fabricate highly efficient thickness-insensitive non-fullerene PSCs. After this treatment, PBDB-T/IT-M-based nonfullerene PSCs can afford an impressive PCE of up to ∼9.4% at an active layer thickness of 250 nm. In addition, the devices with an active layer thickness of 400 nm still maintain a high efficiency close to 9%. The photovoltaic properties and morphology parameters resolved from hard and soft X-ray scattering clearly indicate that thermal annealing plays a key role in improving the film thickness insensitivity for non-fullerene PSCs.
Introduction
Solution-processed bulk-heterojunction (BHJ) organic solar cells have been attracting increased attention in recent years due to their great potential to make large area and flexible solar panels through low-cost solution-coating methods.1–6 In BHJ polymer solar cells (PSCs), the active layer typically incorporates a conjugated polymeric donor and a polymeric or small molecule acceptor material.7–9 Most high-performance PSCs are fabricated with a thin active layer with a thickness of around 100 nm due to the relatively low carrier mobilities of the organic semiconductors. As the active layer thickness of the PSCs increases, the probability of charge recombination also increases, resulting in a reduced fill factor (FF) and consequently inferior photovoltaic performance.10–15 Although thin-film PSCs are promising for lowering production costs, it is challenging to fabricate reproducible PSCs with precise control over commercialization processes. For large area and high-speed roll-to-roll printing, increasing the thickness of the active layer would not only absorb more solar radiation and produce higher photocurrent, but also be more tolerant of thickness variation for high quality or yield control of PSC products. Therefore, the pursuit of thickness-insensitive photovoltaic devices via a facile method is of great importance for the large-scale production of PSCs.Substantial progress in the PCE has been achieved in thick-film polymer–fullerene PSCs mainly due to extensive efforts in improving and developing the thickness-insensitive devices for photoactive layers over 200 nm. In 2005, the Yang group fabricated a polymer-based photovoltaic device that consisted of P3HT![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :PCBM (1:1) with an active layer thickness of 210–230 nm and a very high FF of 67.4% and a PCE of 4.4%.16 Subsequently, the You group reported the first successful application of fluorine to a donor–acceptor conjugated polymer, PBnDT–DTffBT, which exhibited exceptional performance (7.2% for a PBnDT–DTffBT/PC61BM-based device with a 190 nm thick active layer) in BHJ solar cells.17 Recently, Yan and co-workers developed a favorable polymer:fullerene morphology (containing highly crystalline and small polymer domains) in high-performance thick-film PSCs (250–300 nm) for multiple polymer:fullerene combinations. All devices yielded PCEs over 9.5% and the best combination achieved a PCE of 10.8% and fill factor up to 77%.18 As demonstrated, more and more studies on thick film PSCs are focused on the conventional BHJ active layer based on a semi-crystalline polymer donor and a fullerene derivative acceptor.16–26 However, there is a lack of a simple and effective processing method to improve the thickness insensitivity in non-fullerene PSCs.
:PCBM (1:1) with an active layer thickness of 210–230 nm and a very high FF of 67.4% and a PCE of 4.4%.16 Subsequently, the You group reported the first successful application of fluorine to a donor–acceptor conjugated polymer, PBnDT–DTffBT, which exhibited exceptional performance (7.2% for a PBnDT–DTffBT/PC61BM-based device with a 190 nm thick active layer) in BHJ solar cells.17 Recently, Yan and co-workers developed a favorable polymer:fullerene morphology (containing highly crystalline and small polymer domains) in high-performance thick-film PSCs (250–300 nm) for multiple polymer:fullerene combinations. All devices yielded PCEs over 9.5% and the best combination achieved a PCE of 10.8% and fill factor up to 77%.18 As demonstrated, more and more studies on thick film PSCs are focused on the conventional BHJ active layer based on a semi-crystalline polymer donor and a fullerene derivative acceptor.16–26 However, there is a lack of a simple and effective processing method to improve the thickness insensitivity in non-fullerene PSCs.
At present, the photovoltaic performance of PSCs based on non-fullerene acceptors has surpassed the traditional fullerene-based PSCs.27–36 Compared with fullerene acceptors, non-fullerene acceptors have attracted much attention because of their outstanding absorption properties, easily tunable energy levels, and promise for compatibility with high-throughput roll-to-roll printing.37–42 However, the majority of fullerene-free PSCs have optimum PCEs with active layer thicknesses of 100 nm or less. For example, our group demonstrated PBDB-T:ITIC-based fullerene-free PSCs with outstanding PCEs of up to 11.21%, and the optimal thickness is 100 nm.43,44 Our group also rationally designed a new non-fullerene acceptor (IT-M in Fig. 1) with a superior photovoltaic performance. For the PBDB-T:IT-M devices, the highest PCE among single-junction laboratory-scale PSCs was 12.05%.5 Importantly, the Li group fabricated a non-fullerene PSC with a high PCE of 11.77% by using m-ITIC as an acceptor and a medium band gap conjugated polymer, J61, as a donor, and the performance showed less thickness dependence.45 As has been demonstrated in earlier studies, most of the record-performing non-fullerene PSCs were obtained at an optimal thickness of ∼100 nm, while the thickness-dependent behavior of nonfullerene PSCs has not been well studied and explored in terms of molecular packing, mesoscale morphology, and charge transport. Thus, it is very important and meaningful to develop a simple morphology optimization method which could maintain high PCEs across a range of film thicknesses in the non-fullerene PSCs.
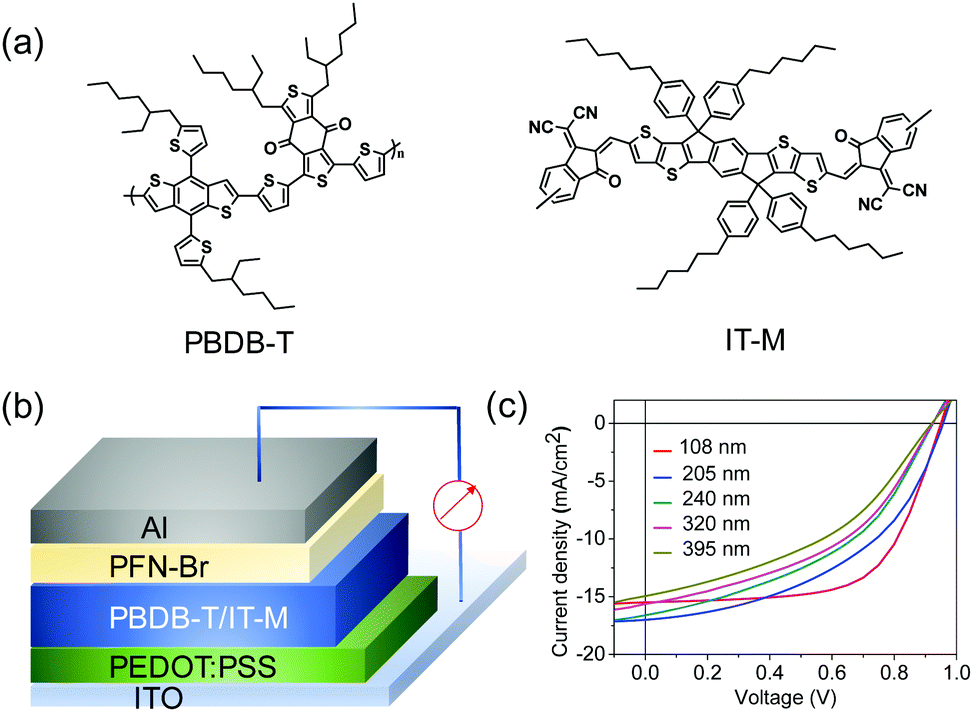 | ||
| Fig. 1 (a) Chemical structures of PBDB-T and IT-M; (b) device configuration of non-fullerene PSCs; (c) J–V curves of as-cast PBDB-T:IT-M devices as a function of active layer thickness. | ||
In this contribution, we present thermal annealing as a simple morphology optimization method to promote the device performance of thick-film non-fullerene PSCs. These devices are based on PBDB-T (poly[(2,6-(4,8-bis(5-(2-ethylhexyl)thiophen-2-yl)-benzo[1,2-b:4,5-b′]dithiophene))-alt-(5,5-(1′,3′-di-2-thienyl-5′,7′-bis(2-ethylhexyl)benzo[1′,2′-c:4′,5′-c′]dithiophene-4,8-dione))]):IT-M and a regular device configuration of ITO (indium tin oxide)/PEDOT:PSS/PBDB-T:IT-M(1:1, w/w)/PFN-Br/Al (see Fig. 1a and b). Encouragingly, a high PCE of up to 9.37% was achieved (82% of the optimum value at ∼100 nm) with an active layer thickness of ∼250 nm. Moreover, the PBDB-T:IT-M devices still reach a PCE of nearly 9% (77% of the optimum value) when the thickness of the active layer is close to 400 nm. Detailed morphology and charge transport studies suggest efficient charge transport, ordered packing and relatively high domain composition variations in non-fullerene systems collectively afford such high PCEs in thick-film devices after thermal annealing. Our findings indicate that the formation of efficient photovoltaic blends with a thick active layer is not limited to fullerene-based systems. Our morphology control approach should be applicable to other polymer:non-fullerene systems to fabricate nearly thickness-insensitive devices.
Results and discussion
Annealing promotes ordering
We first examined the characteristics of devices that were prepared with PBDB-T:IT-M with varying active layer thicknesses. The current density–voltage (J–V) curves under the illumination of AM 1.5 G 100 mW cm−2 are shown in Fig. 1c. The thick-film devices show a dramatic reduction in both FF and PCE compared to the control devices (∼100 nm active layer). For roll-to-roll solution-processing of BHJ PSCs, a large loss of photovoltaic performance resulting from thick active layers would be unfavourable. The ultraviolet-visible (UV-vis) absorption spectra of the as-cast and annealed films are presented in Fig. 2. In comparison to the as-cast devices, the absorption spectra of the thermally annealed devices show a slight red-shift. Moreover, a significant increase in the intensity of the characteristic absorption peak (∼700 nm) of IT-M is observed for the annealed blend films, indicating that thermal annealing most likely influences the ordering of the small molecule acceptor in this blend system. Therefore, we introduce thermal annealing as a simple method for the morphology optimization of the PBDB-T:IT-M films.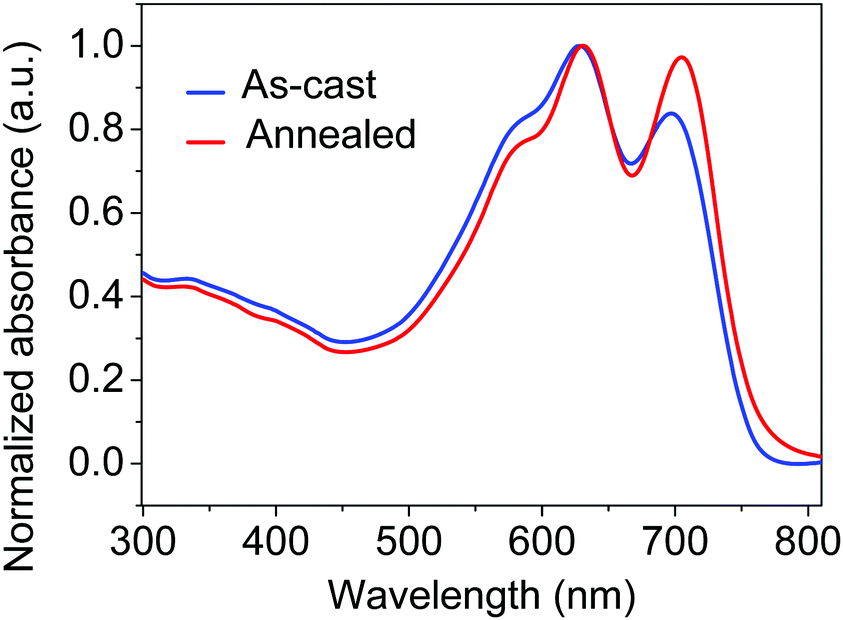 | ||
| Fig. 2 UV-vis absorption spectra of as-cast and annealed PBDB-T:IT-M blend films with a thickness of ∼250 nm. | ||
Thickness-dependent photovoltaic properties
To study the thickness dependence of the photovoltaic performance of PBDB-T:IT-M-based non-fullerene PSCs, we fabricated the devices under optimal processing conditions while varying the thickness of the active layer. A regular device configuration of ITO (indium tin oxide)/PEDOT:PSS/PBDB-T:IT-M (1:1, w/w)/PFN-Br/Al was utilized in this work. The active layers with different thicknesses were prepared in two varieties: as-cast and post-cast thermally annealed. The processing parameters such as D/A ratio (w/w), additive ratio (v/v), and spin coating rate were systematically investigated to obtain the optimal device performance based on the PBDB-T:IT-M system. Here, chlorobenzene was selected as a host solvent to fabricate thick-film PSCs with an optimal D/A ratio of 1:1 and 1% (vol) of 1,8-diiodoctane as the additive. The photovoltaic performances of the thick-film PSCs were measured under illumination of an AM 1.5 G simulated solar light at 100 mW cm−2 and the detailed photovoltaic parameters are summarized in Table 1. The corresponding J–V curves are shown in Fig. 4a and Fig. S1 (ESI†). As shown in Fig. 3, the PCE and FF values of the as-cast and annealed devices are plotted as a function of the thickness of the active layer. Among the device parameters, we noticed that the significant loss in the PCE for thick-film devices is mainly due to a large drop in the FF. For instance, the FF is significantly decreased by ∼1/3 in the as-cast devices when the thickness of the active layer increases from ∼100 nm to ∼250 nm. After the thermal annealing treatment, the thickness dependence of the PCE and FF for the annealed devices is reduced relative to the as-cast devices.
:1) based BHJ solar cells fabricated with different active layer thicknesses
| Conditions | V oc (V) | J sc (mA cm−2) | FF (%) | PCEa (%) | Thickness (nm) |
|---|---|---|---|---|---|
| a The reported data are the average PCEs from ten devices. | |||||
| As-cast | 0.924 ± 0.01 | 16.60 ± 0.16 | 0.44 ± 0.02 | 6.52 ± 0.12 (6.73) | 240 |
| Annealed | 0.930 ± 0.01 | 9.90 ± 0.23 | 0.68 ± 0.01 | 6.17 ± 0.06 (6.28) | 50 |
| 0.961 ± 0.01 | 16.69 ± 0.13 | 0.70 ± 0.02 | 11.34 ± 0.12(11.50) | 100 | |
| 0.940 ± 0.01 | 18.17 ± 0.20 | 0.54 ± 0.03 | 9.21 ± 0.13 (9.35) | 225 | |
| 0.948 ± 0.01 | 18.13 ± 0.17 | 0.54 ± 0.03 | 9.09 ± 0.22 (9.37) | 250 | |
| 0.943 ± 0.01 | 18.44 ± 0.15 | 0.53 ± 0.01 | 9.03 ± 0.25 (9.32) | 305 | |
| 0.942 ± 0.01 | 18.66 ± 0.12 | 0.51 ± 0.02 | 8.73 ± 0.16 (8.90) | 390 | |
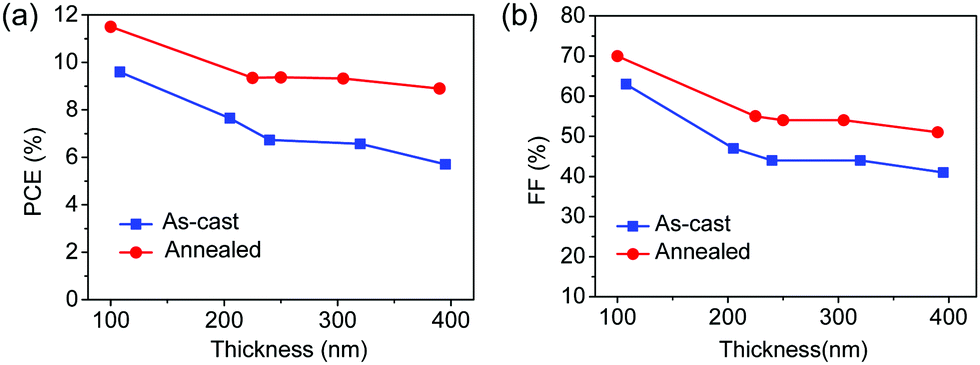 | ||
| Fig. 3 Plots of PCE (a) and FF (b) as a function of the active layer thickness for PBDB-T/IT-M BHJ solar cells under different conditions (as-cast and thermal annealing). | ||
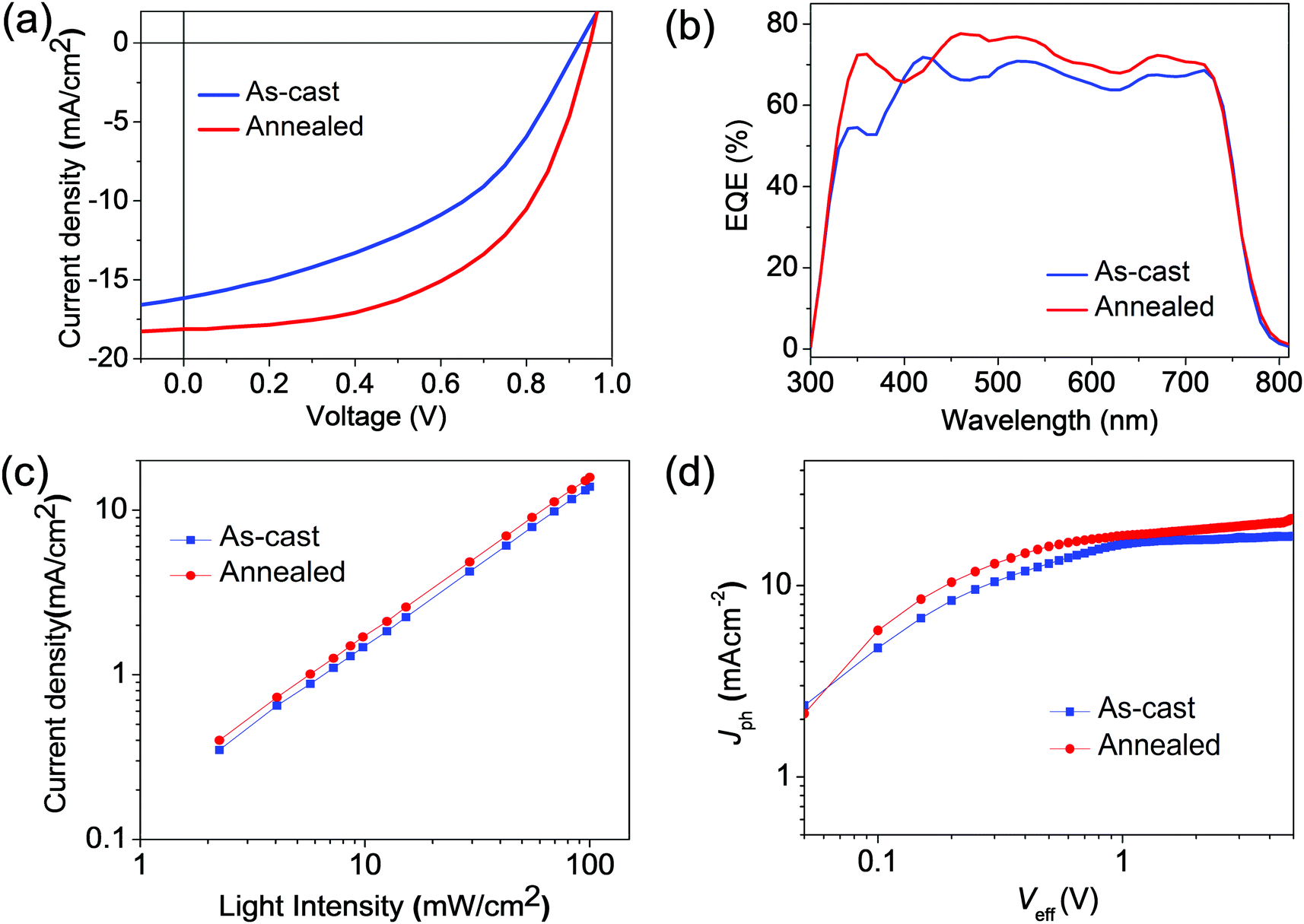 | ||
| Fig. 4 The best-performing thick-film PBDB-T/IT-M solar cells for as-cast and annealed devices: (a) J–V curves; (b) EQE curves; (c) plot of Jscvs. light intensity; (d) Jphversus Veff plots. | ||
We examined ∼250 nm-thick non-fullerene devices to amplify the morphology-performance relations, as the best-performing thick-film (over 200 nm) devices were achieved at a thickness of ∼250 nm (see Fig. 4a). With thermal annealing (100 °C), the thick-film PSCs based on PBDB-T:IT-M displayed a Voc of 0.95 V, a Jsc of 18.13 mA cm−2, and an FF of 0.54, yielding a high PCE of 9.37%. While for the as-cast devices without thermal annealing, the thick-film PSCs showed a lower PCE of 6.73% along with a decreased Voc of 0.92 V, a Jsc of 16.60 mA cm−2, and a low FF of 0.44. Furthermore, we fabricated thick-film PSCs with a larger device area of 1.00 cm2, and the photovoltaic parameters of the best device show a Voc of 0.93 V, a Jsc of 17.4 mA cm−2, and an FF of 0.51, resulting in a PCE of 8.28% (see Fig. S3, ESI†). The external quantum efficiency (EQE) curves of the two thick-film PSCs are displayed in Fig. 4b. Clearly, the thermally annealed devices show higher EQE values in the wavelength region of 300–750 nm, which is consistent with their higher Jsc. To understand the charge recombination of the thick-film PBDB-T:IT-M-based devices, we measured the dependence of Jsc as a function of light intensity (Plight) (Fig. 4c). Generally, a linear dependence of log (Jsc) and log (Plight) with a slope close to 1 suggests weak bimolecular recombination in the PSCs, whereas a slope far less than 1 indicates the partial loss of charge carriers due to bimolecular recombination between free holes and electrons during charge transport. Compared with the as-cast device, the fitted slope (S) of the annealed device is almost the same (0.97) within the experimental error.46,47 These results suggest that both the as-cast and annealed thick-film PBDB-T:IT-M based PSCs have a similar charge recombination behavior. The exciton dissociation probabilities (Pdiss) of these two kinds of devices were measured and analyzed by the curves of photo-generated current density (Jph) versus effective voltage (Veff). As shown in Fig. 4d, under short-circuit conditions, according to the equation Pdiss = Jph/Jsat (Jsat represents the saturation photocurrent density), the exciton dissociation probability was calculated to be 90.7% for the as-cast devices and 91.9% for the thermally annealed devices. This indicates that PBDB-T:IT-M based devices with thermal annealing are more beneficial to exciton dissociation and charge creation.
To further explore the influence of thermal annealing on the charge transport properties of thick films, the space-charge-limited (SCL) hole and electron mobilities were measured with a device structure of ITO/PEDOT:PSS/active layer/Au and ITO/ZnO/active layer/Al, respectively.48 As shown in Fig. S4 (ESI†), the hole mobility (μh) and electron mobility (μe) of the PBDB-T/IT-M based as-cast devices are 2.09 × 10−4 cm2 V−1 s−1 and 1.22 × 10−4 cm2 V−1 s−1 with a μh/μe value of 1.71. In contrast, the devices with thermal annealing exhibit higher hole and electron mobilities (3.41 × 10−4 cm2 V−1 s−1 and 2.06 × 10−4 cm2 V−1 s−1 with μh/μe of 1.65). We also found that the electron mobility of the pure acceptor was significantly increased from 2.17 × 10−5 cm2 V−1 s−1 to 2.20 × 10−4 cm2 V−1 s−1 after thermal annealing. The higher carrier mobility in the active layer of the annealed devices relative to the as-cast devices resulted in the higher Jsc value for the corresponding PSCs. In contrast with fullerene-based thick-film devices, non-fullerene systems commonly show a reduction in charge carrier mobility of almost one or two orders of magnitude.49–51 Here, we interpret these results as follows: the improved carrier transport helps alleviate the accumulation of photo-generated carriers in thick-film devices, which decreases the probability of carrier recombination and consequently enhances the carrier collection efficiency.52–54
It is known that thicker films can enhance light absorption, while we still cannot achieve a higher PCE due to the decrease of the FF when the active layer thickness is over 100 nm.55,56 Due to the increased carrier diffusion length, more and more charge recombination will occur in the active layer. Presently, PSC systems still face a severe challenge to achieve efficient charge carrier transport and suppress recombination in thick-films. For thick-film non-fullerene PSCs based on PBDB-T:IT-M, thermal annealing can obviously maintain a reasonably good FF and enhance the Jsc as the active layer thickness increased up to 400 nm (see Fig. S1, ESI†). In contrast, as shown in Table S1 (ESI†), the photovoltaic performance of the as-cast devices decreased with an increase in active layer thickness. Our results suggest that thermal annealing is a good candidate as a processing step to improve thickness-insensitivity for the large area and high-speed roll-to-roll production of non-fullerene PSCs.
Surface morphology and molecular packing for thick-films
To understand the morphology in real-space, we used tapping-mode atomic force microscopy (AFM) and transmission electron microscopy (TEM) to analyze the annealed and as-cast thick-film (∼250 nm) devices. According to the AFM results (see Fig. 5a and b), the two blend films show a very similar surface topography, and the root-mean-square roughness (Rq) values of the two films are 1.93 nm and 2.11 nm, respectively. As shown in the AFM phase images (Fig. S5, ESI†) of the two blend films, the features of phase separation are clearly observed. In the TEM measurements, the two films also exhibit phase-separated morphologies (see Fig. S6, ESI†), which may be advantageous for realizing efficient exciton dissociations in the device. Based on the AFM and TEM observations, we were not able to quantitatively compare the length scale of phase separation between as-cast and annealed thick-film devices. Considering that AFM cannot reflect the bulk structure and morphology of the active layers in detail, we further analyzed thin films with synchrotron radiation-based techniques including grazing incidence wide-angle X-ray scattering (GIWAXS) and resonant soft X-ray scattering (R-SoXS). GIWAXS and R-SoXS were performed at beamlines 7.3.3 and 11.0.1.2, respectively, at the Advanced Light Source (ALS), Lawrence Berkeley National Laboratory.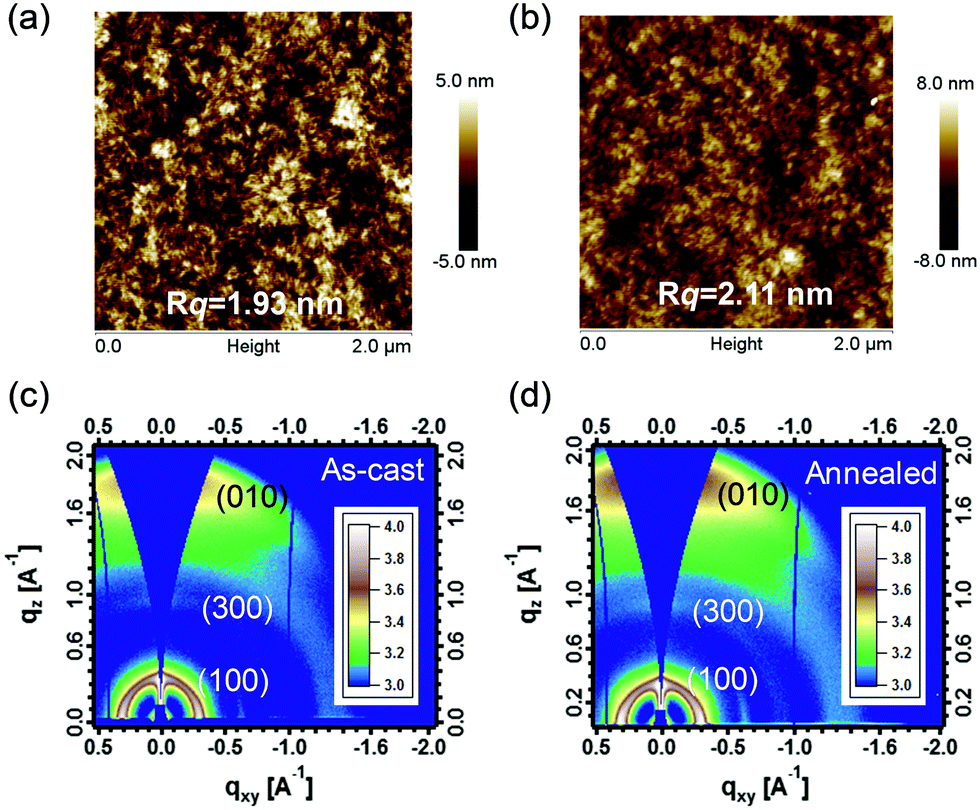 | ||
| Fig. 5 Tapping mode AFM topography of PBDB-T/IT-M based thick-film devices with (a) annealing treatment and (b) as-cast treatment; (c and d) 2D GIWAXS patterns of as-cast and annealed PBDB-T:IT-M thick-films (∼250 nm). | ||
GIWAXS was used to investigate the molecular packing and crystalline texture in the thick films with and without thermal treatment.57 From the 2D patterns and 1D line profiles (see Fig. 5c and d and Fig. S7, ESI†), the annealed thick-film shows a slightly higher scattering intensity although the features/peaks in the out-of-plane (OOP) direction in both the as-cast and annealed films are very similar. The π–π coherence length in the OOP direction is a key measure of ordering in organic semiconductor materials and often related to charge transport in the devices. We note that the OOP (010) peaks of the donor and acceptor have some overlap, and we deconvoluted the individual contributions via multi-peak fitting with Gaussian peaks.
We find that both the OOP π–π coherence lengths of the small molecular acceptors (SMAs) (LA) and the polymer donor (LD) are increased after annealing (see Table 2). We rationalized that thermal annealing provides sufficient driving forces for the polymer and SMA molecules to reorganize. The change of molecular ordering upon annealing is schematically shown in Fig. S8 (ESI†). It is expected that charge transport improves with increasing π–π coherence length. Therefore, the improved OOP π–π coherence lengths of SMA indicate the higher ordering of SMA in the thick film after thermal annealing. This may be a key factor that partially contributes to the higher Jsc and FF. This observation also agrees well with our recent studies.58
| Blends | L D (nm) | L A (nm) | Long period (nm) | Relative ACV |
|---|---|---|---|---|
| As-cast | 1.79 | 3.29 | 51.1 | 0.96 |
| Annealed | 1.89 | 3.74 | 53.5 | 1 |
Bulk morphology for thick-films
Resonant soft X-ray scattering (R-SoXS) was applied to analyze the domain spacing and average composition variation (average domain purity) of the thick-films. The soft X-ray energy was tuned between 270 and 300 eV in transmission R-SoXS experiments. Here, we select an X-ray energy of 283.8 eV to enhance the material contrast. From the scattering profiles plotted in Fig. 6, the two blend films exhibit similar length-scale features and a long period (center-to-center domain spacing) of ∼50 nm was obtained under the assumption of a globally isotropic 3D morphology (Please refer to an appendix in the SI for details). Following our previous analysis,59,60 the average composition variation (ACV) of an organic blend film is proportional to the square-root of the normalized integrated scattering intensity (ISI). Thus, ISI is a measure of the average domain purity over the full q range probed. Here, we found that the relative domain purity is improved by 4.2% after annealing. A higher domain purity can aid in efficient charge creation61 and improve the device FF. Recently, we found that the PBDB-T:IT-M system is likely62 a lower critical solution temperature (LCST) system (interaction parameter χ scales with −1/T). Also, this inference is verified experimentally by explicit miscibility measurements following previously established protocols63 (see Fig. S9, ESI†). We note that annealing at ∼150 °C is a temperature where the residual IT-M in the mixed polymer-rich domains is ∼24% and thus close to the percolation threshold,64 a composition that is likely optimal in terms of the balance between charge creation (Jsc) and charge recombination (FF).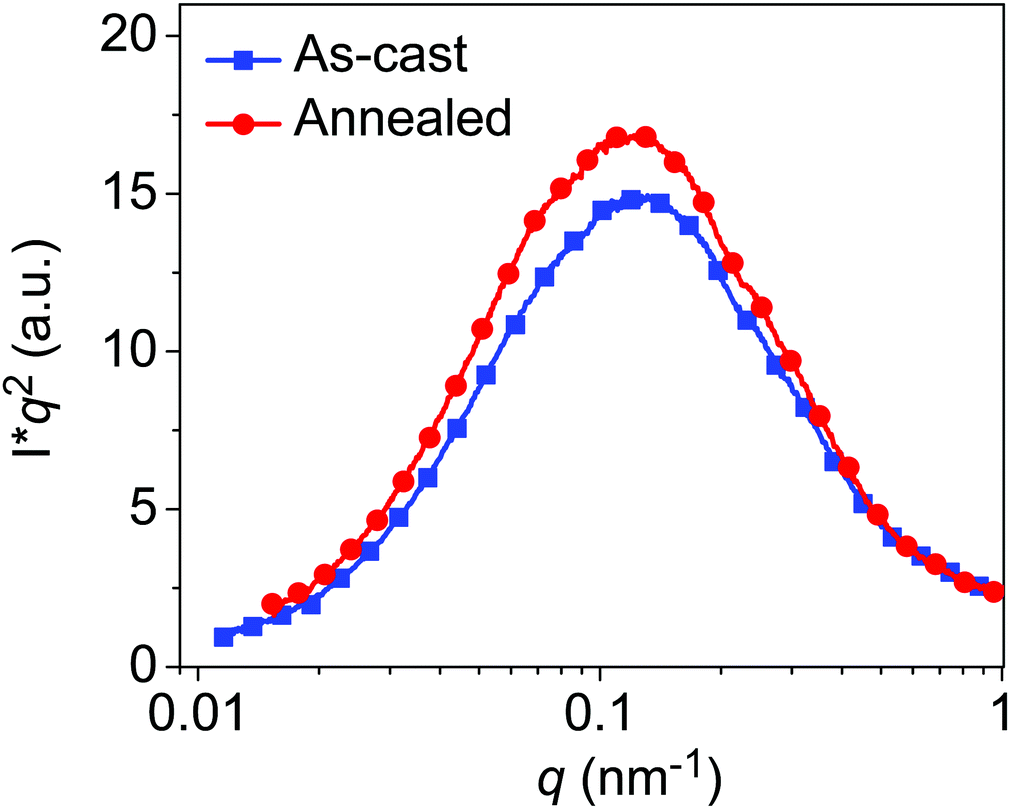 | ||
| Fig. 6 Thickness normalized and Lorentz-corrected R-SoXS profiles of the as-cast and annealed PBDB-T:IT-M thick-films. | ||
Generally, a higher χ parameter enables a larger average domain purity and a higher scattering intensity. Therefore, a higher processing temperature may induce a higher domain purity compared to as-cast devices processed at room temperature.5 In addition, the strong relation observed here is in line with the FF-ACV correlations shown in our prior studies of thin-film devices.60,62,65 These studies suggest that optimization of the molecular ordering and average composition variation is key to achieving high-efficiency in non-fullerene PSC devices.
Conclusions
In summary, we have systematically studied the molecular ordering, mesoscale morphology, and charge recombination in thick-film non-fullerene PSCs based on a high-performance PBDB-T/IT-M system. We found that thermal annealing can be very useful to boost the performance of thick-film PBDB-T/IT-M devices. A high PCE of 9.37% was obtained from PBDB-T:IT-M devices with a film thickness of 250 nm and the devices can maintain a high efficiency of ∼9% when the thickness of the active layer is increased to 400 nm. UV-vis absorption, SCLC mobility, and morphology studies show that the electron mobility of IT-M becomes higher and the blend films possess more ordered packing and a higher average domain purity after thermal annealing. Consequently, these factors improve charge creation and transport, and thus lead to an improved thickness insensitivity of device performance. We note that the photovoltaic performance in our thick-film devices is still limited by the carrier mobility. It will be more advantageous to improve the efficiency of thick film devices if the carrier mobility is improved via future molecular design and device engineering. These results reveal the critical morphological parameters that determine the photovoltaic performance in thick-film devices. This work highlights the importance of controlling molecular ordering and phase separation in achieving high-efficiency thick-film non-fullerene PSC devices, which will be beneficial for commercial roll-to-roll printing technology.Experiments
Fabrication of the polymer solar cells
PBDB-T35 and IT-M5 were synthesized as described in our previous works. The PSC devices were fabricated with a traditional device configuration of ITO (indium tin oxide)/PEDOT:PSS/PBDB-T:IT-M (1:1, w/w)/PFN-Br/Al. At first, the ITO-coated glass substrates were cleaned sequentially by using detergent, deionized water, acetone, and isopropanol. Then UV/ozone treatment was performed on the ITO glass substrates for 20 minutes. A thin layer (nearly 30 nm) of poly(3,4-ethylenedioxythiophene):poly(styrene sulfonate) (PEDOT:PSS) (Heraeus Materials, 4083) was spin coated onto the cleaned ITO surface, and the ITO substrates were annealed in air at 150 °C for 15 minutes. The active-layer solution of PBDB-T:IT-M (1:1, wt/wt) was prepared in chlorobenzene, and the solution was stirred on a hot plate at 40 °C for at least 5 h. The concentration of the PBDB-T:IT-M blend solution (based on the weight of the polymer) was 10 mg mL−1. 1% vol solvent additive (1, 8-diiodoctane) was added to the blend solution before the spin coating process. After spin coating the active layer onto PEDOT:PSS, the devices adopted two different processing methods (with and without thermal annealing) to optimize the morphology of the blend films. Next, a 5 nm thin PFN-Br layer was spin-coated on top of the active layer. Then, the device fabrications were completed by vacuum evaporation of an Al metal electrode. Except for the spin coating of PEDOT:PSS, the other processes were conducted inside a glove box under a nitrogen atmosphere.
Device measurements
The device current–voltage curves were measured under AM 1.5 G (100 mW cm−2) using a Class AAA solar simulator (XES-70S1, SAN-EI Electric Co., Ltd). The light intensity was calibrated to obtain a spectral mismatch to unity via a standard photovoltaic cell (with a KG5 filter). The effective area of the device is 4.03 mm2, which is defined by using the metal mask aligned with an aperture. All the EQE data were measured by using a solar cell spectral response measurement system (QE-R3011, Enli Technology Co. Ltd), which was equipped with a standard silicon solar cell. The film thickness data were determined using a surface profilometer (Dektak XT, Bruker). The SCLC measurements for the PBDB-T:IT-M devices were measured in the configuration of ITO/PEDOT:PSS/active layer (200 nm)/Au for hole-only devices and ITO/ZnO/active layer (200 nm)/Al for electron-only devices. The light-dependent tests for the PBDB-T:IT-M devices were measured by inserting neutral density filters to tune the light intensity during J–V testing. The nanoscale morphology of the blend films was measured by using a Veeco Nanoscope V atomic force microscope (AFM) in tapping mode. The transmission electron microscopy (TEM) characterization was carried out using a JEOL 2200FS instrument with an accelerating voltage of 160 kV. All film samples were spin cast on PEDOT:PSS coated indium tin oxide (ITO) substrates.X-ray scattering measurements
GIWAXS66 and near-edge X-ray absorption fine structure spectroscopy (NEXAFS)67 experiments were respectively done at beamlines 7.3.3 and 5.3.2.2 at the ALS. Resonant soft X-ray scattering (R-SoXS)68 was performed in a transmission geometry with linearly polarized photons under high vacuum (1 × 10−7 Torr) at beamline 11.0.1.2 at the Advanced Light Source (ALS), and the scattering 2D images were recorded by an in-vacuum cooled (−45 °C) CCD detector (Princeton Instruments, PI-MTE, 2048 pixels × 2048 pixels). Azimuthally averaged 1 − D I(q) − q profiles can be obtained by the reduction of 2D images and subsequently normalized for the instantaneous X-ray flux.Acknowledgements
We acknowledge financial support from the Ministry of Science and Technology of China (2014CB643501), NSFC (21325419, 91333204, 21604017 and 51373181), and the Chinese Academy of Sciences (XDB12030200, KJZD-EW-J01). L. Ye and H. Ade were supported by the ONR grant N00141512322 and a University of North Carolina General Administration Research Opportunity Initiative (ROI) grant. X-ray data were acquired at beamlines 11.0.1.2, 7.3.3, and 5.3.2.2 at the Advanced Light Source, LBNL, which is supported by the Director, Office of Science, Office of Basic Energy Sciences, of the U.S. Department of Energy under Contract No. DE-AC02-05CH11231. C. Zhu, A. L. D. Kilcoyne, Y. Yu, and E. Schaible are appreciated for beamline maintenance.Notes and references
- J. Zhao, Y. Li, J. Zhang, L. Zhang, J. Y. L. Lai, K. Jiang, C. Mu, Z. Li, C. L. C. Chan and A. Hunt, J. Mater. Chem. A, 2015, 3, 20108–20112 CAS.
- G. Yu, J. Gao, J. C. Hummelen, F. Wudl and A. J. Heeger, Science, 1995, 270, 1789–1791 CAS.
- C. J. Brabec, N. S. Sariciftci and J. C. Hummelen, Adv. Funct. Mater., 2001, 11, 15–26 CrossRef CAS.
- R. Søndergaard, M. Hösel, D. Angmo, T. T. Larsen-Olsen and F. C. Krebs, Mater. Today, 2012, 15, 36–49 CrossRef.
- S. Li, L. Ye, W. Zhao, S. Zhang, S. Mukherjee, H. Ade and J. Hou, Adv. Mater., 2016, 28, 9423–9429 CrossRef CAS PubMed.
- L. Ye, Y. Xiong, H. Yao, A. Gadisa, H. Zhang, S. Li, M. Ghasemi, N. Balar, A. Hunt, B. T. O'Connor, J. Hou and H. Ade, Chem. Mater., 2016, 28, 7451–7458 CrossRef CAS.
- W. Jiang, L. Ye, X. Li, C. Xiao, F. Tan, W. Zhao, J. Hou and Z. Wang, Chem. Commun., 2014, 50, 1024–1026 RSC.
- L. Lu, T. Zheng, Q. Wu, A. M. Schneider, D. Zhao and L. Yu, Chem. Rev., 2015, 115, 12666 CrossRef CAS PubMed.
- J. Chen and Y. Cao, J. Cheminf., 2010, 42, 1709–1718 Search PubMed.
- M. Shin, H. Kim, J. Park, S. Nam, K. Heo, M. Ree, C. S. Ha and Y. Kim, Adv. Funct. Mater., 2010, 20, 748–754 CrossRef CAS.
- W. Ma, C. Yang, X. Gong, K. Lee and A. J. Heeger, Adv. Funct. Mater., 2005, 15, 1617–1622 CrossRef CAS.
- Y. Y. Lee, K. H. Tu, C. C. Yu, S. S. Li, J. Y. Hwang, C. C. Lin, K. H. Chen, L. C. Chen, H. L. Chen and C. W. Chen, ACS Nano, 2011, 5, 6564–6570 CrossRef CAS PubMed.
- H. Kim, M. Shin and Y. Kim, J. Phys. Chem. C, 2009, 113, 1620–1623 CAS.
- S. R. Mi, H. J. Cha and J. Jin, Curr. Appl. Phys., 2010, 10, S206–S209 CrossRef.
- J. Peet, J. Y. Kim, N. E. Coates, W. L. Ma, D. Moses, A. J. Heeger and G. C. Bazan, Nat. Mater., 2007, 6, 497–500 CrossRef CAS PubMed.
- G. Li, V. Shrotriya, J. Huang, Y. Yao, T. Moriarty, K. Emery and Y. Yang, Nat. Mater., 2005, 4, 864–868 CrossRef CAS.
- H. Zhou, L. Yang, A. C. Stuart, S. C. Price, S. Liu and W. You, Angew. Chem., 2011, 50, 2995–2998 CrossRef CAS PubMed.
- Y. Liu, J. Zhao, Z. Li, C. Mu, W. Ma, H. Hu, K. Jiang, H. Lin, H. Ade and H. Yan, Nat. Commun., 2014, 5, 5293 CrossRef CAS PubMed.
- W. Li, K. H. Hendriks, W. S. C. Roelofs, Y. Kim, M. M. Wienk and R. A. J. Janssen, Adv. Mater., 2013, 25, 3182–3186 CrossRef CAS PubMed.
- N. Gasparini, L. Lucera, M. Salvador, M. Prosa, G. D. Spyropoulos, P. Kubis, H.-J. Egelhaaf, C. J. Brabec and T. Ameri, Energy Environ. Sci., 2017, 10, 885–892 CAS.
- A. Armin, M. Hambsch, P. Wolfer, H. Jin, J. Li, Z. Shi, P. L. Burn and P. Meredith, Adv. Energy Mater., 2015, 5, 1401221 CrossRef.
- X. Zhu, J. Fang, K. Lu, J. Zhang, L. Zhu, Y. Zhao, Z. Shuai and Z. Wei, Chem. Mater., 2014, 26, 6947–6954 CrossRef CAS.
- J. Chen, L. Zhang, X. Jiang, K. Gao, F. Liu, X. Gong, J. Chen and Y. Cao, Adv. Energy Mater., 2017, 7, 1601344 CrossRef.
- Z. Chen, P. Cai, J. Chen, X. Liu, L. Zhang, L. Lan, J. Peng, Y. Ma and Y. Cao, Adv. Mater., 2014, 26, 2586–2591 CrossRef CAS PubMed.
- J. Zhao, Y. Li, A. Hunt, J. Zhang, H. Yao, Z. Li, J. Zhang, F. Huang, H. Ade and H. Yan, Adv. Mater., 2016, 28, 1868–1873 CrossRef CAS PubMed.
- X. Zhu, B. Xia, K. Lu, H. Li, R. Zhou, J. Zhang, Y. Zhang, Z. Shuai and Z. Wei, Chem. Mater., 2016, 28, 943–950 CrossRef CAS.
- L. Ye, W. Jiang, W. Zhao, S. Zhang, D. Qian, Z. Wang and J. Hou, Small, 2014, 10, 4658–4663 CrossRef CAS PubMed.
- E. Zhou, J. Cong, K. Hashimoto and K. Tajima, Adv. Mater., 2013, 25, 6991–6996 CrossRef CAS PubMed.
- M. Schubert, D. Dolfen, J. Frisch, S. Roland, R. Steyrleuthner, B. Stiller, Z. Chen, U. Scherf, N. Koch, A. Facchetti and D. Neher, Adv. Energy Mater., 2012, 2, 369–380 CrossRef CAS.
- S. Li, H. Zhang, W. Zhao, L. Ye, H. Yao, B. Yang, S. Zhang and J. Hou, Adv. Energy Mater., 2016, 6, 1501991 CrossRef.
- C. Lee, H. Kang, W. Lee, T. Kim, K. H. Kim, Y. W. Han, W. Cheng and B. J. Kim, Adv. Mater., 2015, 27, 2466–2471 CrossRef CAS PubMed.
- Y. Lin, J. Wang, Z. G. Zhang, H. Bai, Y. Li, D. Zhu and X. Zhan, Adv. Mater., 2015, 27, 1170–1174 CrossRef CAS PubMed.
- L. Gao, Z. G. Zhang, L. Xue, J. Min, J. Zhang, Z. Wei and Y. Li, Adv. Mater., 2016, 28, 1884 CrossRef CAS PubMed.
- H. Li, Y. J. Hwang, B. A. Courtright, F. N. Eberle, S. Subramaniyan and S. A. Jenekhe, Adv. Mater., 2015, 27, 3266–3272 CrossRef CAS PubMed.
- D. Qian, W. Ma, Z. Li, X. Guo, S. Zhang, L. Ye, H. Ade, Z. A. Tan and J. Hou, J. Am. Chem. Soc., 2013, 135, 8464–8467 CrossRef CAS PubMed.
- Y. Zhou, T. Kurosawa, W. Ma, Y. Guo, L. Fang, K. Vandewal, Y. Diao, C. Wang, Q. Yan and J. Reinspach, Adv. Mater., 2014, 26, 3767–3772 CrossRef CAS PubMed.
- X. Zhan, Z. Tan, B. Domercq, Z. An, X. Zhang, S. Barlow, Y. Li, D. Zhu, B. Kippelen and S. R. Marder, J. Am. Chem. Soc., 2007, 129, 7246 CrossRef CAS PubMed.
- J. W. Jung, J. W. Jo, C. C. Chueh, F. Liu, W. H. Jo, T. P. Russell and A. K. Jen, Adv. Mater., 2015, 27, 3310–3317 CrossRef CAS PubMed.
- S. Holliday, R. S. Ashraf, A. Wadsworth, D. Baran, S. A. Yousaf, C. B. Nielsen, C. H. Tan, S. D. Dimitrov, Z. Shang and N. Gasparini, Nat. Commun., 2016, 7, 11585 CrossRef CAS PubMed.
- Y. Zhong, M. T. Trinh, R. Chen, G. E. Purdum, P. P. Khlyabich, M. Sezen, S. Oh, H. Zhu, B. Fowler and B. Zhang, Nat. Commun., 2015, 6, 8242 CrossRef CAS PubMed.
- H. Li, T. Earmme, G. Ren, A. Saeki, S. Yoshikawa, N. M. Murari, S. Subramaniyan, M. J. Crane, S. Seki and S. A. Jenekhe, J. Am. Chem. Soc., 2016, 136, 14589 CrossRef PubMed.
- S. Li, W. Liu, M. Shi, J. Mai, T. K. Lau, J. H. Wan, X. Lu, C. Z. Li and H. Chen, Energy Environ. Sci., 2015, 9, 604–610 Search PubMed.
- W. Zhao, D. Qian, S. Zhang, S. Li, O. Inganas, F. Gao and J. Hou, Adv. Mater., 2016, 28, 4734–4739 CrossRef CAS PubMed.
- Y. Lin, J. Wang, Z. G. Zhang, H. Bai, Y. Li, D. Zhu and X. Zhan, Adv. Mater., 2015, 27, 1170–1174 CrossRef CAS PubMed.
- Y. Yang, Z. G. Zhang, H. Bin, S. Chen, L. Gao, L. Xue, C. Yang and Y. Li, J. Am. Chem. Soc., 2016, 138, 15011–15018 CrossRef CAS PubMed.
- A. C. Stuart, J. R. Tumbleston, H. Zhou, W. Li, S. Liu, H. Ade and W. You, J. Am. Chem. Soc., 2013, 135, 1806–1815 CrossRef CAS PubMed.
- S. R. Cowan, J. Wang, J. Yi, Y. J. Lee, D. C. Olson and J. W. P. Hsu, J. Appl. Phys., 2013, 113, 154504 CrossRef.
- J. C. Scott, G. G. Malliaras, W. D. Chen, J. C. Breach, J. R. Salem, P. J. Brock, S. B. Sachs and C. E. D. Chidsey, Appl. Phys. Lett., 1999, 74, 1510–1512 CrossRef CAS.
- C. E. Small, S.-W. Tsang, S. Chen, S. Baek, C. M. Amb, J. Subbiah, J. R. Reynolds and F. So, Adv. Energy Mater., 2013, 3, 909–916 CrossRef CAS.
- H. Hu, K. Jiang, G. Yang, J. Liu, Z. Li, H. Lin, Y. Liu, J. Zhao, J. Zhang, F. Huang, Y. Qu, W. Ma and H. Yan, J. Am. Chem. Soc., 2015, 137, 14149–14157 CrossRef CAS PubMed.
- J. W. Jung, T. P. Russell and W. H. Jo, ACS Appl. Mater. Interfaces, 2015, 7, 13666–13674 CAS.
- L. M. Chen, Z. Hong, G. Li and Y. Yang, Adv. Mater., 2009, 21, 1434–1449 CrossRef CAS.
- V. Shrotriya, Y. Yao, G. Li and Y. Yang, Appl. Phys. Lett., 2006, 89, 063505 CrossRef.
- E. C. P. Smits, S. Setayesh, T. D. Anthopoulos, M. Buechel, W. Nijssen, R. Coehoorn, P. W. M. Blom, B. D. Boer and D. M. D. Leeuw, Adv. Mater., 2007, 19, 734–738 CrossRef CAS.
- F. Nickel, C. Sprau, M. F. G. Klein, P. Kapetana, N. Christ, X. Liu, S. Klinkhammer, U. Lemmer and A. Colsmann, Sol. Energy Mater. Sol. Cells, 2012, 104, 18–22 CrossRef CAS.
- A. Armin, M. Velusamy, P. Wolfer, Y. Zhang, P. L. Burn, P. Meredith and A. Pivrikas, ACS Photonics, 2014, 1, 173–181 CrossRef CAS.
- A. Hexemer, W. Bras, J. Glossinger, E. Schaible, E. Gann, R. Kirian, A. Macdowell, M. Church, B. Rude and H. Padmore, J. Phys.: Conf. Ser., 2010, 247, 235–253 CrossRef.
- S. Li, L. Ye, W. Zhao, S. Zhang, H. Ade and J. Hou, Adv. Energy Mater., 2017, 7, 1700183 CrossRef.
- X. Jiao, L. Ye and H. Ade, Adv. Energy Mater., 2017, 1700084 CrossRef.
- L. Ye, X. Jiao, S. Zhang, H. Yao, Y. Qin, H. Ade and J. Hou, Adv. Energy Mater., 2017, 7, 1601138 CrossRef.
- S. Mukherjee, X. Jiao and H. Ade, Adv. Energy Mater., 2016, 6, 1600699 CrossRef.
- L. Ye, W. Zhao, S. Li, S. Mukherjee, J. H. Carpenter, O. Awartani, X. Jiao, J. Hou and H. Ade, Adv. Energy Mater., 2017, 7, 1602000 CrossRef.
- B. A. Collins, E. Gann, L. Guignard, X. He, C. R. McNeill and H. Ade, J. Phys. Chem. Lett., 2010, 1, 3160–3166 CrossRef CAS.
- J. A. Bartelt, Z. M. Beiley, E. T. Hoke, W. R. Mateker, J. D. Douglas, B. A. Collins, J. R. Tumbleston, K. R. Graham, A. Amassian, H. Ade, J. M. J. Frechet, M. F. Toney and M. D. McGehee, Adv. Energy Mater., 2013, 3, 364–374 CrossRef CAS.
- S. Mukherjee, C. M. Proctor, G. C. Bazan, T. Q. Nguyen and H. Ade, Adv. Energy Mater., 2015, 5, 1500877 CrossRef.
- A. Hexemer, W. Bras, J. Glossinger, E. Schaible, E. Gann, R. Kirian, A. MacDowell, M. Church, B. Rude and H. Padmore, J. Phys.: Conf. Ser., 2010, 247, 012007 CrossRef.
- A. L. D. Kilcoyne, T. Tyliszczak, W. F. Steele, S. Fakra, P. Hitchcock, K. Franck, E. Anderson, B. Harteneck, E. G. Rightor, G. E. Mitchell, A. P. Hitchcock, L. Yang, T. Warwick and H. Ade, J. Synchrotron Radiat., 2003, 10, 125–136 CrossRef CAS PubMed.
- E. Gann, A. T. Young, B. A. Collins, H. Yan, J. Nasiatka, H. A. Padmore, H. Ade, A. Hexemer and C. Wang, Rev. Sci. Instrum., 2012, 83, 045110 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional characterization data. See DOI: 10.1039/c7qm00182g |
| This journal is © the Partner Organisations 2017 |