
Hybrid SiO2@POSS nanofiller: a promising reinforcing system for rubber nanocomposites†
Massimiliano
D'Arienzo
*a,
Matteo
Redaelli
a,
Emanuela
Callone
b,
Lucia
Conzatti
c,
Barbara
Di Credico
a,
Sandra
Dirè
b,
Luca
Giannini
d,
Stefano
Polizzi
e,
Ilaria
Schizzi
c,
Roberto
Scotti
a,
Luciano
Tadiello
d and
Franca
Morazzoni
a
aDept. of Materials Science, INSTM, University of Milano-Bicocca, Via R. Cozzi, 55, 20125 Milano, Italy. E-mail: massimiliano.darienzo1@unimib.it; Fax: +39-02-64485400; Tel: +39-02-64485023
b“K. Müller” Magnetic Resonance Lab., Dept. of Industrial Engineering, University of Trento, Via Sommarive, 9, 38123 Trento, Italy
cIstituto per lo Studio delle Macromolecole, ISMAC, CNR, Via De Marini 6, 16149 Genova, Italy
dPirelli Tyre SpA, Viale Sarca, 222, 20126 Milano, Italy
eDipartimento di Scienze Molecolari e Nanosistemi, Università Ca' Foscari Venezia and Centro di Microscopia Elettronica “G. Stevanato”, Via Torino 155/b, 30172 Venezia-Mestre, Italy
First published on 9th March 2017
Abstract
A novel hybrid nanofiller, SiO2@POSS, where the silica nanoparticles (NPs) and the POSS belong to the same functional structure, has been synthesized by grafting different loadings of OctaMethacrylPOSS onto silanized commercial SiO2, using a surface reaction mediated by dicumylperoxide (DCP). The peroxide, besides anchoring the nanocages onto the silica surface, ensures the presence of methacryl functionalities in the final structure, which are still available for cross-linking reactions with a polymer host. The hybrid SiO2@POSS NPs were used to prepare, by ex situ blending, SBR nanocomposites. The dynamic-mechanical analysis performed on the cured SBR/SiO2@POSS composites indicated that the presence of POSS induces a remarkable increase of modulus either at low or at high strain, and a considerable decrease of hysteresis. This has been associated with the peculiar hybrid structure of the SiO2@POSS filler, in which silica NP aggregates are partially interconnected and surrounded by a thin shell of POSS nanounits which, thanks to their high number of reactive functionalities, promote the formation of “sticky regions” among the silica aggregates and, consequently, a tight filler network wherein rubber is immobilized. This grants a relevant reinforcement and increased hysteretic properties, suggesting SiO2@POSS as a promising filler system for decreasing the energy loss under strain and for leading to a potential reduction of filler utilization in rubber composite formulations.
Introduction
The addition of silica nanofillers to elastomers is a common practice in the rubber industry to improve abrasion resistance and wet grip, and to reduce the rolling resistance.1–6 In this context, we reported that the fine tuning of size and shape in silica nanoparticles (NPs), along with their surface organic functionalization, favors filler interaction with the polymer and allows the formation of a homogeneous and continuous percolative filler network which enhances the mechanical properties of the ensuing composites.7,8 Recently, we also demonstrated that particles having an anisotropic shape are able to self-assemble in nanostructures inside the rubber matrix, providing an increase of the rubber immobilized at the filler/rubber interface and a consequent improvement of the mechanical properties.9These results suggest that the utilization of fillers with tailored structures and functionalities, able to simultaneously improve the filler networking and the filler–rubber interaction, increasing the amount of immobilized rubber, is a promising approach to obtain better mechanical properties of the composite and to limit the silica amount during compounding.
Polyhedral oligomeric silsesquioxanes (POSS) seem favorable candidates, thanks to their capability of suitably interacting with the polymer matrix through their organic functionalities.10–15 In fact, POSS constitute a unique family of molecular materials with zero-dimensionality (average size ∼1–3 nm),10,14,15 composed of an inorganic silica-like core (Si8O12) surrounded by eight covalently bonded organic (polar or non polar) groups (R8). The high number of tunable organic functionalities on the same small unit provides a remarkable compatibility with the polymer matrix, due to the enhanced possibility of chemical or physical interactions at the polymer–POSS interface.10,15
These peculiar characteristics stimulated their utilizations in polymer nanocomposites as suitable units for enhancing a number of properties, i.e. tear strength, shear modulus, rigidity, and reduced flammability, while retaining their lightweight and ductile features.10,16,17 Commonly, POSS have been exploited as versatile nano-building blocks which, chemically bonded to the polymeric backbones through “grafting from”, “grafting onto” or via (co)polymerization, act basically as cross-linking agents among the polymer chains.10 For instance, the in situ copolymerization of POSS to produce hybrid organic–inorganic polymers with pendant POSS groups has been widely studied for both thermoplastics and thermosets, showing frequently improved thermal stability and physical properties.18–21 Recently, Matějka et al.22 prepared rubbery epoxy–POSS hybrid composites with advanced reinforcement by incorporating and polymerizing a novel diepoxy–POSS monomer in an epoxy network. Many other studies demonstrated improved properties of polymer–POSS hybrid nanocomposites by exploiting the covalent bond between POSS functional groups and monomer/polymer chains.23 However, these procedures often involve time-consuming syntheses, which restrict the large-scale utilization of POSS in marketable composites.
To overcome these issues, the possibility of preparing POSS–polymer systems by ex situ blending10,30 have been recently explored as an appealing, cost-effective and environmentally friendly alternative for the industrial development of hybrid elastomeric nanocomposites.31 The key point of this approach is to facilitate the chemico-physical interaction between POSS and the matrix, by tuning the chemical structure of the organic groups covalently attached to the Si–O framework.10,24–29 In this frame, Tanaka et al.29 reported the mechanical properties of composites based on polystyrene, poly(methyl methacrylate), or ethylene-(vinyl acetate) incorporating POSS with different functionalities. It appears that unsaturated bonds on the POSS side chains favor a slight improvement of the thermal stability and the elasticity of the polymer matrix, thus suggesting the possibility to design effective POSS filled nanocomposites.
However, during blending, strong self-interaction among POSS cages often occurs, leading to aggregation, phase separation, and in turn to properties comparable or even worse than the more conventionally filled polymers.10,11 To avoid these drawbacks, Kosmalska et al.24 tried to improve the mechanical properties of butadiene–acrylonitrile rubber by using a low percentage (2–10 wt%) of POSS physically mixed to a conventional filler, silica NPs (30 wt%). They found that, after curing, the inclusion of POSS containing unsaturated reactive functionalities (i.e. vinylisobutyl, octavinyl, methacryloisobutyl and octamethacryl) via ex situ blending increases the crosslink density and the aging resistance of the elastomeric matrix, inducing an enhancement of the mechanical properties compared to reference nanoscaled silica.
These issues prompted us to explore the possibility of benefitting from both the POSS cross-linking action and the reinforcement mainly governed by silica, by utilizing SiO2–POSS “hybrid” systems, where silica NPs and POSS are not simply mixed together, but belong to the same functional structure.
In this context, several attempts have been recently made to obtain silica–POSS functional materials.32–34 However, the mechanism proposed for the functionalization with POSS appears rather unclear and convincing evidence about the anchoring of the nanocages onto the oxide surface has not been provided.
Moreover, despite all the above cited endeavours, there are hardly any investigations that report the synthesis and application of SiO2–POSS hybrids as functional nanofillers for rubber composites.
For this reason a new hybrid SiO2–POSS nanofiller, named SiO2@POSS, has been here developed.
SiO2@POSS has been synthesized by silanization of commercial SiO2 with methacrylsilane and successive grafting of small loadings of POSS via a surface reaction assisted by dicumylperoxide (DCP). The amount of peroxide was tailored in order to allow both the anchoring of the nanocages on the silica surface, under activation of the methacrylate groups35 of silane and POSS units, and the preservation in the final hybrid structure of unreacted methacryl functionalities, still available for interaction with the polymer host. It is expected that the filler–polymer interaction may be driven by the large number of methacryl functionalities provided by the POSS nanocages.
POSS grafting has been assessed by Attenuated Total Reflection-Fourier Transform Infrared Spectroscopy (ATR-FTIR) and 13C and 29Si solid-state Nuclear Magnetic Resonance (NMR). Moreover, the presence of POSS nano-units on the SiO2 surface has been observed by High Resolution Transmission Electron Microscopy (HRTEM).
SiO2@POSS hybrid NPs were used to prepare, by ex situ blending, styrene butadiene rubber (SBR) nanocomposites. The morphological and mechanical properties of SiO2/SBR@POSS nanocomposites, both uncured and cured by DCP, were investigated in detail and compared to those of nanocomposites obtained by physical mixing of SiO2, methacrylsilane and OctaMethacrylPOSS in the polymer matrix. This allowed connections among the peculiar hybrid structure of the novel filler, its efficacy in providing stronger interaction with rubber, and the enhancement of mechanical properties to be outlined.
Experimental
Materials
The solvent for swelling measurements was toluene (99.9%) purchased from Aldrich.
Preparation of SiO2@POSS hybrid filler
The SiO2@POSS hybrid filler was prepared by the double-step functionalization procedure described in Scheme 1.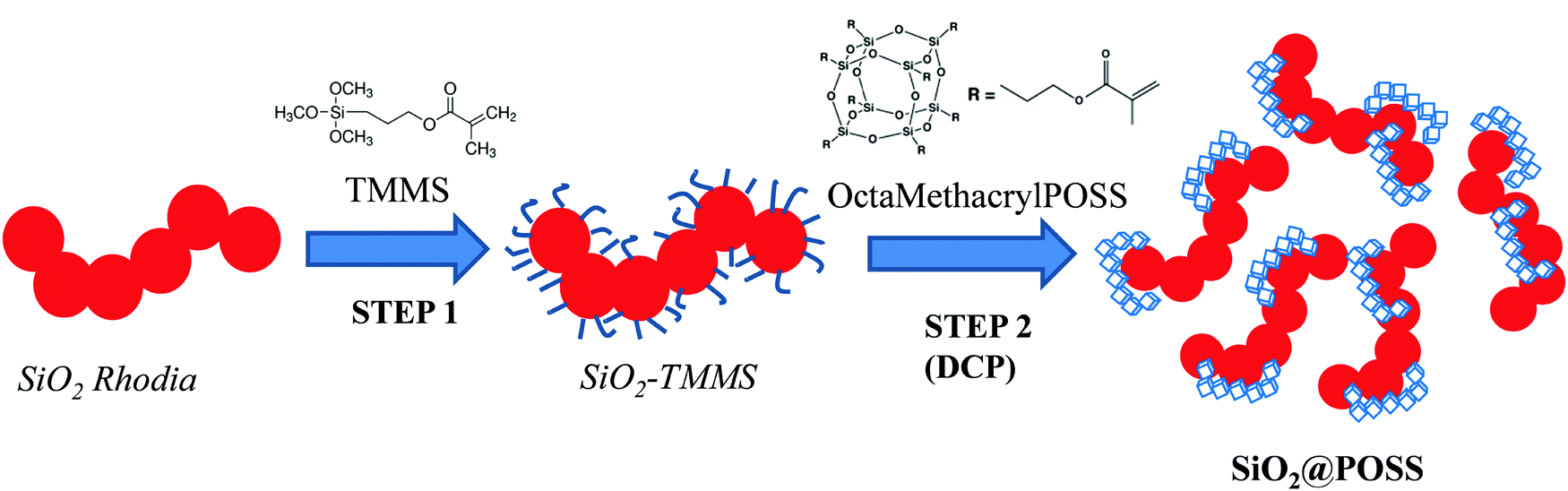 | ||
| Scheme 1 Experimental procedure for the preparation of the SiO2@POSS hybrid filler. | ||
In the first step (STEP 1), silica NPs were functionalized with TMMS according to previously reported procedures.36–38 In detail, the silane was at first dispersed by ultrasound in methanol/water solution (4/1 v/v) and then hydrolyzed at room temperature for 2 h in the presence of a suitable amount of SiO2 (30 phr, i.e. parts of hundreds of rubber). TMMS was used in the amount of 20 wt% with respect to silica. After 2 h, the silane functionalized NPs (SiO2–TMMS) were collected by centrifugation and the powders were washed several times with methanol and finally dried in an oven at 120 °C for 12 h.
In STEP 2 SiO2–TMMS NPs were suspended in toluene (150 mL) and then POSS nanounits (3, 5 or 10 phr) were introduced in the solution. Successively, a suitable amount of DCP (2 wt%, molar ratio POSS/DCP = 20/1) was added and the suspension was refluxed for 2 h. The peroxide promotes the activation of the methacrylate groups of both silane and POSS units, favoring the anchoring of the nanocages onto the silica surface and their partial condensation to form, possibly, nanometric networks (see Scheme 1, STEP 2).34
Finally, the obtained SiO2@POSS powders were filtered, washed several times with toluene and dried in oven at 120 °C in air for 12 h.
Hereafter, the different SiO2@POSS fillers will be labeled as SiO2@POSS-X which indicates 30 phr of SiO2 and where X refers to the different loadings of OctaMethacrylPOSS (X = 0, 3, 5 or 10 phr).
To further prove the validity of the anchoring procedure of POSS units on the filler surface, STEP 1 and STEP 2 were repeated by selecting a different oxide substrate, which does not display IR-absorption bands in the same spectral region of silane and POSS. In detail, commercial crystalline ZnO NPs were utilized for producing ZnO–TMMS and ZnO@POSS-10 hybrid NPs.
Preparation of uncured and cured SBR/SiO2@POSS nanocomposites
In order to prepare uncured rubber nanocomposites, SiO2@POSS hybrid fillers and SiO2–TMMS were mixed by ex situ blending with SBR in a BrabenderPlasti-corder lab station (mixing chamber of 50 mL, filling factor of 0.7) according to the two-step procedure shown in Scheme 2.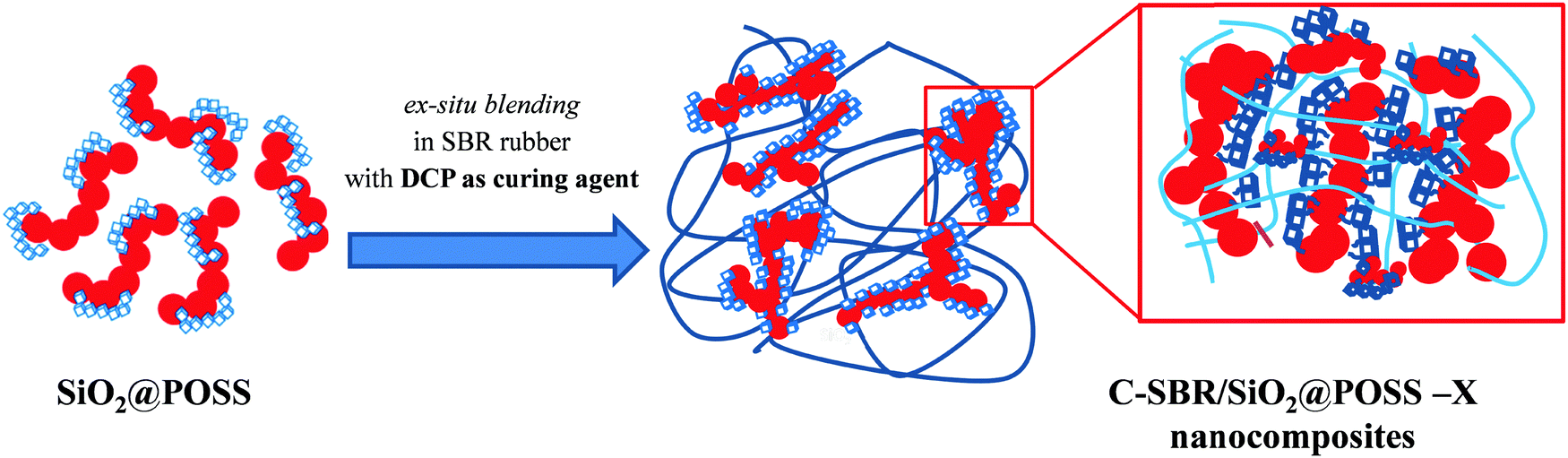 | ||
| Scheme 2 Experimental procedure for the preparation of C-SBR/SiO2@POSS nanocomposites by ex situ blending. | ||
SBR polymer (32.0 ± 0.5 phr) was first introduced into the mixer and plasticized for 30 seconds at 60 RPM at 145 °C, then the filler (SiO2@POSS hybrid fillers or SiO2–TMMS) was introduced, mixed for about 4 minutes and then discharged.
Vulcanization chemicals were then added to the obtained composites in two further steps. Firstly, stearic acid (2 phr), zinc oxide (3.5 phr) and 6-PPD (2 phr) were mixed with the obtained composites at 60 rpm for 5 min at 145 °C. Successively, DCP (1.5 phr) was introduced at a working temperature of 90 °C and by mixing at 60 rpm for 3 min.
The ensuing composites were further milled for 2 min in a two open roll-mill to produce sheets about 0.5 cm thick, suitable for the vulcanization process. Curing profiles were measured with a Moving Die Rheometer (RPA 2000, Alpha Technological). The optimum curing was achieved under the following conditions: ±1° oscillation angle, 170 °C temperature, and 20 min running time. Hereafter, uncured nanocomposites are labelled as SBR/SiO2@POSS-X, where X refers to the different amounts of OctaMethacrylPOSS (X = 0, 3, 5, or 10 phr); accordingly, SBR/SiO2@POSS-0 corresponds to a composite containing exclusively SiO2 (30 phr) functionalized with TMMS (i.e. SiO2–TMMS NPs). The different cured nanocomposites are analogously labelled as C-SBR/SiO2@POSS-X.
In order to find a relation between the peculiar structure of the SiO2@POSS hybrid filler and the features of the resulting composites, the properties of SBR/SiO2@POSS-X, both uncured and cured, were compared to those of nanocomposites prepared by an in situ procedure: SiO2, TMMS and OctaMethacrylPOSS were fed into the same polymer matrix under the same experimental conditions (i.e. curing chemicals, temperature, rotor speed) described above.
Uncured and cured SBR/SiO2 + POSS composites were prepared by blending SBR with a filler mixture composed by SiO2 Rhodia (30 phr), a TMMS coupling agent (2 phr) and different loading of MethacrylPOSS. These composites before and after curing are labelled as SBR/SiO2 + POSS-X and C-SBR/SiO2 + POSS-X, respectively. X refers to the different amounts of OctaMethacrylPOSS (X = 0, 3, 5, or 10 phr). C-SBR/SiO2 + POSS-0 thus corresponds to a composite prepared by mixing SiO2 and TMMS.
Characterization of SiO2@POSS nanofillers
To preliminarily check the silica functionalization with TMMS and, successively, with POSS nanounits ATR-FTIR measurements were performed on a Perkin Elmer Spectrum 100 instrument (1 cm−1 resolution spectra, 650–4000 cm−1 region, 16 scans).To quantitatively assess the TMMS and POSS grafting onto silica surfaces, Thermo Gravimetric Analysis (TGA) measurements before and after the double-step functionalization procedure were performed. TGA thermograms were collected by a Mettler Toledo TGA/DSC1 STARe System, at a constant gas flow (50 cm3 min−1). The sample powders were heated in air from 30 to 1000 °C. The thermal profile was the following: 30–150 °C at 2 °C min−1; dwell at 150 °C for 120 min; 150–1000 °C at 5 °C min−1. TGA curves were used to determine the temperature-dependent mass loss of the functionalizing agents, giving an indication of their interaction with the silica surface.
Nitrogen physisorption measurements on SiO2 Rhodia NPs were carried out by a Quantachrome Autosorb-1 apparatus. The specific surface area (SSABET, BET method) was measured after evacuation at 150 °C for 16 h.
Morphological characterization of SiO2@POSS-X powders was performed on a Jeol 3010 HR-TEM operating at 300 kV with a high-resolution pole piece (0.17 nm point to point resolution) and equipped with a Gatan slow-scan 794 CCD camera. The powders were suspended in isopropanol, and a 5 μL drop of this suspension was deposited on a holey carbon film supported on a 3 mm copper grid for TEM investigation.
Hybrid SiO2@POSS-X nanofillers were also characterized by solid-state NMR in order to confirm the effective surface functionalization. Multinuclear solid state NMR analyses were carried out with a Bruker 400WB spectrometer operating at a proton frequency of 400.13 MHz. MAS NMR spectra were acquired with cross polarization (CP) sequences. 13C frequency: 100.48 MHz, contact time 2000 μs, decoupling length 5.9 μs, recycle delay: 5 s, 2k scans; 29Si frequency: 79.48 MHz, π/2 pulse 3.9 μs, decoupling length 6.3 μs, recycle delay: 10 s, 2k scans. SP sequence: π/4 pulse 3.9 μs, recycle delay 100 s, 2k scans. Samples were packed in 4 mm zirconia rotors, which were spun at 6.5 kHz under air flow. Adamantane and Q8M8 were used as external secondary references. Si units are labeled according to the usual NMR notation: Tn and Qn represent trifunctional SiCO3 and tetrafunctional SiO4 units, respectively and n is the number of oxo-bridges.
Characterization of SiO2@POSS and SiO2 + POSS nanocomposites
The morphological investigation of both uncured and cured SBR/SiO2@POSS-X and SBR/SiO2 + POSS-X composites was carried out by TEM with a Zeiss EM 900 microscope working at an acceleration voltage of 80 kV. Ultrathin sections (about 50 nm thick) of composites were obtained with a Leica EM FCS cryo-ultramicrotome equipped with a diamond knife (samples kept at −130 °C).Swelling and extraction experiments were performed on cured nanocomposites to evaluate the cross-linking density (ν). Samples of 10 × 10 × 2 mm3 (0.20 ± 0.02 g) were immersed in closed vessels filled with 10 mL of toluene at 25 °C for three days in the dark to avoid photodegradation reactions. Toluene swells the composites and extracts the polymer chains not bound to silica or not crosslinked. It was replaced daily by a fresh solvent to eliminate all the extractable chains. Finally, the swollen mass was weighed and dried until it became constant under vacuum at 70 °C for 12 h.
In order to evaluate the cross-linking density of the cured nanocomposites, the volumetric fraction of the swelled rubber Vr was calculated according to the following equation:39
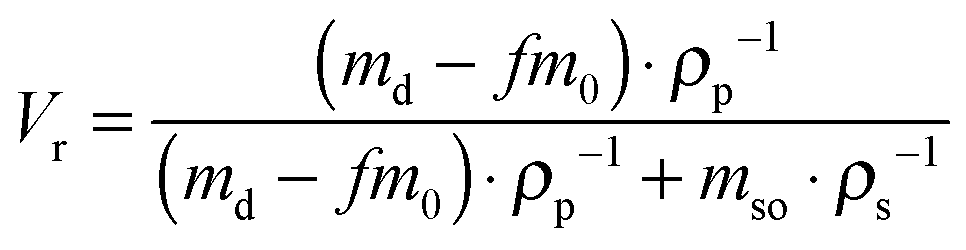 | (1) |
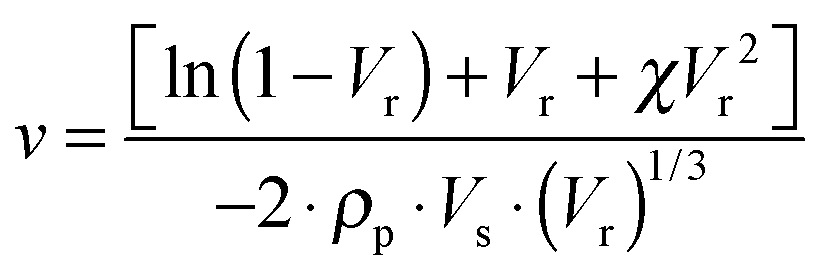 | (2) |
The experimental uncertainty on the calculated ν values does not exceed ±10%.
Dynamic-mechanical analysis
Dynamic mechanical measurements on both uncured and cured SiO2@POSS-X and SiO2 + POSS-X nanocomposites were performed by a Rubber Process Analyzer (RPA2000, Alpha Technologies) by applying a shear stress mode. The strain sweep tests of uncured samples were carried out at 70 °C and 1 Hz from 2 to 100% of elongation. Those of the cured samples were performed at 70 °C and 10 Hz from 2 to 10% of elongation. The same mechanical characterization was also performed on SBR/SiO2@POSS-X and C-SBR/SiO2 + POSS-X composites. Specimens were cut by using a Constant Volume Rubber Sample Cutter (CUTTER 2000, Alpha Technologies); the dimensions were 3.5 cm diameter and ≈0.2 cm thickness, and the weight was 4.5 ± 0.3 g. Two measurements were carried out for each sample, and the average value of the dynamical mechanical characteristics was reported.Results
Spectroscopic and morphological characterization of the hybrid SiO2@POSS nanofiller
The efficacy of the silica functionalization with TMMS and, successively, with POSS nanounits was preliminarily checked by FT-IR spectroscopy (Fig. 1).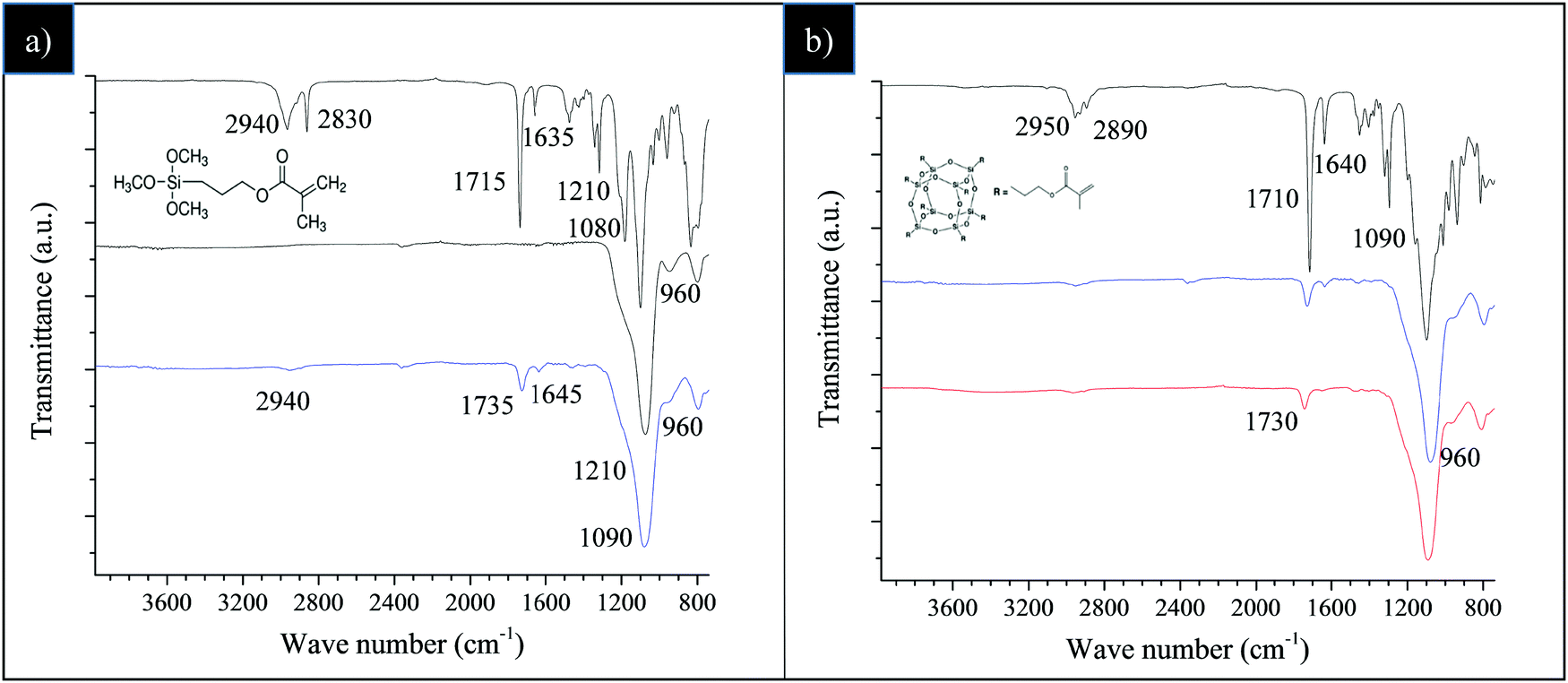 | ||
| Fig. 1 FT-IR spectra of: (a) pristine TMMS (top, bold black line), SiO2 (black line) and SiO2–TMMS (blue line); (b) pure POSS (top, bold black line), SiO2–TMMS (blue line) and SiO2@POSS-10 (red line). | ||
Fig. 1a reports the comparison among the spectra of pure TMMS, bare SiO2 and SiO2–TMMS, obtained after STEP 1 in Scheme 1.
As reported in several previous studies,41–43 the spectrum of TMMS shows the following main bands (Fig. 1a, bold black-line): the silane C–H stretching band at 2940 and 2830 cm−1; the methylene C–H bending band at 1454 cm−1; the –C–CO–O– skeletal vibration band from the methacryloxy group at 1320 and 1210 cm−1; the band of νSi–O–C at 1080 cm−1; the intense carbonyl vibration band of non-hydrolyzed TMMS at 1715 cm−1 and the weak stretching band of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond at 1635 cm−1.
C bond at 1635 cm−1.
For unmodified SiO2 (Fig. 1a, black line), the very broad absorption between 3600–3200 cm−1 attributable to the stretching of OH groups from physically absorbed water, the band of the Si–O–Si stretching vibrations at 1090 cm−1, and the band at 960 cm−1 due to Si–OH stretching are noticeable.
The spectrum of SiO2–TMMS (Fig. 1a, blue line) displays the characteristic absorption bands at 1735 cm−1 and 1645 cm−1 (very weak) deriving from CO and CC stretching of the TMMS methacryl group. Moreover, a weak shoulder at 1210 cm−1 related to the –C–CO–O– skeletal vibration from the methacryloxy group of the silane is detectable.41–43 It can be also observed that the –Si–OH stretching at 960 cm−1 gradually decreases in intensity compared to SiO2, probably due to the formation of Si–O–Si bonds resulting from the condensation reactions between the alkoxy groups of TMMS and the surface hydroxyl groups of silica.
These results suggest the effective silanization of SiO2 particles by TMMS.
To obtain SiO2@POSS, SiO2–TMMS NPs were then reacted with POSS assisted by DCP (STEP 2, of Scheme 1), which should promote the activation of the methacrylate groups of both silane and POSS units, favoring the anchoring of POSS on the oxide surface.
The FT-IR spectra of pristine POSS, SiO2–TMMS and the obtained SiO2@POSS-10 hybrid filler are reported in Fig. 1b. For pure POSS (Fig. 1b, bold black-line), the observed bands have been attributed to the C–H stretching vibration of methyl and methylene groups at 2950 and 2890 cm−1; to the CO and CC stretching of POSS methacryl groups44 at 1710 and 1640 cm−1, respectively; and to the Si–O stretching vibrations typical of the POSS cage at 1090 cm−1. As expected, since the typical absorption bands of the POSS cages overlap the stretching and bending vibrations of silica and TMMS, the spectra of the SiO2@POSS-10 hybrid filler and SiO2–TMMS are very similar (Fig. 1b, red and blue line respectively), in spite of the functionalization with POSS. Analogous results have been attained for SiO2@POSS-3 and SiO2@POSS-5 hybrid nanofillers.
Thus, in order to assess the anchoring of POSS units on the filler surface, STEP 1 and STEP 2 were repeated by selecting a different oxide substrate, which does not display IR-absorption bands in the same spectral region of silane and POSS. We utilized commercial crystalline ZnO NPs,45 since it has been demonstrated that TMMS can be efficiently bonded to ZnO through hydrolysis and condensation reactions similar to those occurring in silica.41,44 The FT-IR spectra of pure ZnO, ZnO–TMMS and ZnO@POSS-10 NPs are reported and described in Fig. S1 of the ESI.†
In detail, the spectrum of ZnO–TMMS (Fig. S1a, ESI†) reveals the characteristic absorption bands of the silane linked on the oxide surface. After reacting with POSS, the methacryloxy group vibrations (νCO, and –C–CO–O– skeletal vibration) and the Si–O–C stretching of ZnO–TMMS increase in intensity (Fig. S1b, ESI†). These results, beyond indicating the effective POSS functionalization of the ZnO NPs, can be considered an indirect probe of the efficacy of the functionalization route in providing the anchoring of POSS nanounits on the silica surface.
To quantitatively assess the silica functionalization, TGA analysis was performed on SiO2–TMMS, SiO2@POSS-10 and on naked silica NPs (Fig. S2, ESI†). In detail, the total amount of TMMS grafted onto SiO2 was evaluated by the net weight loss of SiO2–TMMS between 180 °C and 980 °C, i.e. considering the total weight loss with exclusion of that associated to bare SiO2 (i.e. 3.860 wt%). The quantity of POSS units in SiO2@POSS-10 was similarly calculated subtracting the contributions of both SiO2 and TMMS from the total weight loss.
Moreover, assuming that the functionalizing agents were equally distributed on the silica surface and referring to the BET surface area of SiO2 (SSABET = 168.6 ± 3.4 m2 g−1, see Fig. S3 in ESI†), the number of functionalizing molecules per nm2 (Σ = molecules nm−2) was also estimated (Table 1).
| Compound | Functionalizing agent | M W (g mol−1) | Net weight loss (wt%) | Grafted amount (mmol g−1) | Σ (molecules nm−2) |
|---|---|---|---|---|---|
| SiO2–TMMS | TMMS | 248.4 | 13.398 | 0.539 | 1.927 |
| SiO2@POSS-10 | POSS | 1434.0 | 5.807 | 0.041 | 0.154 |
The obtained results confirm the functionalization of silica with silane in SiO2–TMMS and, though to a lower extent, with POSS nanounits in SiO2@POSS NPs (Table 1).
Further evidence of the POSS functionalization on the SiO2 surface was provided by solid state NMR investigation of SiO2–TMMS and SiO2@POSS samples.
Fig. 2 presents the 29Si CPMAS spectra recorded on SiO2–TMMS, SiO2@POSS-3 and SiO2@POSS-10.
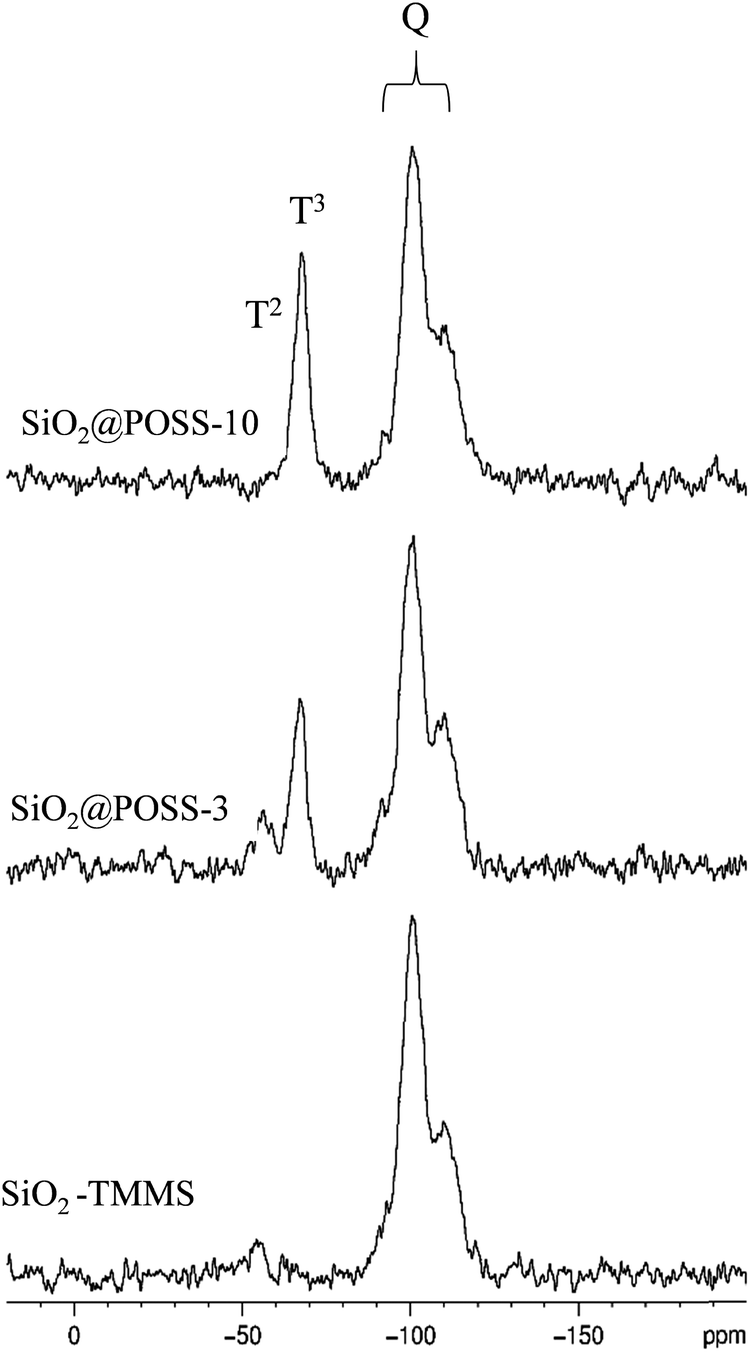 | ||
| Fig. 2 29Si CPMAS NMR spectra of SiO2–TMMS and SiO2@POSS-X (X = 3, 10 phr of POSS). | ||
The Q-unit region of the CPMAS spectrum of the SiO2–TMMS sample (Fig. 2, bottom) displays the complex resonance of the SiO2 phase, resulting from the overlapping of three components at −92.2 (Q2), −100.5 (Q3) and −110.1 (Q4) ppm, respectively. The TMMS functionalization of the SiO2 particles is proved by the resonance at −55.7 ppm due to the T2 units and the very weak signal at −66.5 ppm assigned to the T3 units.
In the CPMAS spectra of SiO2@POSS-3 and SiO2@POSS-10 (Fig. 2), the features of the Q units appear unchanged, whereas the signals of the T units strongly increase in intensity compared to SiO2–TMMS.
It is not possible to distinguish the T units of TMMS from those of POSS since they present the same chemical shift.46 However, the POSS molecules contribute in particular to the T3 resonance, in agreement with their structure. Therefore, the increase of the T3/T2 ratio in SiO2@POSS-10 in comparison to SiO2@POSS-3 and, in particular, to SiO2–TMMS is a clear indication of the effective POSS grafting onto the surface of the functionalized silica NPs.
In order to get information on the interactions among the acrylate functions of TMMS and POSS in the final hybrid nanofillers, 13C CPMAS NMR investigation was performed on SiO2@POSS-X NPs. In particular, the spectrum of SiO2@POSS-10 is shown in Fig. 3. The liquid NMR spectrum of the pristine TMMS (with the labelling scheme and the signal assignment based on previous reports46) is also reported for comparison (Fig. 3, top).
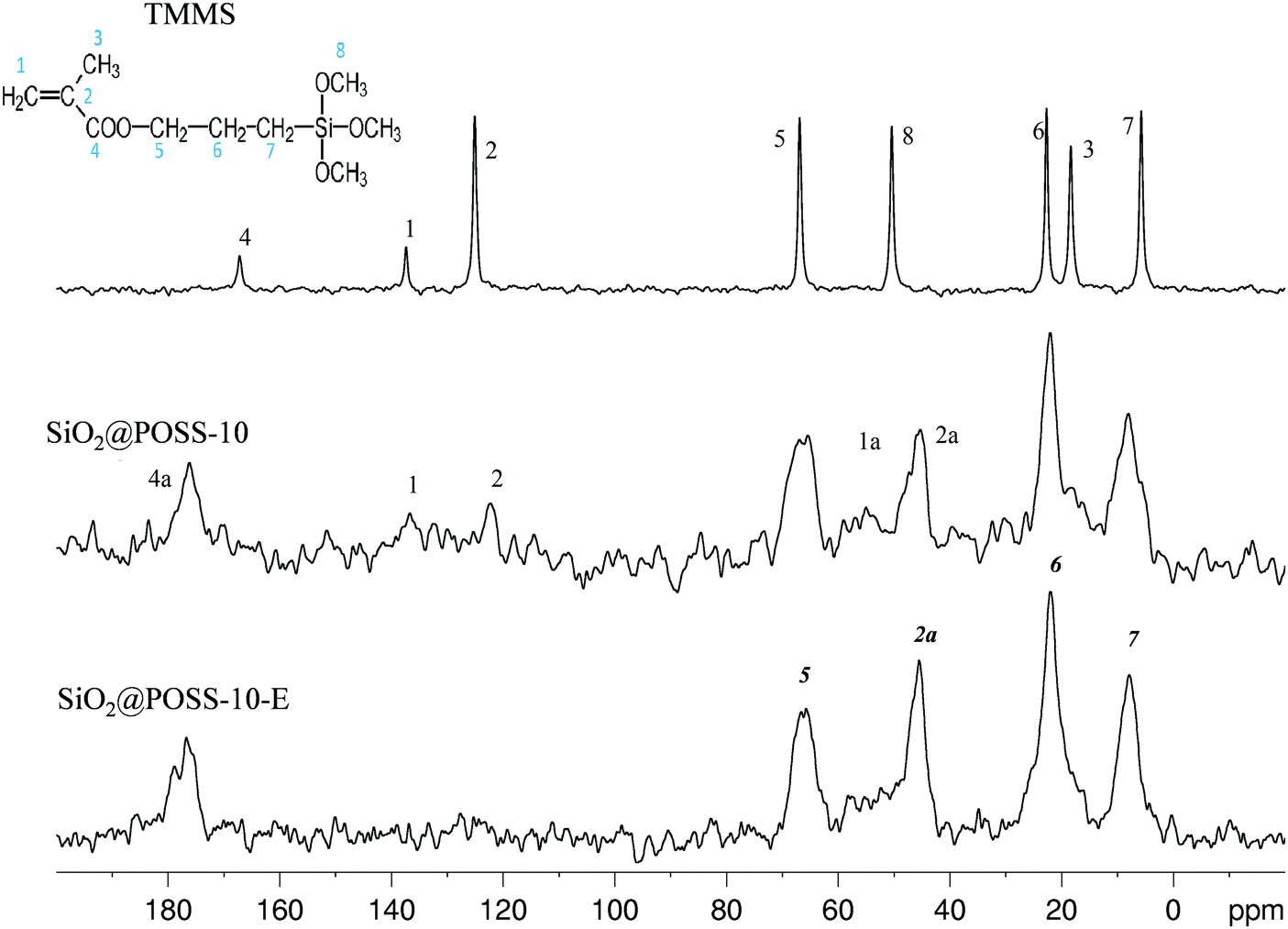 | ||
| Fig. 3 13C CPMAS spectra of silica functionalized with TMMS and POSS. | ||
The polymerization among the acrylate functionalities of TMMS and POSS, activated by DCP, leads to the appearance in the SiO2@POSS-10 spectrum of the peak 2a at 45 ppm and to a broad signal in the range of 52–56 ppm (1a), both attributable to –CH2-based chains formed through polymerization. Moreover, the downfield shift (of about 8 ppm) of the carbonyl peak 4, compared to the resonance found at 167.2 ppm in the unreacted acrylate chain, such as in pristine TMMS, further confirms the successful reaction between organosilane and nanocages. These results further substantiate the anchoring of POSS onto the functionalized silica surfaces.
Despite the poor S/N ratio, in SiO2@POSS-10, residual weak signals in the range of 110–150 ppm may suggest the presence of residual CC bonds (signals at 125.1 and 137.4 ppm, respectively), which could be available for the cross-linking reactions with rubber chains during the curing of the composites. In order to prove this hypothesis, a SiO2@POSS-10 sample prepared by using an excess of DCP, for improving the condensation of the methacrylate functions, was investigated (SiO2@POSS-10-E, Fig. 3 bottom). The obtained spectrum displays an almost flat profile in the CC region, supporting the idea that in the SiO2@POSS-10 hybrid sample, unreacted methacrylate functionalities are still present.
In summary, the NMR study points out the effective grafting of POSS molecules on SiO2–TMMS NPs and the expected presence of residual methacrylate groups available for interaction with rubber.
The morphology of SiO2@POSS NPs was investigated by TEM and compared to that of bare silica (Fig. 4). Images show that pure SiO2 is constituted by large agglomerates where the NPs are highly aggregated, due to their polar surfaces (Fig. 4a). At higher magnifications, the NPs assume an almost elongated shape and an average diameter ranging between 15 and 20 nm (Fig. 4c and e).
 | ||
| Fig. 4 TEM images at different magnifications of: (a), (c) and (e) bare SiO2 NPs; (b), (d) and (f) SiO2@POSS-10 nanofiller. | ||
After functionalization with POSS units (SiO2@POSS-10, as a representative example), the silica NP morphology does not change significantly (Fig. 4b). However, going more in depth into the image observation, the NP boundaries appear less defined than in pure silica (Fig. 4d and f). These effects may be related to the presence of POSS domains, which create a surface shell on the particle surface.
Since both SiO2 and POSS are amorphous, it is tricky to associate the less defined boundaries of the SiO2@POSS-10 images to the presence of POSS nanounits. Thus, in order to observe the effect of the functionalization on the filler surface features, in analogy with the IR investigation, the TEM analysis was carried out also on the ZnO@POSS-10 sample, constituted by highly crystalline ZnO NPs (Fig. 5a) functionalized with POSS nanocages.
 | ||
| Fig. 5 TEM images of (a) pure ZnO and (b) ZnO@POSS-10 NPs. | ||
In this case, the TEM images of ZnO@POSS-10 NPs (Fig. 5b) reveal thin and irregular amorphous layers surrounding and partially connecting the nanocrystals, attributable to the presence of nanometric aggregates of POSS units. This again supports the efficacy of the functionalization procedure in favoring the anchoring of POSS on the oxide surface.
Characterization of SBR/SiO2@POSS and SBR/POSS + SiO2 nanocomposites
The morphology of the nanocomposites and the filler dispersion were investigated by TEM for both cured and uncured samples obtained by incorporating in SBR the hybrid filler SBR/SiO2@POSS-X and for the mixture of SiO2, TMMS and POSS (i.e. SBR/SiO2 + POSS-X). As representative examples, the images collected for uncured SBR/SiO2@POSS-10 and SBR/SiO2 + POSS-10 samples are reported in Fig. 6. SBR/SiO2@POSS-10 is constituted by very compact and large NP agglomerates, which have micrometric dimensions and are inhomogeneously distributed through the rubbery matrix (Fig. 6a). This irregular organization generates regions in which the rubber is constrained (inset Fig. 6a). However, the presence of wide areas lacking in filler particles can be detected (Fig. 6c).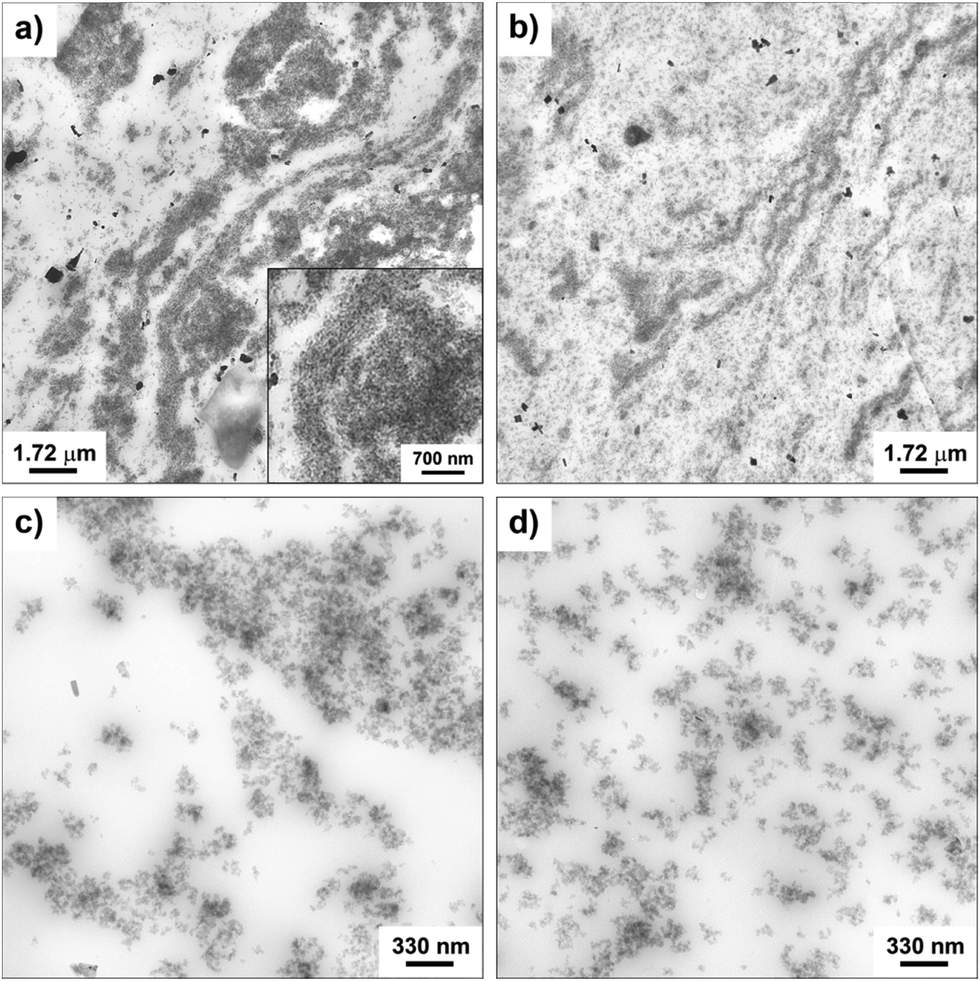 | ||
| Fig. 6 TEM images at different magnifications of uncured: SBR/SiO2@POSS-10 (a and c), and SBR/SiO2 + POSS-10 (b and d) nanocomposites. | ||
Conversely, in SBR/SiO2 + POSS-10, the filler distribution is rather continuous and homogeneous (Fig. 6b), with silica NPs organized in sub-micrometric or even nanometric agglomerates (Fig. 6d).
The better dispersion observed in this sample may be tentatively ascribed to the larger amounts of TMMS and POSS nanounits available during the mixing procedure, in comparison to those effectively grafted onto the surface of silica in SiO2@POSS filler, as assessed by TGA investigation. The TEM images of the cured samples reported in Fig. 7 show a more continuous and homogeneous nanofiller network after curing.
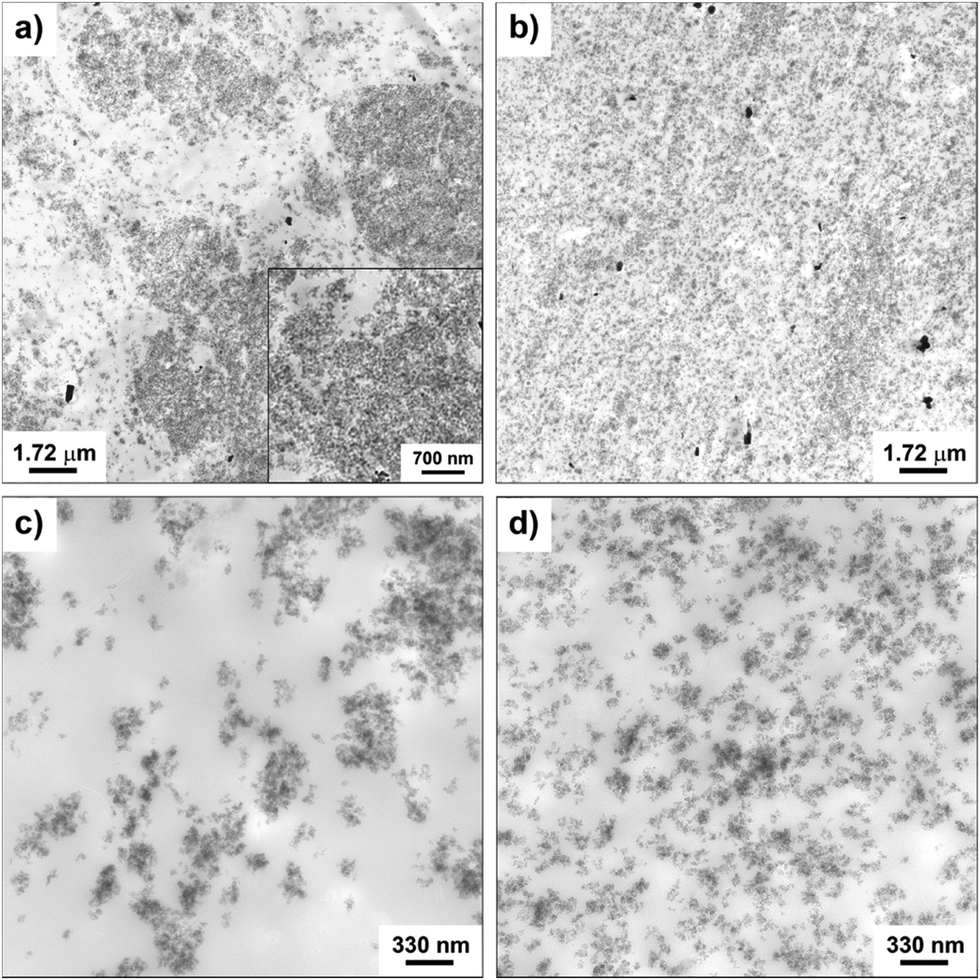 | ||
| Fig. 7 TEM images at different magnifications of: C-SBR/SiO2@POSS-10 (a and c), and C-SBR/SiO2 + POSS-10 (b and d) nanocomposites. | ||
This is particularly evident for C-SBR/SiO2 + POSS-10, where silica NPs form small nanometric aggregates (Fig. 7b), which are very uniformly dispersed in the matrix and only partially interconnected (Fig. 7d). Although an improved particle distribution is detectable also for C-SBR/SiO2@POSS-10, the sample still exhibits micrometric NPs agglomerates and filler-free zones (Fig. 7a and c). However, these agglomerates appear less dense than those showed by the uncured composite (see Fig. 6a), and some thin rubbery regions separating the NPs aggregates are noticeable (inset in Fig. 7a).
This enhancement of the filler dispersion observed after curing may be related to the further mixing steps and to the DCP action,35 which activates the reaction between the methacryl groups of both TMMS and POSS and the rubber vinyl functionalities, leading to an increased cross-linking of elastomer chains and to the possible immobilization of rubber chains in between the NPs aggregates.
Swelling experiments were performed on C-SBR/SiO2@POSS-X and C-SBR/SiO2 + POSS-X composites, in order to evaluate the cross-linking induced by the introduction of POSS nanocages.
The ν value was determined according to the Flory–Rehner eqn (2)39,40 and the results are summarized in Fig. 8. For the C-SBR/SiO2@POSS-X samples, the reticulation degree generally increases with the POSS loading, becoming remarkable for C-SBR/SiO2@POSS-10. Conversely, the ν values for C-SBR/SiO2 + POSS-X nanocomposites for X = 5 and 10 are only slightly higher than those calculated for the reference sample (C-SBR/SiO2 + POSS-0, not including POSS), while they are definitively lower than those of C-SBR/SiO2@POSS-X.
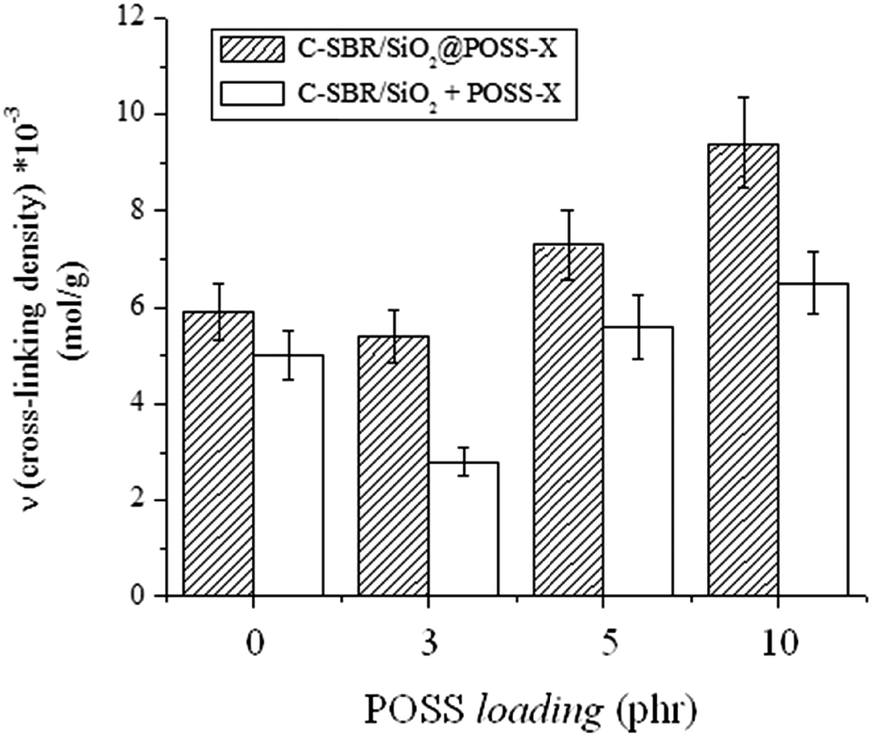 | ||
| Fig. 8 Trend of the ν values for C-SBR/SiO2@POSS-X and C-SBR/SiO2 + POSS-X nanocomposites. | ||
These outcomes may be explained by referring to the peculiar morphological properties of the composites. In C-SBR/SiO2@POSS-X nanocomposites, POSS nanocages are anchored to the SiO2 NPs inducing the formation of a tight filler network, in which thin rubbery regions separate the NP aggregates (see Fig. 7a and c). This may favor an enhanced immobilization of rubber in proximity to the NP surface, resulting in a high cross-linking density.
Instead, when POSS is mixed with silica within the rubber matrix (i.e. C-SBR/SiO2 + POSS-X samples), the synergism between POSS and filler NPs in providing immobilized rubber seems partially hindered, since more dispersed and only partially interconnected aggregates are detected (see Fig. 7b and d). Accordingly, lower cross-linking density values are obtained for these composites.
These results gather more relevance if we consider that the amount of POSS nanounits effectively grafted onto the silica surface in SiO2@POSS (see TGA investigation) is lower in comparison to that present during the mixing procedure for preparing C-SBR/SiO2 + POSS-X samples.
Dynamic-mechanical analysis
The dynamic mechanical characterization of all uncured and cured composites was carried out by RPA. Their performances in terms of reinforcement were determined and compared to those of the composites prepared without POSS (i.e. SBR/SiO2@POSS-0 and SBR/SiO2 + POSS-0 samples), in order to evaluate the effect of POSS nanocages on the final material properties.The trends of the storage modulus (G′) as a function of strain, for uncured SBR/SiO2@POSS-X and C-SBR/SiO2@POSS-X rubber composites, are reported in Fig. 9.
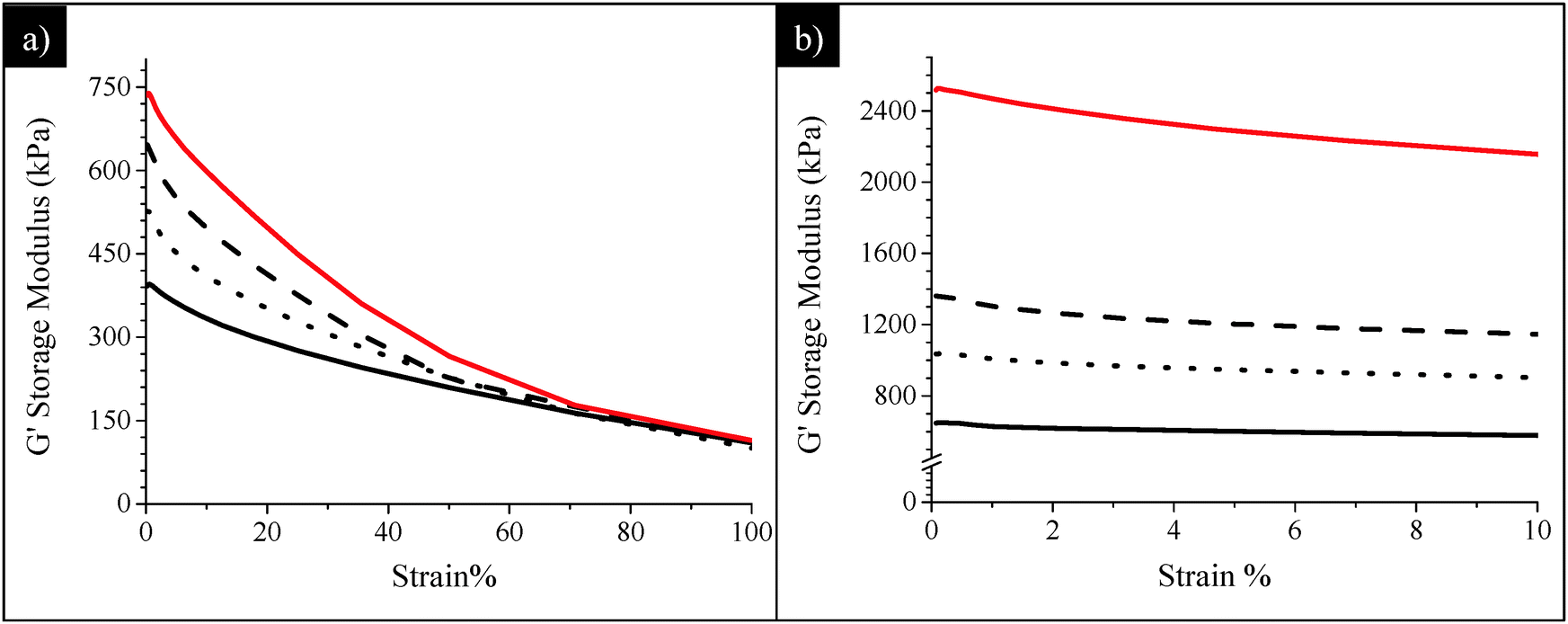 | ||
| Fig. 9 Storage modulus G′ vs. strain plot of (a) SBR-SiO2@POSS-X and (b) C-SBR-SiO2@POSS-X composites: X = 0 (i.e. SiO2 30 phr, black line), X = 3 (dotted line), X = 5 (dashed-line) and X = 10 (red line). | ||
Before curing (Fig. 9a), the anchoring of POSS nanounits onto SiO2 NPs, even in a low amount (3 phr), leads to a significant increase of the modulus at low strain (G0′) if compared to SBR/SiO2@POSS-0. Following the widely accepted mechanistic interpretation that the Payne effect is related to the breakdown of the filler network upon oscillatory shear,8 the higher G0′ values obtained in the presence of SiO2@POSS hybrid particles point to a greater reinforcing effect. This behavior may be connected to the peculiar filler networking in the SBR/SiO2@POSS composites, where compact micrometric agglomerates are inhomogeneously distributed within the rubbery matrix, generating regions in which rubber is constrained (see Fig. 6). These features provide on one hand remarkable filler–filler interactions (i.e. high G0′ values) and, on the other, may favor the interaction between the polymer and the filler albeit weak before curing.
At the highest deformation, the modulus (G∞′) is very similar for all the composites (Fig. 9a), regardless of the POSS loading. This means that the differences in reinforcement are less effective once the network is broken down. Usually, at high strain, only a particular surface reactivity can originate different mechanical characteristics of the samples, as the particles are supposed to be not mutually interacting. In the present case, the trend observed for the SBR/SiO2@POSS-X samples may be connected to the presence of POSS methacryl functionalities which, before the DCP curing activation, are not able to chemically interact with the polymer chains.
The impact of POSS introduction on the mechanical properties of uncured SBR/SiO2@POSS-X and SBR/SiO2 + POSS-X composites was also studied by monitoring the dependence of the loss modulus (G′′) on the strain (Fig. S4, ESI†).47G′′ is connected to the rates of network breakdown and re-assembly. In general, at low strain, the network is still able to reorganize after each deformation, and the composite shows a behavior which affects the G′′ values. Instead, at high deformations, the strain amplitude is high enough to destroy the filler network, and the influence on G′′ becomes negligible.47 Thus, loss modulus vs. strain curves allow the maximum strain at which the composite is able to withstand the mechanical solicitation without fracture of the filler network to be identified. The higher the G′′ values and the strain at which G′′ is maximum, the more compact the network. In the investigated composites, SBR/SiO2@POSS-10 shows a broad maximum at strain located at ∼30% and the highest G′′ values, while SBR/SiO2 + POSS-10 and the composites without POSS display lower loss modulus and maxima located at strain <10% (Fig. S4, ESI†). These issues endorse the ability of SiO2@POSS particles in promoting the formation of a compact particle network, which encloses rubbery regions, as assessed by TEM investigation (Fig. 6a and c).
After curing (Fig. 9b), a marked increase of the modulus either at low or at high strain, i.e. a considerable decrease of the Payne effect, is observed for all the composites. This is much more relevant for the samples containing POSS nanounits and increases as a function of their concentration, resulting in the highest G0′ and G∞′ values for the C-SBR/SiO2@POSS-10 nanocomposite. In general, these issues can be associated with the DCP action, which initiates the reaction between POSS methacryl groups and rubber vinyl functionalities, leading to effective 3-D cross-linking.35 More in depth, the moduli improvement and the reduced hysteresis in C-SBR/SiO2@POSS-X can be discussed in terms of polymer interactions with the silica NP network.
As evidenced by TEM images, the SiO2@POSS filler is constituted by silica aggregates which are partially connected and surrounded by a thin POSS amorphous shell. Thus, it can be argued that the cross-linking action promoted by nanocage functionalities is localized on the surface or even inside the SiO2 aggregates. This may cause an enhanced immobilization of the polymer chains near the NPs’ surface or within their network (see Scheme 2), upgrading the mechanical reinforcement of the composites. These ideas are sustained by the morphology of the C-SBR/SiO2@POSS-10 composite, where thin rubbery regions connect the filler aggregates (see Fig. 7a and c) and also by the higher degree of cross-linking for C-SBR/SiO2 + POSS-X revealed by swelling.
Finally, the mechanical behaviours of SBR/SiO2@POSS-X, both uncured and cured, were compared to those of SBR/SiO2 + POSS-X composites, in order to highlight the role of the hybrid filler NPs in providing enhanced functional properties. In particular, Fig. 10 reports the G′ vs. strain plots for SBR/SiO2@POSS-10 and SBR/SiO2 + POSS-10 nanocomposites. Before curing, the G∞′ values are very similar for all the composites (Fig. 10a). Instead, while no significant differences can be detected between SBR/SiO2 + POSS-10 and its reference sample SBR/SiO2 + POSS-0, a high G0′ is measured for the composite prepared with the hybrid filler. As described above, the performance of SBR/SiO2@POSS-10 can be attributed to the compact filler network generated by SiO2@POSS NPs within the polymer matrix.
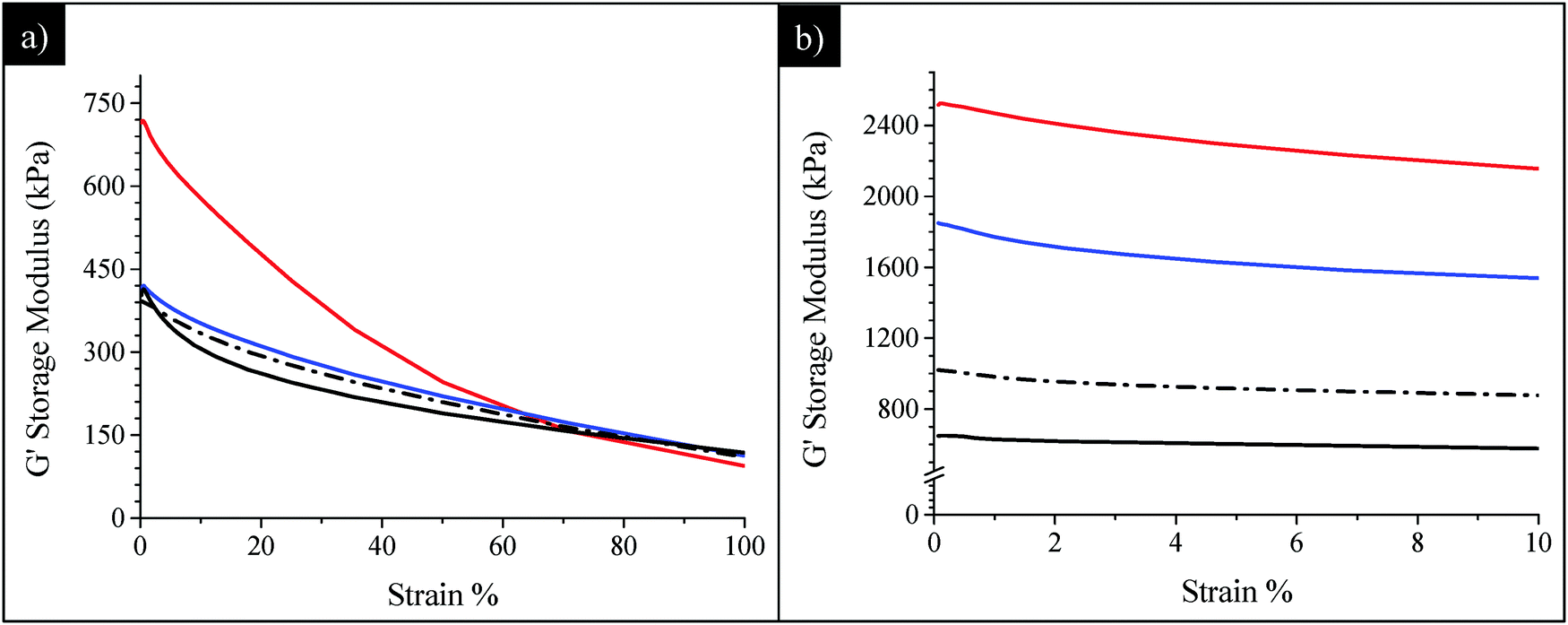 | ||
| Fig. 10 Comparison among (a) uncured and (b) cured SBR/SiO2@POSS-10 (red line), SBR/SiO2 + POSS-10 (blue line), SBR/SiO2@POSS-0 (black line) and SBR/SiO2 + POSS-0 (dot-dashed line) nanocomposites. | ||
The DCP curing leads to an expected decrease of the Payne effect for all the composites, as a result of the modulus increase either at low or at high strain (Fig. 10b). In general, both C-SBR/SiO2@POSS-10 and C-SBR/SiO2 + POSS-10 display outstanding reinforcement and much lower Payne effect in comparison to the composites containing exclusively silica and TMMS. Similar trends have been obtained for the composites with lower POSS loadings. These results point out that the introduction of a low amount of OctaMethacrylPOSS in rubber composites of silica silanised with methacryl silane cured with DCP provides mechanical properties which are significantly superior to those commonly reported in the literature.24
Interestingly, it must be observed that C-SBR/SiO2@POSS-10 exhibits better performance than the C-SBR/SiO2 + POSS-10 nanocomposite. The higher capability of the hybrid nanofiller in upgrading reinforcement and remarkably decreasing the hysteretical losses may derive from the peculiar surface functionalization with POSS nanounits which, through the high number of reactive functionalities, promotes enhanced networking and the formation of “sticky regions” among the silica aggregates, immobilizing both filler and rubber, and affording a relevant increase of the mechanical properties.
In C-SBR/SiO2 + POSS, POSS units are likely grafted to silica NPs, though with a lesser extent than in C-SBR/SiO2@POSS. We suggest that, in this case, POSS nanounits may interact again with the filler exerting their cross-linking close to the filler surfaces and contributing to the networking, but direct interaction among “free” POSS nanounits and polymer chains may also occur. This leads to the presence of more dispersed and less compact filler aggregates (see TEM images), and to a lower cross-linking density, resulting in a partial depletion of the functional properties of the composites.
In summary, the overview of the mechanical behavior of SBR/SiO2@POSS-X and SBR/SiO2 + POSS-X nanocomposites suggests that the utilization of a small amount of POSS nanounits, particularly when anchored onto the silica surface, may be a promising approach for effectively improving the features of rubber composites and for a potential reduction of filler loading.
Conclusions
A novel hybrid nanofiller, SiO2@POSS, where silica NPs and POSS belong to the same functional structure, has been synthesized by grafting different loadings of OctaMethacrylPOSS onto silanized commercial SiO2, using a surface reaction mediated by DCP. As assessed by FT-IR, and solid-state NMR, the peroxide, besides providing the anchoring of the nanocages on the silica surface, ensures the presence of methacryl functionalities still available for cross-linking reactions with a polymer host in the final hybrid structure.These hybrid NPs were used to prepare, by ex situ blending, SBR nanocomposites. The dynamic-mechanical analysis performed on the cured SBR/SiO2@POSS composites, indicated that the presence of POSS, even at very low loadings (5 phr), induces a remarkable increase of the modulus either at low or at high strain and a considerable decrease of the hysteresis. Moreover, by comparing the performances of SBR/SiO2@POSS-X with those of nanocomposites obtained by simple addition of SiO2 and POSS in the polymer matrix, it turned out that the silica grafting with POSS determines a more positive effect on the mechanical properties, even for the uncured systems.
This has been associated with the peculiar structure of the hybrid filler. In fact, SiO2@POSS is constituted by silica NP aggregates partially interconnected and decorated by POSS nanounits which, thanks to the high number of reactive functionalities, promote a cross-linking action at the surface or even inside these aggregates. This fosters, upon introduction of SiO2@POSS in the rubber matrix, the formation of a tight filler network which immobilizes filler and rubber, affords a relevant reinforcement and improves the hysteretic properties. In summary, all the results suggest that SiO2@POSS can be a promising system, which grants the transfer of the advanced properties of POSS nanounits to the final nanocomposites, leading also to a potential reduction of filler utilization in the rubber formulation.
Although this work focuses on rubber nanocomposites, we expect that the presented hybrid filler may provide new scientific and technological opportunities for designing other advanced polymer nanocomposites, which require remarkable mechanical strength and low deformability, simultaneously.
Acknowledgements
This work was carried out in the frame of the European COST action MP1202 “Rational design of hybrid organic–inorganic interfaces: the next step towards advanced functional materials”. Matteo Redaelli thanks Corimav (“Consortium for the Research of Advanced Materials between Pirelli and Milano Bicocca University”) for its support within the PCAM European Doctoral Programme.References
- J. Ramier, C. Gauthier, L. Chazeau, L. Stelandre and L. Guy, J. Polym. Sci., Part B: Polym. Phys., 2007, 45, 286–298 CrossRef CAS.
- G. P. Baeza, A.-C. Genix, C. Degrandcourt, L. Petitjean, J. Gummel, R. Schweins, M. Couty and J. Oberdisse, Macromolecules, 2013, 46, 6621–6633 CrossRef CAS.
- I. Mora-Barrantes, L. Ibarra, A. Rodrıguez, L. Gonzalez and J. L. Valentın, J. Mater. Chem., 2011, 21, 17526–17533 RSC.
- A. K. Bhowmick, Current topics in elastomers research, CRC Press–Taylor & Francis Group, Broken Sound Parkway, NW, 2008 Search PubMed.
- S. Pavlova and P. G. Khalatur, Soft Matter, 2016, 12, 5402–5419 RSC.
- B. Guo, F. Chen, Y. Lei and W. Chen, Polym. J., 2010, 42, 319–325 CrossRef CAS.
- L. Wahba, M. D'Arienzo, R. Donetti, T. Hanel, R. Scotti, L. Tadiello and F. Morazzoni, RSC Adv., 2013, 3, 5832–5844 RSC.
- R. Scotti, L. Conzatti, M. D'Arienzo, B. Di Credico, L. Giannini, T. Hanel, P. Stagnaro, A. Susanna, L. Tadiello and F. Morazzoni, Polymer, 2014, 55, 1497–1506 CrossRef CAS.
- L. Tadiello, M. D'Arienzo, B. Di Credico, T. Hanel, L. Matejka, M. Mauri, F. Morazzoni, R. Simonutti, M. Spirkova and R. Scotti, Soft Matter, 2015, 11, 4022–4033 RSC.
- E. Ayandele, B. Sarkar and P. Alexandridis, Nanomaterials, 2012, 2, 445–475 CrossRef CAS.
- K. N. Raftopoulos and K. Pielichowski, Prog. Polym. Sci., 2016, 52, 136–187 CrossRef CAS.
- S. W. Kuo, ACS Cent. Sci., 2016, 2, 62–64 CrossRef CAS PubMed.
- D. B. Cordes, P. D. Lickiss and F. Rataboul, Chem. Rev., 2010, 110, 2081–2173 CrossRef CAS PubMed.
- S. W. Kuoa and F. C. Chang, Prog. Polym. Sci., 2011, 36, 1649–1696 CrossRef.
- A. Fina, O. Monticelli and G. Camino, J. Mater. Chem., 2010, 20, 9297–9305 RSC.
- W. Zhanga and A. H. E. Müller, Prog. Polym. Sci., 2013, 38, 1121–1162 CrossRef.
- K. Tanaka and Y. Chujo, J. Mater. Chem., 2012, 22, 1733–1750 RSC.
- A. L. Goffin, E. Duquesne, J. M. Raquez, H. E. Miltner, X. Ke, M. Alexandre, G. Van Tendeloo, B. Van Meleb and P. Dubois, J. Mater. Chem., 2010, 20, 9415–9625 RSC.
- J. H. Jeon, K. Tanaka and Y. Chujo, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 3583–3589 CrossRef CAS.
- Z. Zhou, L. Cui, Y. Zhang, Y. Zhang and N. Yin, Eur. Polym. J., 2008, 44, 3057–3066 CrossRef CAS.
- T. S. Haddad and J. D. Lichtenhan, Macromolecules, 1996, 29, 7302–7304 CrossRef CAS.
- L. Matějka, I. A. Kroutilová, J. D. Lichtenhan and T. S. Haddad, Eur. Polym. J., 2014, 52, 117–126 CrossRef.
- O. Bianchi, L. G. Barbosa, G. Machado, L. B. Canto, R. S. Mauler and R. V. B. Oliveira, J. Appl. Polym. Sci., 2013, 128, 811–827 CrossRef CAS.
- A. Kosmalska, A. Strąkowska and M. Zaborski, Mater. Sci. Forum, 2012, 714, 175–181 CrossRef CAS.
- G. Pan, J. E. Mark and D. W. Schaefer, J. Polym. Sci., Part B: Polym. Phys., 2003, 41, 3314–3323 CrossRef CAS.
- V. Joshi, M. Srividhya, M. Dubey, A. K. Ghosh and A. Saxena, J. Appl. Polym. Sci., 2013, 130, 92–99 CrossRef CAS.
- T. Vokatá, M. Twomey, E. Mendez and J. H. Moon, J. Polym. Sci., Part A: Polym. Chem., 2015, 53, 1403–1412 CrossRef.
- J. H. Jeon, K. Tanaka and Y. Chujo, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 3583–3589 CrossRef CAS.
- K. Tanaka, S. Adachi and Y. Chujo, Polym. Chem., 2009, 5690–5697 CrossRef CAS.
- Z. Zhou, Y. Zhang, Z. Zeng and Y. Zhan, J. Appl. Polym. Sci., 2008, 110, 3745–3751 CrossRef CAS.
- L. Liua, M. Tiana, W. Zhanga, L. Zhanga and J. E. Markb, Polymer, 2007, 48, 3201–3212 CrossRef.
- Y. Duan, S. C. Jana, A. M. Reinsel, B. Lama and M. P. Espe, Langmuir, 2012, 28, 15362–15371 CrossRef CAS PubMed.
- F. Ciesielczyk, K. Szwarc-Rzepka and T. Jesionowski, Surf. Interface Anal., 2013, 45, 998–1007 CrossRef CAS.
- K. Szwarc-Rzepka, F. Ciesielczyk and T. Jesionowski, J. Nanomater., 2013, 674237 Search PubMed.
- N. Tsubokawa and H. Ishida, J. Polym. Sci., Part A: Polym. Chem., 1992, 30, 2241–2246 CrossRef CAS.
- M. Nowacka, K. Siwińska-Stefańska and T. Jesionowski, Colloid Polym. Sci., 2013, 29, 603–612 Search PubMed.
- W. K. Shin, J. Cho, G. A. Kannan, Y. S. Lee and D. W. Kim, Sci. Rep., 2016, 6, 26332 CrossRef CAS PubMed.
- M. Sadej, E. Andrzejewska, B. Kurc, H. Gojzewski and T. Jesionowski, J. Polym. Sci., Part A: Polym. Chem., 2014, 52, 3472–3487 CAS.
- B. W. Ellis and G. N. Welding, Rubber Chem. Technol., 1964, 37, 571–575 CrossRef.
- P. J. Flory and J. Rehner, J. Chem. Phys., 1943, 11, 521–526 CrossRef CAS.
- C. Bressy, V. Ngo, F. Ziarelli and A. Margaillan, Langmuir, 2012, 28, 3290–3297 CrossRef CAS PubMed.
- F. Bauer, H. Ernst, U. Decker, M. Findeisen, H. J. Gläsel, H. Langguth, E. Hartmann, R. Mehnert and C. Peuker, Macromol. Chem. Phys., 2000, 2654–2659 CrossRef CAS.
- K. Szwarc-Rzepka, F. Ciesielczyk and T. Jesionowski, J. Nanomater., 2013, 674237 Search PubMed.
- C. G. Allen, D. J. Baker, J. M. Albin, H. E. Oertli, D. T. Gillaspie, D. C. Olson, T. E. Furtak and R. T. Collins, Langmuir, 2008, 24, 13393–13398 CrossRef CAS PubMed.
- M. D'Arienzo, M. Redaelli, B. Di Credico, S. Polizzi, R. Scotti and F. Morazzoni, RSC Adv., 2016, 6, 52987–52997 RSC.
- (a) R. Di Maggio, E. Callone, F. Girardi and S. Diré, J. Appl. Polym. Sci., 2012, 125, 1713–1723 CrossRef CAS; (b) F. Graziola, F. Girardi, R. Di Maggio, E. Callone, E. Miorin, M. Negri, K. Müller and S. Gross, Prog. Org. Coat., 2012, 74, 479–490 CrossRef CAS.
- K. S. W. Sing, D. H. Everett, R. A. W. Haul, L. Moscou, R. A. Pierotti, J. Rouquérol and T. Siemieniewska, Pure Appl. Chem., 1985, 57, 603–619 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7qm00045f |
| This journal is © the Partner Organisations 2017 |