
Ternary silver chlorobromide nanocrystals: intrinsic influence of size and morphology on photocatalytic activity†
Sasitha C.
Abeyweera
and
Yugang
Sun
 *
*
Department of Chemistry, Temple University, 1901 North 13th Street, Philadelphia, Pennsylvania 19122, USA. E-mail: ygsun@temple.edu
First published on 9th March 2017
Abstract
It is found that magnetic stirring can significantly influence diffusion of reaction species in high-viscosity solutions and thus the nucleation process of forming nanocrystals. This simple strategy, for the first time, has been successfully used to control the synthesis of phase-pure ternary silver chlorobromide (AgClxBr1−x) nanoparticles with varying sizes and morphologies while maintaining constant composition through co-precipitation reaction of Ag+ cations with both Cl− and Br− anions at different stirring rates. The as-synthesized AgClxBr1−x nanoparticles offer the unique opportunity to accurately study the dependence of their photocatalytic activity on particle size and morphology by taking photocatalytic decomposition of MB as an example: (1) in a solution with a high concentration of dissolved O2, particle size represents the dominating parameter and larger particles are more active than small particles; (2) with low concentrations of dissolved O2, particle morphology becomes more dominating and nanocubes (mainly bound by {100} surface facets) are more efficient than nanospheres (mainly bound by {111} surface facets) towards decomposition of MB molecules. The results provide an unprecedented insight into the rational design and synthesis of efficient photocatalysts.
Introduction
Optical transparency in the spectral range of 0.6–20 μm makes crystalline silver chlorobromide (AgClxBr1−x) an important component of optical fibers for mid-infrared (IR) signal processing and communications,1 which offer many advantages including better mechanical flexibility and optical properties (e.g., larger transparent window and higher transmittance in the mid-IR range) compared to the fluorides or chalcogenide-based glass fibers.2 For instance, tapered AgClxBr1−x fibers have been integrated with scanning near-field optical microscopy (SNOM) as probes to extend the optical response range of the SNOM into the mid-IR range, suitable for thermal/spectral imaging of chemical and biological samples that are either placed in air or immersed in water.3,4 The optical response of silver halides including AgClxBr1−x crystals can be changed when their lateral dimensions are reduced to the nanometer scale5–8 and/or they are incorporated with light absorbing species (e.g., metallic Ag, Au, etc.),9–15 which can then be employed for different potential applications as photocatalysts.16–20 For example, our group and other groups have recently demonstrated that the ternary AgClxBr1−x nanocrystals usually exhibit enhanced photocatalytic activity (e.g., photodecomposition of organic pollutants,21 photoreduction of CO222) in the visible light region when compared with their binary counterparts, AgCl and AgBr nanoparticles, which are semiconductors with both a direct bandgap (241 nm for AgCl and 289 nm for AgBr)23 and an indirect bandgap (382 nm for AgCl and 463 nm for AgBr)24 in the ultraviolet (UV) region.Systematic investigations reveal that the bandgaps of AgClxBr1−x nanocrystals are tunable in the range of the AgCl bandgap and the AgBr bandgap as the ratio of Cl−/Br− varies.22 The mixing of Cl 3p and Br 4d levels increases the density of states (DOS) in AgClxBr1−x nanocrystals compared to the binary AgCl and AgBr, leading to possible absorption in the visible region even though the absorption coefficient is low. Such band structural changes make the ternary AgClxBr1−x nanocrystals promising as visible-light photocatalysts. Cai et al. evaluated the photocatalytic activities of AgClxBr1−x nanocrystals with different Cl−/Br− ratios towards degradation of methyl orange and reduction of CO2; the results show that different AgClxBr1−x nanocrystals exhibit different photocatalytic performances as a function of the Cl−/Br− ratio.22 As a result, controlled synthesis of AgClxBr1−x nanocrystals with a pure ternary alloy phase and varying Cl−/Br− ratios has been extensively explored in the past several years.21,22,25 The most successful strategy relies on co-precipitation of Cl− and Br− ions with Ag+ ions by avoiding phase separation that tends to form AgX (X = Cl− or Br−) nanocrystals with non-uniform compositions. Due to the fast rate of the precipitation reaction between Ag+ ions and halide anions in aqueous solution, the reaction has to be confined in limited space, such as surfactant micelles26–29 and microemulsions,30,31 to ensure the formation of phase-pure AgClxBr1−x nanocrystals with small sizes. However, the resulting AgClxBr1−x nanoparticles usually exhibit random morphologies or relatively broad size distributions possibly due to the lack of ligand preference on selective crystal surfaces and the lack of control over reaction kinetics. Benefiting from in situ time-resolved, high-energy synchrotron X-ray diffraction that provides unprecedented understanding of the nucleation of growth processes involved in the formation of AgClxBr1−x nanocrystals, we recently succeeded in synthesizing AgClxBr1−x nanocubes with various compositions in ethylene glycol. The success is ascribed to the high viscosity of ethylene glycol that can be tuned by controlling the temperature to determine the solubility of the precursors and AgClxBr1−x and the diffusion rate of ionic species, which enables good separation and controllability over the nucleation and growth of AgClxBr1−x nanocrystals.32 Despite the great success in synthesizing phase-pure AgClxBr1−x nanocrystals and evaluating the dependence of their photocatalytic activity on the Cl−/Br− ratio, the influence of surface crystalline facets and size, which determines the recombination rate and migration distance of excited charges, of AgClxBr1−x nanocrystals on their photocatalytic activity has not been investigated yet due to the lack of appropriate AgClxBr1−x nanoparticles.
In this work, we report the synthesis of AgClxBr1−x nanocubes with different sizes and same-sized nanocrystals with different morphologies while their compositions are maintained constant. Stir rate, which is usually overlooked, can significantly influence the diffusion of reaction species in solution and thus impacts the nucleation and growth processes to form AgClxBr1−x nanocrystals with different parameters even when the reactants are the same. By simply tuning the stir rate, we have successfully synthesized AgCl0.5Br0.5 nanoparticles with different morphologies and sizes, which exhibit different optical absorption coefficients in the visible spectral region. Using photocatalytic decomposition of methylene blue (MB) as a model reaction, the influence of the size and morphology of the AgCl0.5Br0.5 nanoparticles on their photocatalytic responses has been studied.
Experimental
Synthesis of AgCl0.5Br0.5 nanoparticles
In a typical synthesis, 10.3 mg (0.18 mmol) of NaCl (Fisher Chemical), 18.3 mg (0.18 mmol) of NaBr (Acros Organics) and 2.5 g of poly(vinylpyrrolidone) (PVP, Mw ∼ 40![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000, Polyscience Inc.) were added to 12 mL of ethylene glycol (EG, Fisher Chemical) preloaded in a 50 mL three-neck flask. The mixture was then heated to 60 °C and the temperature was maintained until all powders are dissolved under vigorous magnetic stirring (with a stir bar of 19.1 × 9.5 mm) and a nitrogen atmosphere. The stir rate was then adjusted to a specific value. To the solution was added 1 mL EG solution of 0.34 mol L−1 AgNO3 (Acros Organics) at an injection rate of 1 mL min−1 using a syringe pump. The stir rate was maintained throughout the reaction and the reaction system was enclosed by filling the three necks with septa caps. The reaction lasted 1 hour to ensure the complete precipitation reaction between Ag+ and halide ions. The flask was fully wrapped with aluminum foil to prevent the possible influence of light on the reactions. AgCl0.5Br0.5 nanoparticles with varying sizes and morphologies were synthesized with different stir rates.
000, Polyscience Inc.) were added to 12 mL of ethylene glycol (EG, Fisher Chemical) preloaded in a 50 mL three-neck flask. The mixture was then heated to 60 °C and the temperature was maintained until all powders are dissolved under vigorous magnetic stirring (with a stir bar of 19.1 × 9.5 mm) and a nitrogen atmosphere. The stir rate was then adjusted to a specific value. To the solution was added 1 mL EG solution of 0.34 mol L−1 AgNO3 (Acros Organics) at an injection rate of 1 mL min−1 using a syringe pump. The stir rate was maintained throughout the reaction and the reaction system was enclosed by filling the three necks with septa caps. The reaction lasted 1 hour to ensure the complete precipitation reaction between Ag+ and halide ions. The flask was fully wrapped with aluminum foil to prevent the possible influence of light on the reactions. AgCl0.5Br0.5 nanoparticles with varying sizes and morphologies were synthesized with different stir rates.
Characterization of AgCl0.5Br0.5 nanoparticles
Scanning electron microscopy (SEM) images of the synthesized AgCl0.5Br0.5 nanoparticles were recorded using a FEI Quanta 400F scanning electron microscope operated under a high vacuum mode. Each SEM sample was prepared by the following procedure: 0.5 mL of the reaction solution was mixed with 1.5 mL of ethanol followed by centrifugation at 13400 rpm (revolutions per minute) for 5 minutes. After decanting the supernatant, the remaining solid at the bottom of the centrifugation tube was re-dispersed by adding 1.8 mL of ethanol. The dispersion was centrifuged at 13400 rpm for 5 minutes. After decanting the supernatant, the remaining solid at the bottom of the centrifugation tube was re-dispersed by adding 1.8 mL of ethanol. Centrifuging the dispersion at 13400 rpm for 5 minutes, removing the supernatant, and re-dispersing the collected nanoparticles with 0.5 mL of ethanol resulted in a dispersion ready for preparing SEM samples. A pipette was used to take 50 μL of the dispersion, which was then delivered to the surface of a clean Si wafer. The supported dispersion was then dried in a dark fume hood at room temperature. A similar process was used to prepare samples for X-ray diffraction (XRD) measurements, which was performed using a Bruker D8 X-ray powder diffractometer with a Cu Kα target (λ = 1.540 Å). The XRD patterns were recorded in a 2θ range of 20°–80° at a scan rate of 0.5 degree min−1. A Malvern Zetasizer particle size analyzer was used to measure the hydrodynamic size/size distribution and zeta potential of the AgCl0.5Br0.5 nanoparticles, which were prepared by mixing 0.10 mL of each reaction solution with 1.90 mL of water. UV-visible absorption spectra of the nanoparticle dispersions were taken using a photospectrometer (Thermo Scientific, Evolution 220) with an integration sphere. Each nanoparticle dispersion was prepared by diluting 0.5 mL of the reaction solution with 1.5 mL of water.
Evaluation of photocatalytic activity
In a typical measurement, 5 mL of a reaction solution containing appropriate AgCl0.5Br0.5 nanoparticles was washed with copious ethanol solution followed by centrifugation and was added to 15 mL of an aqueous solution of methylene blue (MB, 0.2 g L−1). The mixed solution was stored in the dark for 30 minutes to reach the adsorption/desorption equilibrium of MB molecules on the AgCl0.5Br0.5 nanoparticles. The solution was then illuminated with a collimated visible light source (350–750 nm, Xenon light source 300W – MAX-330, Asahi Spectra, Japan) while it was stirred at 720 rpm. Aliquots of 1 mL solution were then taken out every 15 minutes. To each aliquot was added 1 mL of water, followed by centrifuging the dispersion to remove the nanoparticles. A UV-vis absorption spectrum of the collected MB solution was measured in a quartz cuvette with 10 mm path length on an Evolution 220 photospectrometer.Results and discussion
When an EG solution of AgNO3 is injected into an EG solution containing NaCl, NaBr, and PVP, the Ag+ ions dissociated from the AgNO3 precipitate with halide ions (Cl− and Br−) to form phase-pure ternary AgClxBr1−x nanocrystals when the injection rate of AgNO3 solution is fast enough.32 PVP serves as the capping agent to stabilize the as-grown AgClxBr1−x nanocrystals. Since mixing the Ag+ ions and halide ions is critical for determining the precipitation reaction kinetics, the stir rate that influences the diffusion of ionic species represents an efficient parameter to tune the nucleation and growth kinetics of the forming AgClxBr1−x nanocrystals, leading to the formation of ternary silver halide nanocrystals with different sizes and morphologies while maintaining the same composition.Fig. 1 compares SEM images of the AgClxBr1−x nanoparticles synthesized from the reaction containing halide anions with a Cl−/Br− ratio of 1 under different stir rates, showing their variation in size and morphology. Due to the high viscosity of EG solution containing polymeric PVP molecules with a very high concentration, the initially injected AgNO3 solution cannot quickly disperse and uniformly mix with the halide anions at low stir rate, resulting in a very high concentration of Ag+ ions locally. The localized high cation concentration leads to fast nucleation with the formation of more nuclei, based on which the resulting AgClxBr1−x nanoparticles exhibit small sizes. For example, the nanoparticles synthesized at 60 rpm have an average size of 43 nm with random morphologies (Fig. 1a). Increasing the stir rate can help disperse Ag+ ions and alleviate the locally high concentration of Ag+ ions in the nucleation process, which leads to the formation of a lower number of nuclei and larger AgClxBr1−x nanoparticles correspondingly. Fig. 1b and c present SEM images of the nanoparticles synthesized at 100 rpm and 200 rpm, which are spherical particles with an average size of 57 nm and cube particles with an average size of 217 nm, respectively. The instant non-uniform distribution of Ag+ ions in the reaction solution also accounts for the somehow non-uniform size of the AgClxBr1−x nanoparticles (Fig. 1a–c). When the stir rate is higher than a critical value (i.e., 200 rpm), the injected AgNO3 solution can instantaneously mix with the halide solution to promote nucleation in the whole reaction solution, leading to the formation of more nuclei and smaller AgClxBr1−x nanoparticles accordingly (Fig. 1d for the sample synthesized at 400 rpm). Further increasing the stir rate will not significantly influence the diffusion of Ag+ ions, instead the very high stir rates can increase the collision possibilities between Ag+ ions and halide ions to enable the formation of more nuclei. As a consequence, the resulting AgClxBr1−x nanoparticles become smaller in size. It is worth pointing out that high stir rate is beneficial for synthesizing AgClxBr1−x nanoparticles with uniform size and cubic morphology due to the uniform mixing of reaction ions. Fig. 1e and f present the SEM images of the samples synthesized at 600 rpm and 800 rpm, showing their high morphological and dimensional uniformity with average sizes of 39 nm and 42 nm, respectively. The influence of stir rate on the size of AgClxBr1−x nanoparticles is summarized in Fig. 1g, revealing a volcano-type dependence that consists of varied hydrodynamic particle sizes (Fig. S1, ESI†).
 | ||
| Fig. 1 SEM images of AgCl0.5Br0.5 nanoparticles synthesized at different stirring rates: (a) 60 rpm, (b) 100 rpm, (c) 200 rpm, (d) 400 rpm, (e) 600 rpm, and (f) 800 rpm. The scale bar in (a) also applies to (b)–(f). (g) Statistic average size of the AgCl0.5Br0.5 nanoparticles (i.e., diameter of spherical nanoparticles and edge length of cubic nanoparticles). The error bars represent the size distributions. (h) XRD patterns of the synthesized AgCl0.5Br0.5 nanoparticles. To highlight the weak reflection peaks, the y-axis is plotted as the one-third power of the intensity (I1/3) of individual XRD patterns. The standard XRD patterns of AgBr (JCPDS no. 79-0149) and AgCl (JCPDS no. 85-1355) are plotted with sticks at the bottom and top, respectively, to serve as a reference. | ||
Regardless of the stir rate, the as-synthesized AgClxBr1−x nanoparticles exhibit very similar XRD patterns in terms of peak numbers, peak positions, peak symmetry, and relative peak intensity (Fig. 1h), indicating the same composition and face-centered cubic (f.c.c.) lattice of AgClxBr1−x nanoparticles. The peaks centered at 27.4°, 31.5°, 45.2°, 56.2°, and 65.9° correspond to the (111), (200), (220), (222) and (400) reflections while reflections of other crystalline planes (e.g., (311), (331), (420), (422)) are barely observed. The absence of diffraction peaks of either AgCl or AgBr crystals confirms the formation of ternary AgClxBr1−x alloy nanocrystals. Using the inter-planar distance determined from the (200) reflection peak position, the value of x in the AgClxBr1−x nanoparticles can be calculated according to Vegard's law.33 The values of x are very close to 0.5 for all of the nanoparticles shown in Fig. 1a–f, confirming that the stir rate does not influence the composition and crystalline structure of the synthesized AgClxBr1−x nanoparticles. For simplicity, the composition of the as-synthesized nanoparticles shown in Fig. 1 is denoted as AgCl0.5Br0.5 in the following content. The formation of a ternary alloy is also confirmed from the uniform distribution of all elements including Ag, Br and Cl in the nanoparticles, as characterized with energy-dispersive X-ray scattering (EDX) mapping (Fig. S2, ESI†). As demonstrated in our previous work,25 the ratio of Cl−/Br− in AgClxBr1−x nanoparticles can be easily tuned by controlling the ratio between the concentrations of NaCl and NaBr in the reaction solution.
The consistent composition of the AgCl0.5Br0.5 nanoparticles synthesized at different stir rates enables the possibility to evaluate the influence of size and morphology on the optical properties of the ternary silver halide nanoparticles with exclusion of compositional influence. We choose the nanoparticles synthesized at 100 rpm and 600 rpm, both of which show a similar size of around 50 nm but different morphologies, i.e., nanospheres with an average diameter of 57 nm and nanocubes with an average edge length of 43 nm, respectively, to study the influence of morphology (or surface crystalline facets) on their properties. Comparison of the nanoparticles synthesized at 200 rpm and 600 rpm, both of which show the same cubic morphology but different edge lengths, i.e., 217 nm and 43 nm, respectively, can provide the dependence of properties on particle size. In order to eliminate size-dependent light scattering, a photospectrometer equipped with an integrating sphere is used to precisely measure the optical absorption efficiency of different AgCl0.5Br0.5 nanoparticles, i.e., 57 nm nanospheres, 43 nm nanocubes, and 217 nm nanocubes (Fig. 2). It is clear that nanocubes with larger sizes exhibit stronger absorption than the smaller nanocubes with surfaces mainly bound by {100} facets (Fig. S3a, ESI†)34 across the whole visible spectral range of 350–800 nm (red curve versus black curve, Fig. 2). The nanospheres with surfaces mainly bound by {111} facets (Fig. S3b, ESI†)35 exhibit stronger absorption than the nanocubes with slightly smaller size (43 nm for nanocubes versus 57 nm for nanospheres). The comparisons highlight that a large volume of individual AgCl0.5Br0.5 nanoparticles benefits the absorption efficiency across the whole visible spectral region regardless of nanoparticle morphology. In the short wavelength range (350–450 nm), the absorption of 57 nm nanospheres is stronger than that of 217 nm nanocubes, while the order is switched in the long wavelength range (>450 nm). This comparison implies that exposing {111} facets is feasible to enhance the absorption of AgCl0.5Br0.5 nanoparticles at short wavelengths.
 | ||
| Fig. 2 Absorption spectra of the aqueous dispersions of the synthesized AgCl0.5Br0.5 nanoparticles (e.g., 57 nm spheres, 43 nm cubes, and 216 nm cubes). The spectra were collected with a photodetector in an integration sphere to more accurately measure optical absorption by eliminating scattering signals. | ||
The light absorption of these AgCl0.5Br0.5 nanoparticles in the visible region enables them to behave as photocatalysts to decompose methylene blue (MB) dye molecules under visible light illumination. The photocatalytic activity of the three AgCl0.5Br0.5 nanoparticles shown in Fig. 2 are evaluated by illuminating the MB solutions containing different AgCl0.5Br0.5 nanoparticles with a light source, which produces collimated visible light in the range of 350–800 nm (Fig. S4, ESI†). The solutions are first stored in the dark for 30 minutes to achieve adsorption/desorption equilibrium of MB molecules on the AgCl0.5Br0.5 nanoparticles. Once the solutions are illuminated, the characteristic absorption peaks at 663 nm and 605 nm, corresponding to the monomers and dimers of the MB molecules, respectively, continuously decrease in intensity with time (Fig. 3a and Fig. S5a, b, ESI†). The decomposition of the monomers is faster than that of the dimers. The decrease of MB concentration does not follow simple reaction kinetics as the reaction proceeds (Fig. S6, ESI†). The reaction kinetics can be plotted as the natural logarithm of the ratio between the concentration of the MB monomers (C, which can be calculated from the corresponding absorption spectra according to the Beer–Lambert law36) and the concentration of the MB monomers prior to the decomposition reaction (C0) as a function of the reaction time (t). As highlighted in Fig. 3b, the relationship exhibits a two-segmented linear dependence regardless of the photocatalysts, indicating that the photocatalytic decomposition reaction follows first-order kinetics:
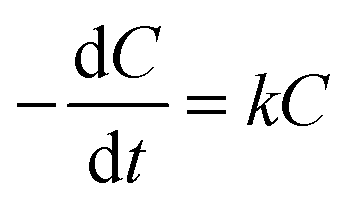
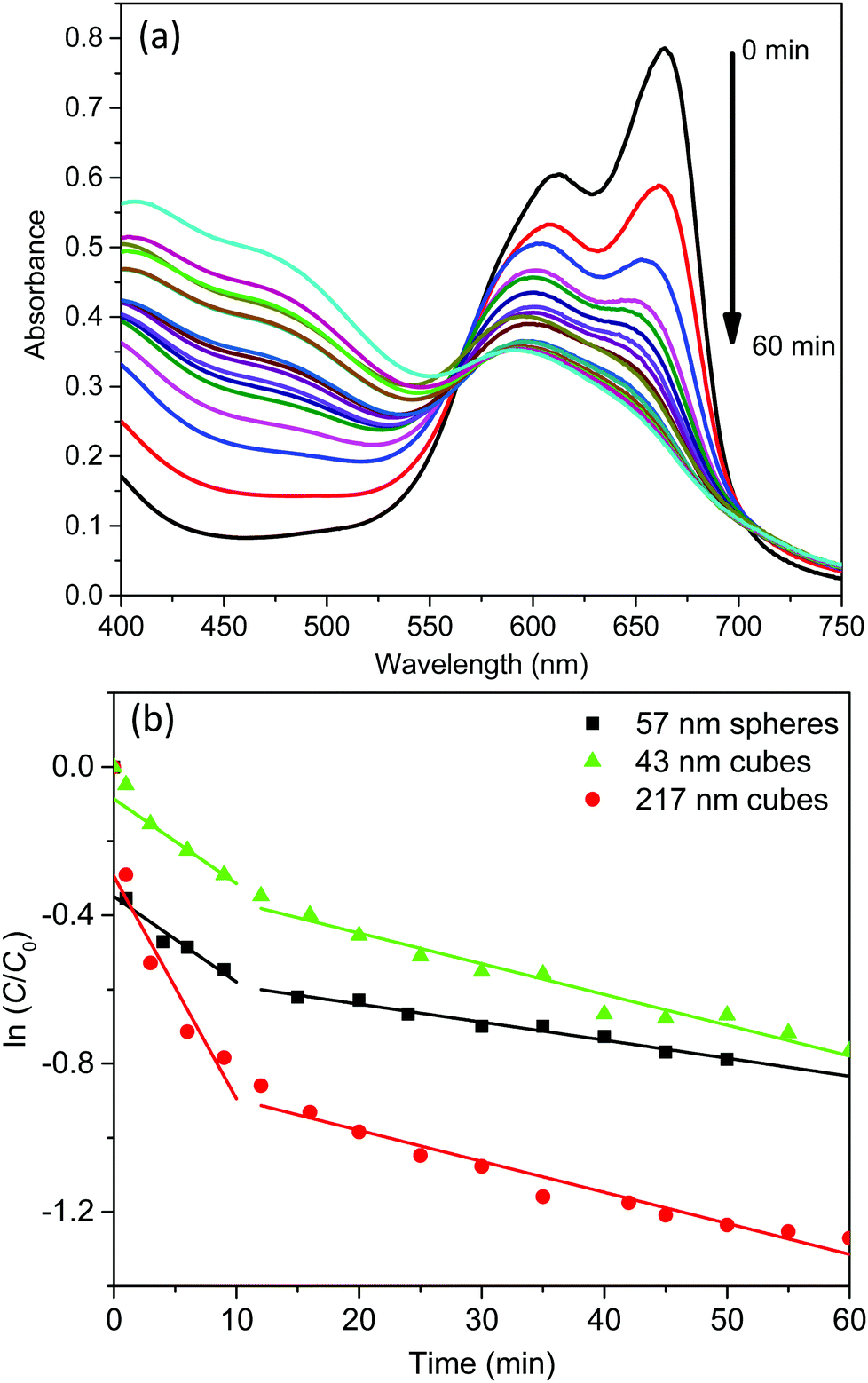 | ||
| Fig. 3 (a) Time-dependent UV-vis absorption spectra of MB in the course of photocatalytic degradation using 217 nm AgCl0.5Br0.5 nanocubes as catalysts. (b) Linear fitting of ln(C/C0) as a function of time for MB when different AgCl0.5Br0.5 nanoparticles were used as catalysts. | ||
Under photo-illumination, the AgCl0.5Br0.5 nanoparticles absorb photons to excite electrons that can migrate to the nanoparticle surface to reduce dissolved O2. The resulting superoxide radicals (O2˙−)37,38 exhibit very strong oxidizing power to oxidize MB quickly. At the same time, the leftover hole in the AgCl0.5Br0.5 nanoparticles can also migrate to the nanoparticle surface to oxidize MB. Both oxidation reactions are responsible for the photocatalytic decomposition of MB. When a reaction starts, the concentration of dissolved O2 is high in the reaction solution to benefit the overall decomposition kinetics, which is responsible for the larger values of k1. Photocatalytic decomposition of MB can quickly consume the dissolved O2 within ∼10 minutes. At a longer time (t > 10 min), O2 in the air will diffuse and dissolve in the reaction solution to support the continuous photocatalytic reaction with slow kinetics due to the lowered concentration of dissolved O2. At the initial stage (i.e., t < 1 min), MB decomposes much faster with the use of 57 nm nanospheres and 217 nm nanocubes compared to using 43 nm nanocubes as photocatalysts. Considering the higher absorption efficiency in 57 nm nanospheres and 217 nm nanocubes than that in 43 nm nanocubes (Fig. 2), it is reasonable to conclude that the very initial MB decomposition kinetics is mainly determined by the number of photons absorbed in the AgCl0.5Br0.5 nanoparticles. Once the reaction is conditioned to reach the first-order reaction region, the rate constants become less dependent on the optical absorption efficiency of AgCl0.5Br0.5 nanoparticles. Instead, the short-time rate constants (k1) at t < 10 min are mainly determined by particle size and the long-time rate constants (k1) at t > 10 min are primarily influenced by particle shape. As summarized in Table 1, 57 nm nanospheres and 43 nm nanocubes exhibit similar short-time rate constants (k1, 0.023 versus 0.023 min−1), which are smaller than that of the 217 nm nanocubes (0.060 min−1). These results indicate that increasing size of AgCl0.5Br0.5 nanoparticles is beneficial for photocatalytic decomposition of MB in the solution with high concentration of dissolved O2. Besides the useful reactions responsible for MB decomposition, the excited electrons and holes can also recombine and be annihilated by surface defect traps on nanoparticle surfaces to lose their power for decomposing MB. Since smaller AgCl0.5Br0.5 nanoparticles exhibit larger surface-to-volume ratios (e.g., 0.11 nm−1 and 0.14 nm−1 for 57 nm nanospheres and 43 nm nanocubes) than the 217 nm nanocubes with a surface-to-volume ratio of 0.03 nm−1, electron–hole recombination and annihilation become more competitive on small nanoparticles, leading to slower decomposition kinetics of MB. The comparable rate constants (k1) of 57 nm nanospheres and 43 nm nanocubes imply that the type of surface crystalline facets (determined by nanoparticle shape) plays a minor role when the solution contains high concentrations of dissolved O2. At longer reaction time when the concentration of dissolved O2 is low, the rate constant (k2) becomes shape-dependent but size-independent: both 43 nm and 217 nm nanocubes exhibit the very similar k2 values (i.e., 0.0083 and 0.0084 min−1) while the 57 nm nanospheres exhibit a lower k2 of 0.0049 min−1. These comparisons indicate that electron–hole recombination and annihilation on nanoparticle surfaces become less predominant when low concentrations of dissolved O2 are present in the MB solutions. In contrast, the type of surface facets associated with the shape of the nanoparticles, which might influence surface reaction turnover frequency, becomes more important to determine the MB decomposition kinetics. {100} facets of AgCl0.5Br0.5 nanocubes are more preferable for MB decomposition compared to {111} facets of AgCl0.5Br0.5 nanospheres.
| C 1min/C0 | Short-time rate constant, k1, at 1 min < t < 10 min (min−1) | Long-time rate constant, k2, at t > 10 min (min−1) | |
|---|---|---|---|
| 57 nm spheres | 0.702 | 0.023 | 0.0049 |
| 43 nm cubes | 0.953 | 0.023 | 0.0083 |
| 217 nm cubes | 0.748 | 0.060 | 0.0084 |
In summary, photocatalytic decomposition of MB using AgCl0.5Br0.5 nanoparticles as photocatalysts follows kinetics that can be separated into three regions: (i) at t < 1 min, the nanoparticles with stronger absorption efficiency are more efficient to decompose MB; (ii) at 1 min < t < 10 min with high concentrations of dissolved O2 in the reaction solution, MB decomposition follows fast first-order reaction kinetics with the rate constant depending on particle size – a higher rate constant for larger nanoparticles; and (iii) at t > 10 min with low concentrations of dissolved O2 in the reaction solution, MB decomposition follows slow first-order reaction kinetics with the rate constant depending on particle morphology – the rate constant for nanocubes bound by {100} facets is higher than that for nanospheres bound by {111} facets.
Conclusions
Diffusion of reaction species (e.g., Ag+ ions and halide anions in this work) in high-viscosity solutions (e.g., ethylene glycol containing high concentrations of PVP) becomes important to influence the nucleation of forming nanocrystals when the corresponding reaction (e.g., precipitation between Ag+ ions and halide ions) is fast. Magnetic stirring is usually used to improve the diffusion of reaction species, thus affecting nucleation/growth kinetics involved in the formation of nanocrystals. When an EG solution of AgNO3 is injected into a viscous EG solution containing NaCl, NaBr, and PVP to trigger the fast precipitation reaction for forming AgClxBr1−x nanocrystals, controlling the stir rate represents a facile and effective way to rationally manage the nanocrystal nucleation process. At low stir rates, nucleation of AgClxBr1−x nanocrystals starts before the injected AgNO3 solution is diffused to uniformly distribute in the reaction solution. The local high concentration of AgNO3 leads to the formation of a high number of nanocrystal nuclei, resulting in AgClxBr1−x nanoparticles with small sizes. Increasing the stir rate can help diffuse the injected AgNO3 solution quickly and lower the local AgNO3 concentration for nanocrystal nucleation, leading to reduction in the number of nanocrystal nuclei and increasing the nanoparticle sizes accordingly. When the stir rate is high enough (>400 rpm), the injected AgNO3 solution can uniformly diffuse in the reaction solution before the nucleation process starts. As a result, the size of the AgClxBr1−x nanoparticles is barely influenced by stir rates higher than 400 rpm. In addition, the AgClxBr1−x nanoparticles formed at high stir rates exhibit uniform cubic morphology while those formed at low stir rates exhibit rounded quasi-spherical morphologies. Tuning the magnetic stirring rate provides a simple but powerful approach to rationally control nucleation and enable the synthesis of AgClxBr1−x nanoparticles with controlled sizes and morphologies, which provide unique platforms to directly correlate their optical properties with their size and morphology.In the visible spectral region, the absorption efficiency of AgCl0.5Br0.5 nanoparticles is dependent on both their size and morphology: increasing particle size makes them more absorbing; forming spheres bound by {111} surface facets benefits the absorption in short wavelengths compared to the comparably-sized cubes bound by {100} surface facets. The absorbed photons in AgCl0.5Br0.5 nanoparticles can excite energetic charge carriers to drive photocatalytic decomposition of MB dye molecules. The corresponding photocatalytic activity is also demonstrated to depend on the size and morphology (or surface crystalline facets) of the AgCl0.5Br0.5 nanoparticles. Particle size is more important to determine the photocatalytic activity at high concentrations of dissolved O2, showing that larger particles are more active than small particles. When the concentration of dissolved O2 is low, the photocatalytic activity of AgCl0.5Br0.5 nanoparticles is more related to particle morphology, highlighting that nanocubes are more efficient than nanospheres. This unprecedented understanding of the relationship of optical properties of ternary AgClxBr1−x nanoparticles with their size and morphology sheds light on the rational design of efficient photocatalysts.
Acknowledgements
We gratefully acknowledge startup support of Temple University and Temple Materials Institute (TMI) for providing electron microscopy and XRD facilities.Notes and references
- T. Lewi, J. Ofek and A. Katzir, Appl. Phys. Lett., 2013, 102, 101104 CrossRef.
- T. Lewi and A. Katzir, Opt. Lett., 2012, 37, 2733–2735 CrossRef CAS PubMed.
- K. König, J. Microsc., 2000, 200, 83–104 CrossRef.
- M. Platkov, A. Tsun, L. Nagli and A. Katzir, Appl. Phys. Lett., 2008, 92, 104104 CrossRef.
- C. An, J. Wang, W. Jiang, M. Zhang, X. Ming, S. Wang and Q. Zhang, Nanoscale, 2012, 4, 5646–5650 RSC.
- H. Wang, J. Gao, T. Guo, R. Wang, L. Guo, Y. Liu and J. Li, Chem. Commun., 2012, 48, 275–277 RSC.
- Y. S. Lee, H.-J. Lee and W. S. Choi, Langmuir, 2014, 30, 9584–9590 CrossRef CAS PubMed.
- K. P. Johansson, A. P. Marchetti and G. L. McLendon, J. Phys. Chem., 1992, 96, 2873–2879 CrossRef CAS.
- P. Wang, B. Huang, X. Zhang, X. Qin, Y. Dai, Z. Wang and Z. Lou, ChemCatChem, 2011, 3, 360–364 CrossRef CAS.
- C. An, J. Wang, C. Qin, W. Jiang, S. Wang, Y. Li and Q. Zhang, J. Mater. Chem., 2012, 22, 13153–13158 RSC.
- Q. Cao, X. Liu, K. Yuan, J. Yu, Q. Liu, J.-J. Delaunay and R. Che, Appl. Catal., B, 2017, 201, 607–616 CrossRef CAS.
- C. Hu, T. Peng, X. Hu, Y. Nie, X. Zhou, J. Qu and H. He, J. Am. Chem. Soc., 2010, 132, 857–862 CrossRef CAS PubMed.
- P. Wang, B. Huang, X. Zhang, X. Qin, H. Jin, Y. Dai, Z. Wang, J. Wei, J. Zhan, S. Wang, J. Wang and M.-H. Whangbo, Chem. – Eur. J., 2009, 15, 1821–1824 CrossRef CAS PubMed.
- S. Zhang, J. Li, X. Wang, Y. Huang, M. Zeng and J. Xu, ACS Appl. Mater. Interfaces, 2014, 6, 22116–22125 CAS.
- C. An, J. Wang, C. Qin, W. Jiang, S. Wang, Y. Li and Q. Zhang, J. Mater. Chem., 2012, 22, 13153–13158 RSC.
- J. Qi, K. Zhao, G. Li, Y. Gao, H. Zhao, R. Yu and Z. Tang, Nanoscale, 2014, 6, 4072–4077 RSC.
- K. Zhao, J. Qi, H. Yin, Z. Wang, S. Zhao, X. Ma, J. Wan, L. Chang, Y. Gao, R. Yu and Z. Tang, J. Mater. Chem. A, 2015, 3, 20465–20470 CAS.
- K. Zhao, S. Zhao, J. Qi, H. Yin, C. Gao, A. M. Khattak, Y. Wu, A. Iqbal, L. Wu, Y. Gao, R. Yu and Z. Tang, Inorg. Chem. Front., 2016, 3, 488–493 RSC.
- C. Gao, Q. Meng, K. Zhao, H. Yin, D. Wang, J. Guo, S. Zhao, L. Chang, M. He, Q. Li, H. Zhao, X. Huang, Y. Gao and Z. Tang, Adv. Mater., 2016, 28, 6485–6490 CrossRef CAS PubMed.
- J. Du, J. Qi, D. Wang and Z. Tang, Energy Environ. Sci., 2012, 5, 6914–6918 CAS.
- Z. Li and Y. Sun, J. Mater. Chem. A, 2013, 1, 6786–6793 CAS.
- B. Cai, J. Wang, D. Han, S. Gan, Q. Zhang, Z. Wu and L. Niu, Nanoscale, 2013, 5, 10989–10995 RSC.
- S. Glaus and G. Calzaferri, Photochem. Photobiol. Sci., 2003, 2, 398–401 CAS.
- P. M. Scop, Phys. Rev., 1965, 139, A934–A940 CrossRef.
- Z. Li, J. S. Okasinski, D. J. Gosztola, Y. Ren and Y. Sun, J. Mater. Chem. C, 2015, 3, 58–65 RSC.
- M. Zhu, P. Chen and M. Liu, J. Mater. Chem., 2011, 21, 16413–16419 RSC.
- E. Rodil, L. Aldous, C. Hardacre and M. C. Lagunas, Nanotechnology, 2008, 19, 105603 CrossRef PubMed.
- S. Kim, H. Chung, J. H. Kwon, H. G. Yoon and W. Kim, Bull. Korean Chem. Soc., 2010, 31, 2918–2922 CrossRef CAS.
- M. Zhu, P. Chen and M. Liu, ACS Nano, 2011, 5, 4529–4536 CrossRef CAS PubMed.
- B. Cai, X. Lv, S. Gan, M. Zhou, W. Ma, T. Wu, F. Li, D. Han and L. Niu, Nanoscale, 2013, 5, 1910–1916 RSC.
- M. M. Husein, E. Rodil and J. H. Vera, J. Colloid Interface Sci., 2005, 288, 457–467 CrossRef CAS PubMed.
- Q. Liu, Z. Li, J. S. Okasinski, Y. Ren and Y. Sun, J. Mater. Chem. C, 2015, 3, 7492–7498 RSC.
- L. Vegard, Z. Phys., 1921, 5, 17–26 CrossRef CAS.
- H. Wang, X. Lang, R. Hao, L. Guo, J. Li, L. Wang and X. Han, Nano Energy, 2016, 19, 8–16 CrossRef CAS.
- B. Yin, X. Huang, R. Mishra and B. Sadtler, Chem. Mater., 2017, 29, 1014–1021 CrossRef CAS.
- P. Atkins and J. de Paula, Atkins' Physical Chemistry, Oxford University Press, Oxford, UK, 8th edn, 2010, ch. 13, p. 432 Search PubMed.
- Y. Fan, W. Ma, D. Han, S. Gan, X. Dong and L. Niu, Adv. Mater., 2015, 27, 3767–3773 CrossRef CAS PubMed.
- Y. Fan, D. Han, B. Cai, W. Ma, M. Javed, S. Gan, T. Wu, M. Siddiq, X. Dong and L. Niu, J. Mater. Chem. A, 2014, 2, 13565–13570 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Size of nanoparticles derived from DLS measurements, emission spectrum of light source, and time-dependent UV-vis spectra of MB with different catalysts. See DOI: 10.1039/c7qm00046d |
| This journal is © the Partner Organisations 2017 |