
Research progress regarding Si-based anode materials towards practical application in high energy density Li-ion batteries†
Jin-Yi
Li
ab,
Quan
Xu
ab,
Ge
Li
ab,
Ya-Xia
Yin
ab,
Li-Jun
Wan
*a and
Yu-Guo
Guo
 *ab
*ab
aCAS Key Laboratory of Molecular Nanostructure and Nanotechnology, and Beijing National Laboratory for Molecular Sciences, Institute of Chemistry, Chinese Academy of Sciences (CAS), Beijing 100190, P. R. China. E-mail: ygguo@iccas.ac.cn; wanlijun@iccas.ac.cn
bSchool of Chemistry and Chemical Engineering, University of Chinese Academy of Sciences, Beijing 100049, P. R. China
First published on 20th February 2017
Abstract
Silicon has been considered as one of the most promising high-capacity anode materials because of its environmentally friendly character, natural abundance, and attractive operating voltage. However, successful implementation of Si-based anodes in Li-ion batteries is seriously hindered by their huge volume variation and low electric conductivity. The rational design of Si and effective combination of nanosized Si with carbonaceous materials represent the most effective approaches to overcome the challenges towards practical application of Si-based anodes. In this review, the mechanisms of Li–Si alloying and cell failure are briefly analyzed to comprehend the inherent impediments of Si-based anodes. Furthermore, nano-structured Si materials are summarized and nano/micro-structured Si/C and SiOx/C composites with 3D conductive networks and stable interfaces are discussed in detail. Accessory battery components that influence electrochemical performance are also reviewed. As for practical application, full batteries with Si-based anodes are discussed. Finally, the key aspects of Si-based materials are emphasized and prospective strategies for promoting the practical applications of Si-based anodes in high energy density Li-ion batteries are proposed.
 Li-Jun Wan | Li-Jun Wan is an Academician of the CAS and a Professor of Chemistry at the ICCAS. He received his BS and MS degrees in Materials Science from the Dalian University of Technology in 1982 and 1987, respectively, and his PhD in Materials Chemistry from Tohoku University of Japan in 1996. His research interests focus on the physical chemistry of single molecules and molecular assemblies, nanomaterials for applications in energy and environmental science, and scanning probe microscopy. |
 Yu-Guo Guo | Yu-Guo Guo is a Professor of Chemistry and Materials Science at ICCAS. He received his PhD in Physical Chemistry from ICCAS in 2004. He worked at the Max Planck Institute for Solid State Research at Stuttgart (Germany), first as a Guest Scientist and then as a Staff Scientist from 2004 to 2007. He joined ICCAS as a full Professor in 2007. His research interests focus on energy materials and electrochemical energy-storage devices, such as Li-ion and Li-metal batteries. |
1 Introduction
Lithium-ion batteries (LIBs) have been the most attractive energy storage systems due to their excellent environmental compatibility, long cycle life, low self-discharge rate and the ability to deliver high rates of power.1–16 And they have become ubiquitous power sources for mobile applications and electric vehicles. Driven by the ever-increasing power consumption, extensive interest has been stimulated towards developing high energy density LIBs to satisfy the power demands, such as in electronic devices and hybrid electric vehicles.17–27Conventional graphite has been widely used as an anode material in commercial LIBs. However, the limited capacity of graphite anodes (372 mA h g−1)28 could not meet the urgent requirements of high energy density. Among all emerging anode materials proposed for LIBs, Si has been considered as one of the most promising anode materials to replace graphite because of its environmentally friendly properties (non-toxic), natural abundance (second largest resource), superior theoretical capacity (3587 mA h g−1, 9786 mA h cm−3 for forming Li15Si4 at room temperature) and attractive operating voltage.29–48 However, commercial application of Si has been hindered by the poor electrical conductivity (<10−3 S cm−1 at 25 °C) and large volume variation (∼300%) upon (de)lithiation. Huge volume variation causes high internal stress, electrode pulverization, and an unstable solid electrolyte interphase (SEI). Thus, a series of nano-structured Si materials have been extensively studied to overcome the aforementioned problems by alleviating the volume change as well as providing short Li ion transport path. However, electrochemical aggregation of nanosized Si materials and the low intrinsic electric conductivity of Si represent significant challenges for rate capability and capacity utilization. Hence, the incorporation of Si and SiOx with a second phase has proven to greatly improve the electrochemical performance. Si-based intermetallic compounds,49–52 for instance, amorphous Si alloys and surface modification of Si with metal, have exhibited enhanced rate capability and capacity retention.53–56 However, phase separation and heterogeneous expansion as well as high cost impede the application of intermetallic compounds. Among them, carbonaceous matrices are regarded as the most optimal phase for incorporation with Si because of their low cost, high electric conductivity, light weight and ductile properties to accommodate volume variation as well as to form a stable solid electrolyte interphase. However, nano-structured Si anodes face more fundamental problems, such as high specific surface area which leads to more irreversible reactions and low tap density. Given these tradeoffs, nano/micro-structured Si-based materials have been synthesized by anchoring nanosized Si with void spaces in the carbon matrices and the matrices have been constructed into micron sized anode materials recently.
In this review, we briefly present the challenges and opportunities of silicon anode materials for LIBs. A series of nano-structured Si materials and nano/micro-structured Si/C and SiOx/C anodes are systematically summarized to address the known issues. In addition, we demonstrate accessory battery components that affect the electrochemical performance, including multidimensional conductive additives,57–59 polymeric binders,60–72 electrolyte and electrolyte additives,73–94 as well as prelithiation.95–103 Then practical application of Si-based anodes in practical LIBs and mechanisms of full-cell failure are discussed.104–117 Consequently, we summarize the key aspects of Si-based anodes and propose prospective strategies for promoting the practical applications of Si-based anodes in high energy density LIBs.
2 Scientific challenges and opportunities of silicon anodes
2.1 Crystal structure of silicon
Silicon is a chemical element with symbol Si and atomic number 14. Despite being the eighth most common element in the universe by mass, silicon very rarely occurs as a pure free element in the Earth's crust. Purified silicon, which has a relatively low electric conductivity at room temperature (<10−3 S cm−1), has been widely applied in solar cells and semiconductor electronics, where most computers, cellular phones, and modern technology depend on it.118–126 Therefore, large-scale production of silicon is a mature technology. Based on its crystal structure, silicon can be categorized into non-crystalline amorphous Si (a-Si) and crystalline Si, including monocrystalline silicon (mono-c-Si) and polycrystalline silicon (poly-c-Si), as shown in Fig. 1. Silicon is a fourfold coordinated atom that is tetrahedrally bonded to four neighboring silicon atoms. In c-Si, this tetrahedral structure continues over a long range, thereby forming a well-ordered crystal lattice. The crystal lattice of mono-c-Si is continuous, unbroken to its edges and free of any grain boundaries. Poly-c-Si is composed of several smaller c-Si grains of varied crystallographic orientation, which are typically >1 mm in size. This material can be easily synthesized by allowing liquid silicon to cool using a seed crystal of the desired crystal structure. Additionally, other methods for forming smaller-grained poly-c-Si exist such as high temperature chemical vapor deposition (CVD). | ||
| Fig. 1 Crystal structure of silicon. | ||
a-Si is the non-crystalline form of silicon, which has no long range order. Moreover, not all the atoms within amorphous silicon are fourfold coordinated. Due to the disordered nature of the material some atoms have a dangling bond, representing defects in the continuous random network and may cause anomalous electric behavior. Therefore, the application of amorphous silicon to thin film solar cells and high-capacity anodes for LIBs has been well studied.127–136
Besides a-Si, investigation into the potential of the use of metal–metalloid alloys has led to the recognition of several potentially attractive amorphous silicon anode materials, including boron-silicide, several silicides, and SiO.131 In all of these cases, an irreversible reaction occurs during the first lithiation resulting in the formation of fine particles of amorphous elemental silicon in a matrix related to the precursor. Upon further cycling, lithium reversibly reacts with this amorphous silicon, thereby reaching desirable values of specific capacities and long cycling stability. As an alternative anode to graphite, SiOx (0 < x < 2) appears promising for practical application because of its high capacity and relatively low volume change upon cycling as compared with pure Si. However, the main challenge associated with SiOx is its poor initial Coulombic efficiency (ICE), which reaches only 50–60%.
The chemical structure of SiOx (x = 1) was investigated with TEM, electron scattering, electron energy-loss spectroscopy (EELS) and electron spectroscopic imaging (ESI). SiO is not a homogeneous single-phase compound but appears to be a nanomixture of amorphous Si and SiO2, and the interphase boundary layer between Si and SiO2 makes up 20–25% of the whole structure137,138 (Fig. 2a–c). Moreover, the structure of SiOx can be changed through various methods including heat treatment, etching, and ball milling. Amorphous silicon monoxide undergoes an atomic-scale disproportionation by forming silicon and silicon-dioxide-like regions (usually above 800 °C) under an inert atmosphere. The generated c-Si nanodomains are confirmed by XRD. With the increasing temperature, the crystalline Si grows. Bu SiO will lose electrochemical activity to a certain extent. Disproportionation of SiO provides a facile approach to obtain c-Si nanodomains embedded in the structure with improved electrochemical performance.
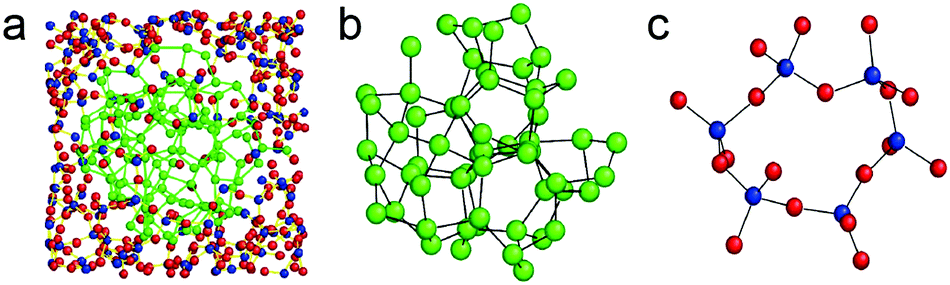 | ||
| Fig. 2 (a) Reconstructed heterostructure model of amorphous SiO. The inner part corresponds to an amorphous Si cluster and the outer part is an amorphous SiO2 matrix. The blue, red and green circles denote Si and O in amorphous SiO2 and Si in the Si cluster, respectively; (b) molecular dynamics model of pure amorphous Si; (c) molecular dynamics model of pure amorphous SiO2. Reprinted with permission from ref. 138. | ||
As reported in literature studies on the electrochemical properties among different types of Si, poly-c-Si shows poorer electric conductivity than mono-c-Si because of the grain boundary. Amorphous silicon can weaken cracking and pulverization compared to the crystalline state. Furthermore, the volume expansion of mono-c-Si upon lithiation is more anisotropic than the other crystal structure of silicon.
2.2 Alloying mechanisms of Li–Si
In the 1970s, a Li–Si alloy (Li2Si, Li21Si8, Li15Si4, Li22Si5) was first reported as an anode material in high-temperature molten salt lithium batteries.139–141 The Li–Si alloy was subsequently studied and the equilibrium Li–Si phase diagram was developed. With the continuing development of in situ techniques, more subtle information was obtained. The mechanism of electrochemical lithiation of c-Si anodes has been investigated with in situ Scanning Electron Microscopy (SEM), Transmission Electron Microscopy (TEM), Nuclear Magnetic Resonance (NMR) and X-ray Diffraction (XRD) by several research groups.118–126 Unlike intercalation electrodes, alloyed compound electrodes experience large volume variation during (de)lithiation and may undergo multiple crystallographic phase transitions (Fig. 3g), which shorten the cycle life and lead to cell failure. It was found that c-Si underwent electrochemically-driven amorphization, which experienced single-crystalline-to-amorphous phase transformation during the first lithiation cycle and remained amorphous afterwards at room temperature.121–125 However, shear-driven amorphization was observed by He et al.126 based on the in situ high-resolution TEM (HRTEM) in their previous work. This process occurred in a dominant shear band starting with the diamond-cubic (dc) to diamond-hexagonal (dh) phase transition before proceeding by dislocation nucleation and accumulation in the newly formed dh-Si phase, thereby forming an a-Si band, which was embedded with dh-Si nanodomains (Fig. 3c and d). Furthermore, Eric et al.125 combined Focused Ion Beam-Time of Flight-Secondary Ion Mass Spectrometry (FIB-ToF-SIMS) imaging with X-ray excited Auger electron spectroscopy (AES) quantitative analysis to precisely describe the lithiation mechanisms of silicon nanoparticles during the first cycle (Fig. 3a and b). At the very beginning, SEI formation consumes lithium during the first 5% of lithiation. Then a sharp interface was visualized between a Li3.1Si shell and a pure silicon core, thereby indicating a two-phase mechanism.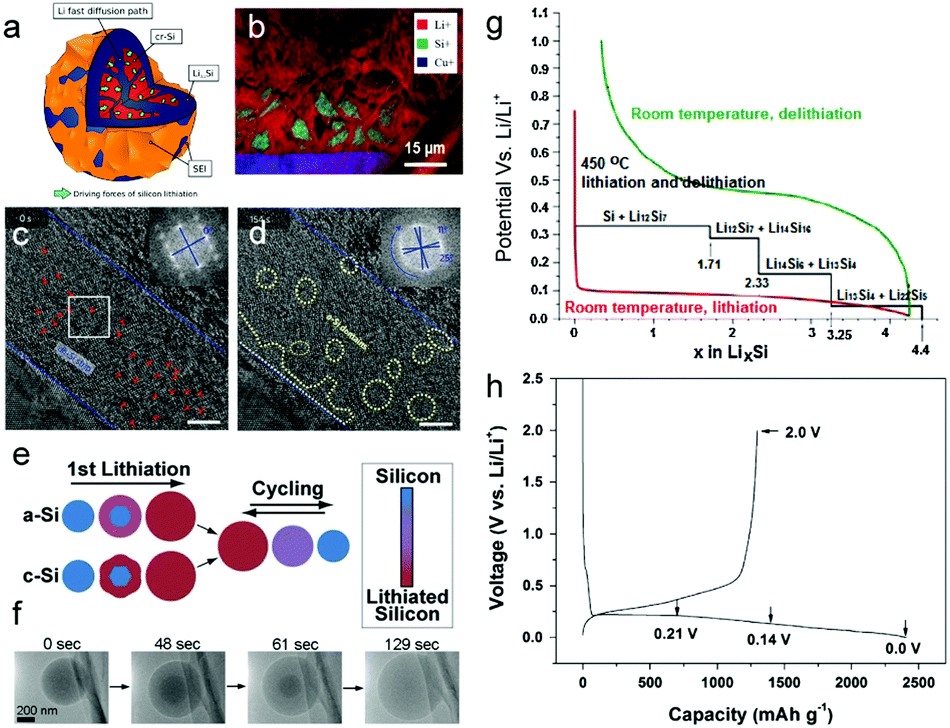 | ||
| Fig. 3 (a) Schematic view of a particle during first lithiation; (b) ToF-SIMS chemical mapping of Li+ (red), Si+ (green) and Cu+ (blue) of the FIB cross section of a delithiated electrode after 5 cycles performed at a limited capacity of 1000 mA h g−1; (c) HRTEM image of the dh-Si strip before the formation of amorphous domains; (d) HRTEM image showing the formation of nanosized amorphous domains (marked by dotted circles). Scale bars in c and d, 5 nm. Insets (c and d): Fast Fourier transform of the strip, with blue crosses tracking the rotation of the dh-Si lattice, and the dh-Si lattice in the strip pulverized and rotated during the shear-driven amorphization process; (e) schematic of the first lithiation and subsequent cycling of a-Si and c-Si; (f) time series of the lithiation of a single a-Si sphere; (g) Si electrochemical lithiation and delithiation curve at room temperature and high temperature. Black line: theoretical voltage curve at 450 °C. Red and green line: lithiation and delithiation of crystalline Si at room temperature, respectively; (h) voltage profiles of the SiO electrode for the first cycle. Reprinted with permission from ref. 29, 125, 126, 129, 132 and 143. | ||
Lithiation of a-Si has been widely thought to be a single-phase mechanism with gentle Li profiles, thus offering significant potential for mitigating pulverization and capacity fade. However, Cui129 and Zhu132et al. observed a sharp phase boundary between the a-Si reactant and an a-LixSi (x ∼ 2.5) product with in situ TEM, confirming a two-phase mechanism that occurred during alloying (Fig. 3e and f).
Fig. 3h illustrates the voltage profile of the SiO electrode at a constant current of 50 mA g−1 for the first cycle.122,142,143 The first discharge (Li insertion) and charge (Li extraction) capacities of pure SiO were about 2410 and 1300 mA h g−1, respectively, with a low ICE of ∼54%.
2.3 Cell failure mechanisms of silicon
Silicon-based anodes are one of the most promising anode materials for LIBs because of the high lithium storage capacity of silicon. However, the rapid capacity fading problem of silicon anodes prevents their practical use in current LIBs. Understanding the mechanisms of (de)lithiation and cell failure in silicon anodes is significant to enhance their electrochemical properties. Fig. 4 illustrates the key failure modes in the fading mechanism that originate from the huge volume variation of Si upon (de)lithiation (Li15Si4 290%, Li22Si5 320%).29,33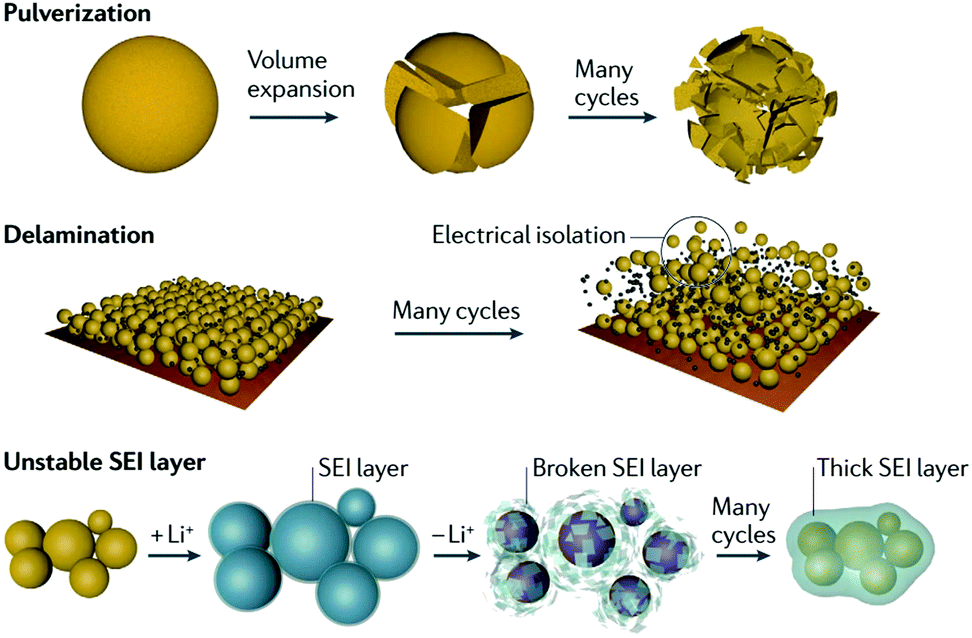 | ||
| Fig. 4 Cell failure mechanism of silicon. Reprinted with permission from ref. 33. | ||
For creating a stable SEI layer, SEI growth mechanisms and models may provide more guidance. Recently, a SEI model was proposed from the investigation by atomic force microscopy (AFM).149 The results showed that self-limiting SEI growth on Si at higher potentials (0.3–1.2 V), referred to as mesoporous organic SEI, was believed to consist primarily of carbon-rich electrolyte decomposition products. The formation of inorganic SEI at lower potentials (≤0.3 V) was widely believed that these materials limit electron conductivity, while Li2CO3 and Li2O permit fast Li+ transport. So further SEI growth is largely limited by electron availability at the internal interface. In the meantime, the unrepeated formation of SEI will consume more Li+, leading to most of the irreversible capacity and low ICE. Thus, mitigating the irreversible capacity of Si-based anodes is highly essential to improve the electrochemical performance. Lately, Grey et al.150 proposed a model for lithiation capacity loss that can be roughly approximated into two stages. In the first stage, the capacity loss was correlated with the growth of SEI and increasing electrode tortuosity. The increased electrode density observed by FIB/SEM was ascribed to a combination of SEI formation on the Si particles, particle agglomeration, and SEI exfoliation from particle surfaces filling the pores of the electrode. In the second stage, the decreasing lithiation capacity was attributed to sluggish kinetic diffusion of Li+.
Eventually, initial cycling determines critical electrochemical and mechanical properties of the SEI and the SEI growth model provides new insight into engineering better SEI with improved properties by modifying the charge–discharge program. And the model for irreversible capacity will aid optimization of electrochemical cycling, electrode formulation, and the use of electrolyte additives.
3 Nano-structured Si anode materials
Based on the fundamental understanding of the alloying and cell failure mechanisms of Si anodes for LIBs, strategies have already been explored to achieve long cycle life. Nano-structured Si materials indicate a potential approach to alleviate the huge volume variation of Si-based anodes. The electrochemical reactions are also facilitated by fast lithium diffusion and high surface-to-volume ratio of the nanostructure. Thus, a number of nano-structured Si materials have been extensively investigated. Fig. 5 displays the changes of Si nanostructures and morphology during (de)lithiation, including 0D solid nanoparticles154–174 or nanospheres with pores or voids,175–179 1D nanowires,180–190 nanorods,191–195 and nanotubes,196–198 2D thin films127–136 and nanosheets,199–205 and 3D porous nanostructures.206–220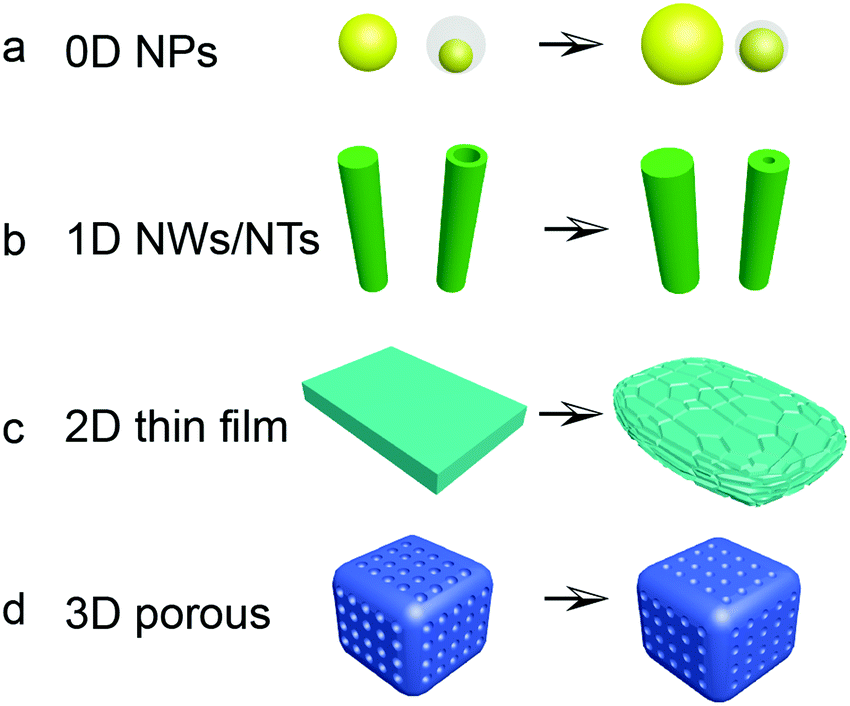 | ||
| Fig. 5 Schematic of Si nanostructures and morphology change after cycling: (a) 0D Si nanoparticles and yolk–shell nanostructure; (b) 1D Si nanowires and nanotubes; (c) 2D thin films; (d) 3D porous structure. | ||
For 0D nanoparticles, it has been proven that nanoparticles with smaller diameters show a higher specific capacity and better capacity retention under the same circumstance because of the reduced tendency for particle cracking.145 However, the particle-based anode material is also hindered by electrochemical aggregation and electrode destruction after repeated expansion/contraction. To address these issues, nanoparticles are dispersed into matrices so that stress generated from volume expansion during lithiation could be quickly released. The matrix does not experience huge volumetric change and may buffer the huge expansion of silicon, maintaining the structural integrity of the electrode and enhancing the electric conductivity and cycling stability. Graphene, sp2 hybridized carbon that is a single-atom-thick, has been identified as an attractive supporting material that may address the huge volume expansion and low electric conductivity associated with Si anodes. Zhou et al.173 prepared Si NP@G using a self-assembly method. The Si NP@G showed quite stable cycling performance with a capacity of 1205 mA h g−1 after 150 cycles in the voltage range of 0.05–1.0 V vs. Li/Li+ at a current density of 100 mA g−1. Similar to graphene-wrapped Si, yolk–shell structures with Si nanoparticles inside have been extensively studied to address the issues of volume variation and unstable interface (Fig. 5a). This structure provided space for expansion of the Si core during lithiation and a carbon shell conducive to a stable SEI. In addition, hollow and mesoporous nanospheres with pores and void spaces were developed for further improvement. Magnesiothermic reduction of mesoporous silica hollow spheres is reported as a simple method for the preparation of Si hollow spheres compared with the CVD process with toxic SiH4. However, both methods are not feasible for large-scale production.
Other than 0D nanoparticles, 1D silicon nanowires, nanorods and nanotubes have shown stable cycling stability with high reversible specific capacity. 1D Si materials can accommodate volume variation without fracturing and form a direct current pathway when they are grown directly on the current collector (Fig. 5b). Cao et al.195 reported a new concept of Cu–Si nanocable arrays that were directly anchored on the Cu current collector for effective electron conduction to enhance the rate performance. Furthermore, the space between the nanocables can accommodate huge Si volume variation and an additional coating could stabilize the Si/electrolyte interface for long cycle life. Besides, the axial Si nanotubes provide accessible channels for electrolyte transportation and shortened lithium diffusion path from double walls, and offer additional space for volume variation during cycling. However, the high specific surface of the 1D nanostructure may lead to more irreversible reaction, resulting in serious unstable SEI formation. Then the unique double wall structural design was proposed and attributed to yield highly desirable electrochemical parameters (Fig. 5b). The outside coating layer is mechanically rigid, preventing Si from expanding outward while still permitting Li ions to pass through. Furthermore, the electrolyte does not penetrate into the NTs due to the high aspect ratio of the NTs. Therefore, the outer surface of the NTs interfaced with the electrolyte is static, thereby allowing for the generation of a stable SEI on the outer surface.
2D thin silicon films and nanosheets have been demonstrated as high capacity and long cycle life anodes for LIBs. Furthermore, a silicon thin film is considered as an excellent model material for scientific research as it is free of binders and conductive additives. Thin films can be mainly fabricated by chemical vapor decomposition (CVD) and physical vapor deposition (PVD). However, the thin film expands during lithiation along the perpendicular direction, while contraction during delithiation occurs both perpendicular to and in plane with the substrate, leading to cracking of the film after the first cycle (Fig. 5c). The reversible capacity and cycling stability decrease with increasing thickness of the films. Thus Si thin films encounter the predicament of limited capacity with thin thickness and mass production.
A 3D porous Si-based material, which refers to a continuous Si nanostructure with pores and voids, is the most effective approach to improve the electrochemical performance of Si-based anode materials by providing a fast lithium diffusion path as well as alleviating volume variation during cycling with anchored pores and void spaces (Fig. 5d). To fabricate 3D porous Si structures, a metal-assisted chemical etching method and a self-template method are frequently used. Compared with the above-mentioned non-template method, the template assisted approach is another way to fabricate porous structures. Monodispersed SiO2 nanospheres were widely used as a sacrificial hard template to form porous Si structures with a tunable particle size.
Each of the reviewed nano-structured Si materials has unique advantages and disadvantages. As for the synthetic methods, Si NPs can be prepared by CVD, plasma technology, mechanical ball-milling, molten salts and reduction of nanosized SiO2. However, the low yield of 1D NWs, NTs, NRs, 2D thin films and other porous nano-structured morphologies share the same dilemma of expensive and complex processes and impracticality for mass production. To date, the 0D nanoparticulate morphology synthesized via ball milling and pyrolysis has the advantages of large-scale production, low-cost and easy doping. Nevertheless, more low cost and scalable preparation techniques of high quality Si nanostructures need to be developed for the upcoming commercial Si-based anodes.
4 Nano/micro-structured Si-based anode materials
Nano-structured Si anodes have fruitfully addressed some serious challenges in terms of preventing particle fracture and facilitating a fast Li ion diffusion path. However, fundamental disadvantages associated with nano-structured designs are difficult to overcome, including the high specific surface area (SSA) and low tap density. High interparticle resistance and uncontrolled reactions induced by large surface contact with the electrolyte seriously reduces the Coulombic efficiency of nano-structured Si anodes. Besides, nano-structured materials with inappropriate voids and empty spaces lead to low tap density and generate a longer electron pathway. Hence, rational void design and valid structure of anode materials are urgently needed. Carbon frameworks hosting Si form a valid structure of Si anodes for enhanced electric conductivity to accommodate volume variation and contribute to a stable SEI. Given these tradeoffs, nano/micro-structured Si-based materials with a 3D conductive network and stable interface were extensively investigated. Such a design has multiple benefits: (i) 3D conductive networks constructed with carbon to provide fast electron/Li ion transport; (ii) avoidance of structural degradation and full utilization of active materials with nano-structured Si and hierarchical buffer structures; (iii) carbon shells for a stable interface; and (iv) effective solution of the dilemmas associated with high SSA and low tap density. Herein, the nano/micro-structured Si-based materials can be categorized into the following types: (i) nano-Si/pyrolysis carbon microparticles;221–237 (ii) nano-Si/graphitic carbon microparticles;106,238–257 and (iii) SiOx/C microparticles.258–2614.1 Nano-Si/pyrolysis carbon microparticles
Nano-Si/pyrolysis carbon microparticles usually exhibit high reversible capacities (>1000 mA h g−1) and superior cycling stability due to their well-established voids/pores and uniform carbon coating. As shown in Fig. 6a, Zhang et al.237 fabricated silicon/nitrogen-doped carbon/carbon nanotube (SNCC) nano/micro-hierarchical structured spheres via an electrospray approach. The silicon nanoparticles, prepared from rice husk, are homogenously encapsulated in the highly conductive porous carbon matrix built by carbon nanotubes and nitrogen-doped carbon, which contributes to fast electronic transport, prevents silicon from aggregating and maintains structural integrity upon (de)lithiation. The as-prepared SNCC spheres delivered a reversible specific capacity of 1380 mA h g−1 at a current density of 0.5 A g−1, and still maintain 1031 mA h g−1 after 100 cycles. The SNCC possessed a narrow pore size distribution of 2 nm and a specific surface area of 78.5 m2 g−1 whereas the SSA of silicon from rice husk reached 288.4 m2 g−1. The superior electrochemical performance was attributed to the low SAA of the micro-sized carbon matrix. The hierarchical structure with pores accommodate volume variation. In addition, the pomegranate-inspired Si/C clusters were designed and fabricated through micro-emulsion and step-growth polymerization methods.230 The sufficient and well-defined internal void space of the pomegranate-inspired Si/C clusters could maintain the structural integrity of Si/C microparticles (Fig. 6b). Besides, the multifunctional carbon component acted as an electrolyte-blocking layer that contributed to stable SEI formation on the outer surface of the microparticles. Based on the hierarchical arrangement, the prepared Si/C anodes exhibited improved cyclic stability and high average Coulombic efficiency at a mass loading of 0.2 mA h cm−2. However, the low tap density and low mass loading led to low volumetric density. Therefore, Lin et al.227 proposed Si/C microparticles with a high tap density as fabricated via a scalable mechanical pressing process with extraordinary industrial compatibility (Fig. 6c). This research demonstrated an effective approach to prepare ultrahigh density packing of solids and provided more opportunities for modification.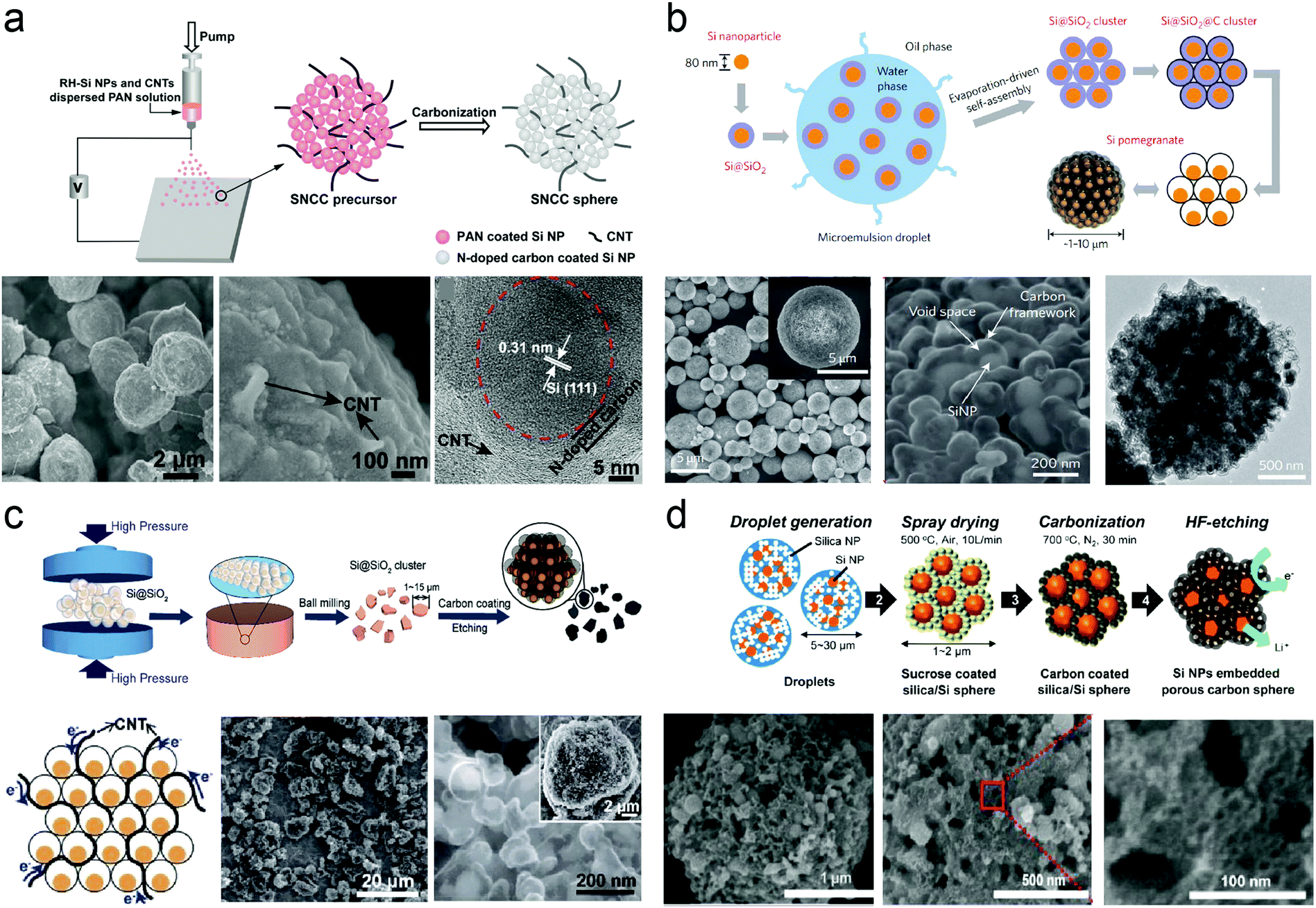 | ||
| Fig. 6 (a) Schematic illustration of the preparation process for SNCC nano/micro-structured spheres and SEM micrographs recorded at different magnifications, and HRTEM image of SNCC spheres; (b) schematic of the fabrication process for silicon pomegranates, SEM images of silicon pomegranates showing the micrometer-sized spherical morphology and the local structure, and TEM image of the carbon framework after etching away silicon using NaOH; (c) schematic illustration of the mechanical-based Si/C composite fabrication process and produced pellets under various pressures; (d) schematic of the ultrasonic spray drying process and SEM images of porous Si/C materials. Reprinted with permission from ref. 230, 224, 227 and 237. | ||
The synthesized Si/C materials presented high tap density (0.91 g cm−3) and superior electric contact. With well-established void space and an engineered electrolyte blocking layer, stable cycling of Si/C anodes (95% capacity retention after 1400 cycles) could be achieved at a mass loading of 0.2 mg cm−2. Furthermore, the obtained Si/C anodes with a mass loading of 2.02 mg cm−2 retained the areal capacity of 2.3 mA h cm−2 for 100 cycles without obvious capacity decay at a current density of 0.5 mA cm−2 due to improved electric conductivity. The authors suggested that high interparticle resistance could be effectively addressed by dense packing and good interparticle connection induced by high-pressure dry pressing. These aforementioned strategies attracted a great deal of attention from both researchers and industry, and supplied insights on the preparation of nano/micro-structured microparticles, although the electrospun and micro-emulsion methods were generally impractical for large-scale manufacturing. Hence, a vast majority of practical routes attempting to solve these disadvantages were proposed. For example, Jung et al.224 adopted an industrially established spray drying process to scalably produce Si/C microparticles (Fig. 6d). The facile spray drying process only required 2 s for the formation of each particle to produce Si/C materials with porous structure as well as large quantity syntheses. The silica functioned not only as colloidal seeds for the formation of dense and uniform Si/C microparticles as well as templates for pore generation via a HF etching process. The Si/C anodes displayed superior cycling stability (94% capacity retention after 50 cycles) and excellent rate capabilities from 0.1C to 10C. The above-mentioned studies made significant breakthrough on the scalable production of Si/C materials and improved tap density, which established a solid foundation to promote the practical application of Si/C anodes.
Generally, microparticles composed of nano-Si and pyrolysis carbon anodes could achieve excellent electrochemical performance in terms of cycling stability and high reversible capacity after HF etching and the carbon-coated process in half cells. Unfortunately, the following disadvantages of assembled Si/C microparticles will seriously hinder their practical applications: (i) HF etching was inevitably performed to remove the SiO2 sacrificial layer for engineering void space which could provide the empty space for huge volume variation of Si. Although, HF etching was essential to the high performance of Si/C materials, this process was dangerous and not feasible for large-scale application; (ii) the assembled Si/C anodes presented low Coulombic efficiency which irreversibly consumes lithium ions, thereby resulting in rapid capacity fading in practical application and continuous consumption of lithium ions provided by the cathode and electrolyte. However, the disadvantages of low Coulombic efficiency are always ignored due to the Li-metal electrode in the half cell providing excess Li; (iii) high mass loading was indispensable to improve the energy density of LIBs while the mass loading associated with energy stored in Si-based electrodes was relatively low (<1 mg cm−2). However, the nano-Si deep inside Si/C microparticles was difficult to be activated at high mass loading due to the poor electric conductivity of pyrolysis carbon. A 3D conductive network could address this issue; and (iv) a roll-to-roll process can reduce the electrolyte and binder dosage. This process plays an important role in practical LIBs to ensure high energy density. Pyrolysis carbon could not withstand the high pressure during the roll-to-roll process while graphite carbon materials show high anti-pressure ability. Hence, graphite carbon is commonly used to improve the structure integrity.
4.2 Nano-Si/graphitic carbon microparticles
Amorphous carbon is generally obtained from the pyrolysis of a polymer and acetylene and has poor electric conductivity and low specific capacity which could not be heavily used in Si/C composites. However, nano-Si and carbonaceous materials easily generate carborundum (SiC) at high temperatures (>1000 °C) because of the high reactivity of nanomaterials. Therefore, graphitic carbon materials are applied as the host materials to construct Si/C composites due to the low-cost, low volume variation, excellent electric conductivity and high CE. Amorphous carbon is best at forming a stable SEI layer as the coating layer on the outer surface of micron particles. In practical LIBs, volume deformation of the electrodes has strict requirement. According to the calculations and experienced estimations of Hong Li's group,106 the volume deformation of anode materials should be no more than 70%, preferably below 30%, to satisfy the requirements of pouch cells. The specific capacity of Si/C anodes needs to be calculated based on the relationship between volume variation (290%) and theoretical capacity (3590 mA h g−1 for forming Li15Si4). Consequently, the specific capacity of Si should be more than 867 mA h g−1. Besides, the roll-to-roll process is an essential factor to ensure the high energy density of LIBs, thus requiring graphite to maintain the structural integrity of Si/C anodes. The CE represented the reversibility of lithium ion transfer between the cathode and anode. In a full cell, the lithium ions provided by the cathode and electrolyte are limited. Therefore, the CE should be maintained as high as possible to improve the cycle life of full cells. Herein, we will start with the structural design of Si/C anode materials composed of nano-Si and graphitic carbon. In summary, the Si/C anodes in practical application should meet the following key aspects: volume deformation (<70%), areal capacity loading (>3 mA h cm−2), pressing density (>1.3 g cm−3), and average CE (>99.8%). To achieve the key requirements, the rational design of Si/C composites is composed of a graphitic carbon/nano-Si core and an amorphous carbon shell. This design has multiple advantages: (i) graphite is beneficial to maintain the structural integrity, lower SSA, improve tap density and serve as a buffer structure; (ii) the Si component ensures the high capacity of the Si/C anodes; (iii) amorphous carbon contributes to a stable SEI layer; and (iv) the inner graphitic carbon and outer amorphous carbon coating construct 3D conductive networks, which fully utilize the active Si materials and improve the electric conductivity of the whole electrode. There are mainly two structural designs for Si/C composites: nano-Si@C on the surface of spherical graphite and nano-Si@C uniformly dispersed in the Si/C composites. | ||
| Fig. 7 Schematic and structure characteristic of (a) Si attached and (b) deposited on the surface of spherical graphite; (c) watermelon-inspired Si/C microspheres and (d) walnut-structured nano-micro silicon/carbon. Reprinted with permission from ref. 106, 243, 244 and 257. | ||
4.3 SiOx/C microparticles
The electrochemical performance of SiOx is strongly related to the value of x. The final products of SiOx generated during lithiation include Li–Si alloy, Li2O and lithium silicates. And the irreversible capacity for SiOx in the first cycle was attributed to Li2O and lithium silicates.128,129 Thus, the value of x in SiOx has great influence on the lithiation products. It is inferred that when the x value is higher, formation of the irreversible product increases and the irreversible product acts as a buffer layer to accommodate volume expansion, contributing to improved cycling stability. However, the specific capacity decreases at the same time because the amount of active Si is reduced while x increases. Takezawa et al.270 successfully synthesized composition-controlled a-SiOx (0.17 ≤ x ≤ 1.34) films by the reactive evaporation method. Lithiated products were identified by XPS as fully lithiated Si, Li2O, Li silicates (Li4SiO4, Li2SiO3), and SiO2. As a result, excellent cyclability was achieved when the x value was 1.02, or 1.34. Moreover, Suh et al.259 synthesized granulated SiOx nanoparticles via a thermal plasma and water granulation process and determination of the optimum value of x. A high initial capacity of 1196 mA h g−1 was achieved when the value of x was 1.06 with a Coulombic efficiency of 75%.Despite the high capacity and low volume variation of SiOx, large initial capacity loss and unsatisfactory electrochemical performance still hinder the application when directly used as anode materials for LIBs. Further combination of 3 wt% SiOx with graphite for use as an anode gave rise to high capacity (397 mA h g−1) and a high Coulombic efficiency of 99.99% after 200 cycles. Combining with graphite is a potential approach to improve the electrochemical performance. Ren et al.271 synthesized a SiO/G/CNT composite by ball milling followed by the CVD method (Fig. 8e–h). The SiO/G/CNT composite exhibited a higher discharge capacity and a better durability than the SiO/G composite. The main reasons for the better electrochemical performance could be attributed to the excellent conductivity and ductility of the CNT matrix, and the effective dispersion of SiO/G. Besides, researchers have successfully improved the electrochemical performance via creating porous structures, surface modification and combination with a second phase. Yi et al.233 prepared a micro-sized Si–C composite composed of interconnected Si and carbon nanoscale building blocks through disproportion of SiO followed by HF etching (Fig. 8a–d). The Si–C composite exhibits a reversible capacity of 1459 mA h g−1 after 200 cycles at 1 A g−1 with a capacity retention of 97.8% and has a high tap density of 0.78 g cm−3. The excellent performance is attributed to the nanoscale size of primary particles and interconnected carbon and Si networks which can maintain internal electrical contact and sustain cycling stability.
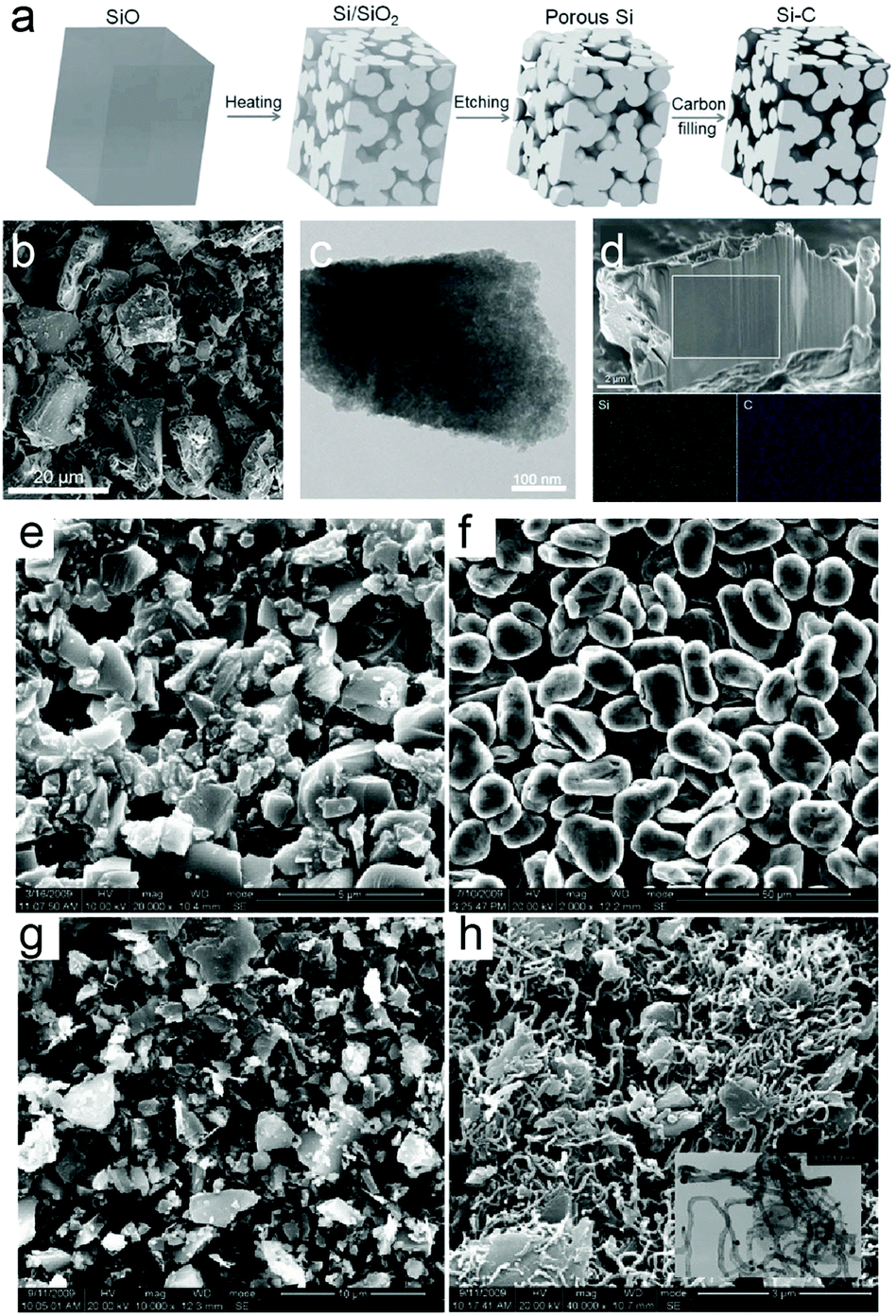 | ||
| Fig. 8 (a) Schematic of the preparation process from the SiO precursor to the Si–C composite; (b) SEM image; (c) TEM image of the obtained product; (d) cross-section SEM and EDS mapping of Si and C in the area marked by the white square. SEM micrographs of samples: (e) SiO, (f) graphite, (g) SiO/G, and (h) CNT-coated SiO/G; inserted TEM image of SiO/G/CNTs. Reprinted with permission from ref. 233 and 271. | ||
To date, SiOx has been used in commercial LIBs in a blended form with graphite. However, the SiOx content is less than 10 wt%, which reflects the infancy of Si-based anode technology. These following issues still need to be settled: (i) large lithium consumption during the first cycle results in low ICE and affects the energy density. (ii) Lucubrate electrochemical mechanism of SiOx upon lithiation. (iii) Optimum value of x to compromise both high capacity and cycling stability. (iv) Compatible technology of SiOx for upcoming applications of Si-based anodes in high energy density LIBs.
5 Accessory battery components
In addition to the abovementioned structure and morphology, there are several battery accessories that affect the electrochemical performance of Si-based anodes, including conductive additives,57–59 polymeric binders,60–72 electrolyte and electrolyte additives,73–94 aside from prelithiation95–103 effects.5.1 Conductive additives
As aforementioned, poor conductivity and electrode pulverization have a significant impact on electrochemical performance. Conductive additives can effectively improve conductivity and reduce polarization by forming electron-conducting pathways. Carbon black is a typical 0D conductive additive with the average particle size of spherical acetylene black being around 50 nm. However, 0D carbon black only provides point-to-point contact with active Si materials. In contrast, 1D CNTs with point-to-line contact exhibit good conductivity and maintain electrode integrity. The flexible graphene network effectively enhances the strength, flexibility and conductivity of the electrode. However, graphene and CNTs share the same problems of low Coulombic efficiency and relatively high cost for practical application. To construct 3D conductive networks on the electrode for Si-based anodes, carbon black combined with CNTs and graphene or RGO with an optimized distribution size can effectively improve the electrical conductivity and reduce the pulverization of Si-based anodes.5.2 Polymeric binders
Polymeric binders are usually employed to bind the active materials and conductive additives together onto the current collector. Poly(vinylidene)difluoride (PVDF) has been the most widely used binder in commercial electrodes. However, PVDF will seriously swell in EC/DMC solvents to form an adjunct SEI. Moreover, the weak interactions of PVDF cannot withstand the grand volume variation of Si-based anodes during (de)lithiation, thereby leading to fast cell failure in a few cycles.To enhance the cycling stability and the capacity retention of Si-based anodes, several novel binder systems have been explored, such as electronically conducting polymers,60 polymers with cross-link chains,64,66,67 self-healing polymeric matrices,62,72 and carbohydrate-based polymers.61,68–70 These binders effectively maintain the electrode structure during the repeated volume variation of Si, mainly via 3D interchain interactions (Fig. 9a and b). Current binders have dramatically improved the cycle life of Si-based anodes, but still cannot satisfy the demand of the cycling performance of electrodes. A suitable binder for Si-based anodes for practical applications should conquer the following difficulties to improve the performance: (i) supramolecular chemistry that can incorporate non-covalent interactions, such as hydrogen bonding, ion–dipole and π–π interactions; (ii) stretchable elastic polymers with simultaneous Si-binder and binder–current collector interactions; (iii) optimized polymers for fast Li-ion transport, high Coulombic efficiency and stable SEI. (iv) Natural abundance, low cost and sufficient affinity at lower binder content comparable to the present industrial conditions (<5 wt%).
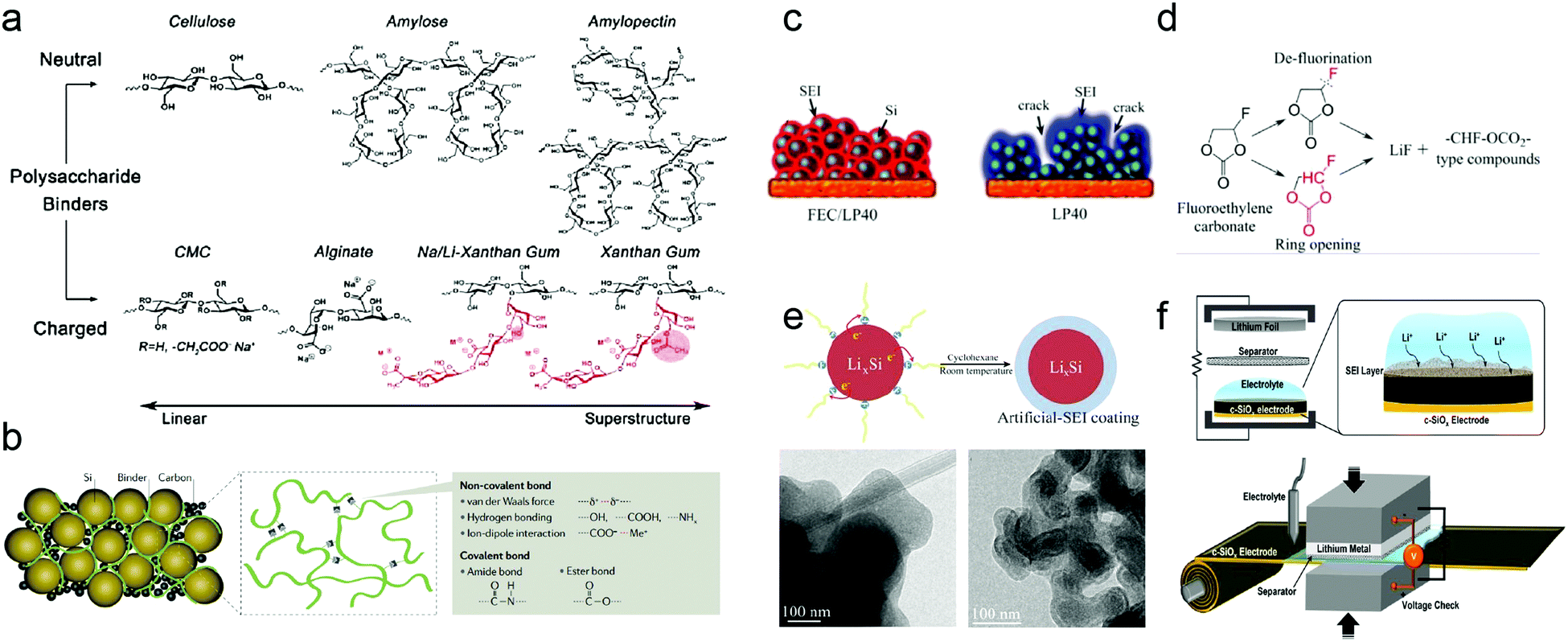 | ||
| Fig. 9 (a) Chemical structures of various polysaccharide binders classified based on their electrostatic charge and superstructures; (b) various binder concepts for Si anodes. SEI, solid electrolyte interphase; (c) schematic representation of SEI formation on a silicon anode which is cycled long-term with different electrolytes; (d) possible FEC decomposition reactions and products; (e) schematic diagram of the artificial SEI coating formed by reduction of 1-fluorodecane on the surface of LixSi NPs in cyclohexane. TEM images of LixSi NPs before and after coating; (f) graphical illustration of the prelithiation process of the c-SiOx electrode. Reprinted with permission from ref. 33, 67, 80, 97 and 102. | ||
5.3 Electrolyte and electrolyte additives
As discussed above, large volume variation challenges the formation of a stable SEI on Si-based anode materials. Smart structures,272,273 binders, and conductive additives successfully alleviate the stress caused by (de)lithiation and contribute to a stable SEI. However, electrolyte composition can greatly influence both the morphology and composition of the SEI layer. Focus has been directed towards modifying the electrolytes with additives and replacing the carbonate-based electrolytes with new solvent compositions and Si-based room temperature ionic liquids.73–76Vinylene carbonate,87–90 tris(pentafluorophenyl)borane,94 succinic anhydride,92,93 and fluoroethylene carbonate (FEC),80–86 undergoing reductive deposition at the surface of Si-based materials at higher potential than commonly used EC, have been added to improve cycling stability by forming a relatively uniform SEI with lower electrochemical resistance. SEI formed in the electrolyte with VC additives has been proved impermeable to the electrolyte and the impedance was almost invariant upon cycling. As an electrolyte additive, FEC efficiently improved the Coulombic efficiency and capacity retention of Si-based anodes. A conformal SEI, which sufficiently limited the emergence of large cracks and preserved the original surface morphology, formed on the silicon electrode in the presence of FEC.80Fig. 9c and d illustrates SEI formation on a silicon anode cycled with different electrolytes and possible FEC decomposition reactions and products, respectively.
5.4 Prelithiation
Si-based anodes commonly show low ICE and unsatisfactory cycle performance resulting from the large consumption of Li ions in the initial cycle and its gradual loss in subsequent cycles, which are attributed to SEI formation and irreversible reactions. To improve the ICE and cycle performance of the Si-based anode, prelithiation to compensate for the lithium loss demonstrates a promising approach. Therefore, prelithiation of the Si-based anodes has received sufficient attention.In view of the current investigations, the prelithiation methods of the Si-based anode can be categorized into chemical lithiation and electrochemical lithiation. In chemical lithiation, the active material is lithiated by the lithiation reagent. Zhao et al.97 demonstrated that LixSi NPs synthesized by thermal alloying can serve as a high-capacity prelithiation reagent. The reduction of 1-fluorodecane on the LixSi surface was exploited to form a continuous and dense coating, the coating serves as an effective passivation layer in the ambient environment as an artificial-SEI-protective layer (Fig. 9e). The first cycle CE of the Si NPs, which is coated with a LixSi artificial-SEI-protective layer, increased from 76.1% to 96.8%, confirming effective compensation for the large irreversible capacity loss. SiOx has been successfully commercialized in practical LIBs, because the silicon oxide matrix accommodates the volume change of embedded Si nanodomains. However, the poor Coulombic efficiencies in the early period of cycling limit the SiOx content. Therefore, Kim et al.102 proposed a subtle prelithiation scheme based on electrical shorting with lithium metal foil. The accurate shorting time and voltage monitoring allow fine-tuning of the degree of prelithiation, to a level where the Coulombic efficiencies in the first three cycles reach 94.9%, 95.7%, and 97.2%, respectively (Fig. 9f).
However, the electrochemical lithiation is conducted at the lab scale and the complex process is impractical for industrial applications. Chemical lithiation in practical production is hindered by the challenging synthesis of the uniform lithiation reagent and the highly active lithiation reagent as well as the unstable lithiation product, which cause safety concerns. Hence, more theoretical and practical research should be conducted towards industrial application of prelithiation for Si-based anodes, especially for SiOx anodes.
6 Practical full batteries with Si-based anodes
Towards practical application in high energy density LIBs, full batteries with Si-based anodes have been explored.104–117 After years of R&D, further application of Si-based anodes should focus on the energy density of LIBs in practical application instead of observed capacity and capacity retention in coin cells. Taking the fully-discharged compaction density of Si-based anodes into consideration, Li et al.104–106 calculated theoretical promotion of volumetric and gravimetric energy density pairing Si/C or graphite with LiCoO2, whose results are listed in Table 1. For example, Si/C (600 mA h g−1, average delithiation potential 0.3 V vs. Li+/Li) with a fully-discharged compaction density of 1.10 g cm−3 exhibited almost 50% improvement in gravimetric density compared to graphite anodes theoretically and 20% actual improvement due to unbalanced capacity of LiCoO2 cathodes and Si anodes. When pairing cathode with high capacity Si-based anodes, high capacity Ni-rich (e.g. LiNi0.8Co0.15Al0.05O2, 200 mA h g−1, average discharge potential 3.8 V vs. Li+/Li) and Li-rich (e.g. Li1.2Mn0.54Ni0.13Co0.13O2, 300 mA h g−1, average discharge potential 3.5 V vs. Li+/Li) cathode materials may be the preferred option in improving the energy density of LIBs compared with the commercial layered cathode materials LiCoO2 (∼145 mA h g−1, average discharge potential 3.8 V vs. Li+/Li) and LiNi1/3Co1/3Mn1/3O2 (∼153 mA h g−1, average discharge potential 3.7 V vs. Li+/Li). A high energy density LIB of 374 W h kg−1 was successfully developed.105 If the full-discharged compaction density of the anodes is controlled to a higher level, energy density promotion for high capacity Si-based anodes will be more significant. Moreover, prelithiation of Si/C and SiOx/C may increase the energy density further through lowering the loading amount of the cathode.| Specific capacity/mA h g−1 | Discharge voltage/V | Fully-discharged compaction density/g cm−3 | Initial Coulombic efficiency/% | Volumetric energy density/W h L−1 | Gravimetric energy density/W h kg−1 | |
|---|---|---|---|---|---|---|
| Graphite | 365 | 3.8 | 1.35 | 92 | 650 | 240 |
| Si/C | 450 | 3.72 | 1.25 | 89 | 702 | 280 |
| 500 | 3.69 | 1.15 | 88 | 705 | 306 | |
| 600 | 3.62 | 1.10 | 88 | 715 | 360 | |
High energy density LIBs with Si-based anodes in practical application have to face the difficulty that the long lifespan of full cells with Si-based anodes is still challenging. Therefore, it is vital to enclose the failure mechanisms for practical LIBs with silicon-based anodes which appear very different from that known for half-cells considering the amount of cyclable lithium. In full batteries, the amount of lithium is limited by the cathode and electrolyte while lithium metal in half cells provide a large excess of lithium. Thus, the lack of cyclable lithium results in the full cell failure. This mechanism was proved using a silicon anode paired with a LiNi1/3Mn1/3Co1/3O2 cathode in a full cell which was studied recently.108 This work provided an in-depth characterization of SEI formation using a combination of 7Li, 19F MAS NMR, XPS, TOF-SIMS and STEM-EELS. Researchers clarified that it was due to a lack of cyclable lithium because of parasitic reactions occurring before the accumulation of electrolyte degradation products that clogged the porosity of the composite electrode or disconnected the active material particles. Furthermore, non-coherent coulombic efficiencies, interfacial reaction behaviour and unstable solid electrolyte interphase have been proposed to be responsible for the full cell failure.111,112
From the above analysis of silicon-based anodes for full cells, it seems still challenging to attain the high energy density and long cycle life required for practical LIBs. Much effort should be spent to match suitable high capacity cathodes with silicon-based anodes, to increase the fully-discharged compaction density, to form a stable SEI on the surface and to mitigate the irreversibility of silicon-based anodes. Furthermore, continuous work still needs to be conducted on the failure mechanism of full cells and compatible technology with practical LIBs.
7 Summary and perspective
In summary, Si-based materials have been regarded as the most promising anode candidates for replacing graphite in high energy density LIBs. However, the huge volume variation during (de)lithiation and low electric conductivity of Si are the main impediments to successful implementation of Si-based anodes in practical applications. In this review, the mechanisms of Li–Si alloying and cell failure are analyzed to comprehend the inherent impediments at the beginning. To address these issues, we have systematically summarized a series of nano-structured Si materials that could significantly improve the electrochemical performance of Si-based anodes. However new fundamental problems associated with nanomaterials are yet to be overcome in terms of high SSA, low tap density and complex expensive technology to realize large-scale production. Over the last decade, tremendous progress has been made in addressing the restrictions by constructing micro/nano-structured Si/C and SiOx/C materials (Table S1, ESI†). Besides the above-mentioned structural design, excellent electrochemical performance requires the development of accessory battery components, including polymeric binders, multidimensional conductive additives, electrolytes and electrolyte additives for stable SEI formation as well as prelithiation. As discussed for full batteries, practical application of Si-based anodes is still challenging but full of opportunities. High ICE, areal capacity and fully-discharged compaction density are highly essential to attain high energy density for utilizing the high capacity of Si/SiOx. Recently, new processes, such as prelithiation technology, have been successfully utilized to eliminate the irreversible capacity loss during the first cycle in practical LIBs, greatly improving the energy density of LIBs with silicon anodes.To ensure high energy density toward practical applications, future exploration of Si-based materials should focus on nano/micro composites with a hierarchical buffer structure to obtain high reversible capacity and excellent cycling stability. Si-based anodes should meet the following key aspects: volume deformation of electrodes (<70%), areal capacity loading (>3 mA h cm−2), pressing density (>1.3 g cm−3) and average CE (>99.8%). To achieve this goal, possible approaches are as follows: (i) low cost, large-scale production of high-quality nanosized Si with controlled size to gain both electrochemical and mechanical properties, and SiOx technology with optimum values of x to achieve both high capacity and cycling stability. (ii) Rational void space for volumetric expansion of Si particles to control the total deformation of the electrode; (iii) dense core of graphitic carbon for utilizing Si and an amorphous carbon shell with suitable thickness to reduce the specific surface area and facilitate the formation of a stable SEI. A graphitic and amorphous carbon 3D conductive network will improve the electric conductivity. (iv) Spherical particles with optimized particle size distribution to improve the compaction density of anode materials; (v) in-depth research on full batteries with Si-based anodes and compatible technologies will boost the practical application of Si-based anodes. It is quite promising that continuous development of the strategies for promoting electrochemical performance will enable the production of high energy storage devices based on Si-based anodes in the near future.
Acknowledgements
This work was supported by the Ministry of Science and Technology of the People's Republic of China (Grant 2016YFB0100100), the National Natural Science Foundation of China (Grant No. 51225204, 21303222, and 21127901), and the “Strategic Priority Research Program” of the Chinese Academy of Sciences (Grant No. XDA09010100) and CAS.References
- J. M. Tarascon and M. Armand, Nature, 2001, 414, 359–367 CrossRef CAS PubMed
.
- P. Poizot, S. Laruelle, S. Grugeon, L. Dupont and J. Tarascon, Nature, 2000, 407, 496–499 CrossRef CAS PubMed
- J. R. Owen, Chem. Soc. Rev., 1997, 26, 259–267 RSC
- K. Xu, Chem. Rev., 2004, 104, 4303–4417 CrossRef CAS PubMed
- M. S. Whittingham, Chem. Rev., 2004, 104, 4271–4301 CrossRef CAS PubMed
- J. B. Goodenough and Y. Kim, Chem. Mater., 2009, 22, 587–603 CrossRef
- M. Winter, J. O. Besenhard, M. E. Spahr and P. Novak, Adv. Mater., 1998, 10, 725–763 CrossRef CAS
- Y. Wang and G. Z. Cao, Adv. Mater., 2008, 20, 2251–2269 CrossRef CAS
- G. Liu, S. Xun, N. Vukmirovic, X. Song, P. Olalde-Velasco, H. Zheng, V. S. Battaglia, L. Wang and W. Yang, Adv. Mater., 2011, 23, 4679–4683 CrossRef CAS PubMed
- H. Li, Z. X. Wang, L. Q. Chen and X. J. Huang, Adv. Mater., 2009, 21, 4593–4607 CrossRef CAS
- P. G. Bruce, B. Scrosati and J. M. Tarascon, Angew. Chem., Int. Ed., 2008, 47, 2930–2946 CrossRef CAS PubMed
- Y. X. Yin, S. Xin, Y. G. Guo and L. J. Wan, Angew. Chem., Int. Ed., 2013, 52, 13186–13200 CrossRef CAS PubMed
- C. P. Yang, Y. X. Yin, Y. G. Guo and L. J. Wan, J. Am. Chem. Soc., 2015, 137, 2215–2218 CrossRef CAS PubMed
- C. P. Yang, S. Xin, Y. X. Yin, H. Ye, J. Zhang and Y. G. Guo, Angew. Chem., Int. Ed., 2013, 52, 8363–8367 CrossRef CAS PubMed
- X. L. Wu, L. Y. Jiang, F. F. Cao, Y. G. Guo and L. J. Wan, Adv. Mater., 2009, 21, 2710–2714 CrossRef CAS
- X. Huang, X. Qi, F. Boey and H. Zhang, Chem. Soc. Rev., 2012, 41, 666–686 RSC
- J. Wang, H. Tang, L. Zhang, H. Ren, R. Yu, Q. Jin, J. Qi, D. Mao, M. Yang and D. Wang, Nat. Energy, 2016, 1, 16050 CrossRef CAS
- B. Wang, X.-L. Wu, C.-Y. Shu, Y.-G. Guo and C.-R. Wang, J. Mater. Chem., 2010, 20, 10661–10664 RSC
- H. Ren, R. Yu, J. Wang, Q. Jin, M. Yang, D. Mao, D. Kisailus, H. Zhao and D. Wang, Nano Lett., 2014, 14, 6679–6684 CrossRef CAS PubMed
- H. Ren, J. Sun, R. Yu, M. Yang, L. Gu, P. Liu, H. Zhao, D. Kisailus and D. Wang, Chem. Sci., 2015, 7, 793–798 RSC
- D. J. Xue, J. H. Tan, J. S. Hu, W. P. Hu, Y. G. Guo and L. J. Wan, Adv. Mater., 2012, 24, 4528–4533 CrossRef CAS PubMed
- S. Xin, Y. X. Yin, Y. G. Guo and L. J. Wan, Adv. Mater., 2014, 26, 1261–1265 CrossRef CAS PubMed
- Y. Q. Wang, L. Guo, Y. G. Guo, H. Li, X. Q. He, S. Tsukimoto, Y. Ikuhara and L. J. Wan, J. Am. Chem. Soc., 2012, 134, 7874–7879 CrossRef CAS PubMed
- J. J. Wang, F. F. Cao, L. Jiang, Y. G. Guo, W. P. Hu and L. J. Wan, J. Am. Chem. Soc., 2009, 131, 15602–15603 CrossRef CAS PubMed
- N. W. Li, Y. X. Yin, C. P. Yang and Y. G. Guo, Adv. Mater., 2016, 28, 1853–1858 CrossRef CAS PubMed
- C. Wu, J. Maier and Y. Yu, Adv. Mater., 2016, 28, 174–180 CrossRef CAS PubMed
- W. Li, Z. Yang, M. Li, Y. Jiang, X. Wei, X. Zhong, L. Gu and Y. Yu, Nano Lett., 2016, 16, 1546–1553 CrossRef CAS PubMed
- R. Yazami and P. Touzain, J. Power Sources, 1983, 9, 365–371 CrossRef CAS
- H. Wu and Y. Cui, Nano Today, 2012, 7, 414–429 CrossRef CAS
- J. R. Szczech and S. Jin, Energy Environ. Sci., 2011, 4, 56–72 CAS
- I. H. Son, J. Hwan Park, S. Kwon, S. Park, M. H. Rummeli, A. Bachmatiuk, H. J. Song, J. Ku, J. W. Choi, J. M. Choi, S. G. Doo and H. Chang, Nat. Commun., 2015, 6, 7393 CrossRef CAS PubMed
- D. L. Ma, Z. Y. Cao and A. M. Hu, Nano-Micro Lett., 2014, 6, 347–358 CrossRef
- J. W. Choi and D. Aurbach, Nat. Rev. Mater., 2016, 1, 16013 CrossRef CAS
- A. S. Arico, P. Bruce, B. Scrosati, J. M. Tarascon and W. van Schalkwijk, Nat. Mater., 2005, 4, 366–377 CrossRef CAS PubMed
- X. L. Wu, Y. G. Guo and L. J. Wan, Chem. – Asian J., 2013, 8, 1948–1958 CrossRef CAS PubMed
- L. Liu, J. Lyu, T. Li and T. Zhao, Nanoscale, 2016, 8, 701–722 RSC
- J. K. Lee, C. Oh, N. Kim, J. Y. Hwang and Y. K. Sun, J. Mater. Chem. A, 2016, 4, 5366–5384 CAS
- M. Ko, P. Oh, S. Chae, W. Cho and J. Cho, Small, 2015, 11, 4058–4073 CrossRef CAS PubMed
- B. Liang, Y. P. Liu and Y. H. Xu, J. Power Sources, 2014, 267, 469–490 CrossRef CAS
- S. C. Lai, J. Electrochem. Soc., 1976, 123, 1196–1197 CrossRef CAS
- U. Kasavajjula, C. S. Wang and A. J. Appleby, J. Power Sources, 2007, 163, 1003–1039 CrossRef CAS
- H. Wu, G. Y. Zheng, N. A. Liu, T. J. Carney, Y. Yang and Y. Cui, Nano Lett., 2012, 12, 904–909 CrossRef CAS PubMed
- X. Li and L. Zhi, Nanoscale, 2013, 5, 8864–8873 RSC
- M. Ko, S. Chae and J. Cho, ChemElectroChem, 2015, 2, 1645–1651 CrossRef CAS PubMed
- Y. Wang, Y. He, R. Xiao, H. Li, K. E. Aifantis and X. Huang, J. Power Sources, 2012, 202, 236–245 CrossRef CAS
- W. R. Liu, Y. C. Yen, H. C. Wu, M. Winter and N. L. Wu, J. Appl. Electrochem., 2009, 39, 1643–1649 CrossRef CAS
- M. Obrovac and V. Chevrier, Chem. Rev., 2014, 114, 11444–11502 CrossRef CAS PubMed
- C.-M. Park, J.-H. Kim, H. Kim and H.-J. Sohn, Chem. Soc. Rev., 2010, 39, 3115–3141 RSC
- S. Yoon, S.-I. Lee, H. Kim and H.-J. Sohn, J. Power Sources, 2006, 161, 1319–1323 CrossRef CAS
- G. Wang, L. Sun, D. Bradhurst, S. Zhong, S. Dou and H. Liu, J. Power Sources, 2000, 88, 278–281 CrossRef CAS
- H.-Y. Lee and S.-M. Lee, J. Power Sources, 2002, 112, 649–654 CrossRef CAS
- I.-C. Kim, D. Byun, S. Lee and J. K. Lee, Electrochim. Acta, 2006, 52, 1532–1537 CrossRef CAS
- T. Hatchard, M. Obrovac and J. Dahn, J. Electrochem. Soc., 2006, 153, A282–A287 CrossRef CAS
- T. Hatchard, J. Dahn, S. Trussler, M. Fleischauer, A. Bonakdarpour, J. Mueller-Neuhaus and K. Hewitt, Thin Solid Films, 2003, 443, 144–150 CrossRef CAS
- M. Fleischauer, M. Obrovac and J. Dahn, J. Electrochem. Soc., 2006, 153, A1201–A1205 CrossRef CAS
- M. Fleischauer and J. Dahn, J. Electrochem. Soc., 2004, 151, A1216–A1221 CrossRef CAS
- R. Yi, J. Zai, F. Dai, M. L. Gordin and D. Wang, Nano Energy, 2014, 6, 211–218 CrossRef CAS
- Z. D. Huang, B. Zhang, Y. B. He, S. W. Oh, Y. Yu and J. K. Kim, J. Electrochem. Soc., 2012, 159, A2024–A2028 CrossRef CAS
- Y. Fan, Q. Zhang, Q. Xiao, X. Wang and K. Huang, Carbon, 2013, 59, 264–269 CrossRef CAS
- H. Wu, G. Yu, L. Pan, N. Liu, M. T. McDowell, Z. Bao and Y. Cui, Nat. Commun., 2013, 4, 1943 Search PubMed
- I. Kovalenko, B. Zdyrko, A. Magasinski, B. Hertzberg, Z. Milicev, R. Burtovyy, I. Luzinov and G. Yushin, Science, 2011, 334, 75–79 CrossRef CAS PubMed
- C. Wang, H. Wu, Z. Chen, M. T. McDowell, Y. Cui and Z. Bao, Nat. Chem., 2013, 5, 1042–1048 CrossRef CAS PubMed
- J. Li, R. B. Lewis and J. R. Dahn, Electrochem. Solid-State Lett., 2007, 10, A17–A20 CrossRef CAS
- B. Koo, H. Kim, Y. Cho, K. T. Lee, N. S. Choi and J. Cho, Angew. Chem., Int. Ed., 2012, 51, 8762–8767 CrossRef CAS PubMed
- M. H. Ryou, J. Kim, I. Lee, S. Kim, Y. K. Jeong, S. Hong, J. H. Ryu, T. S. Kim, J. K. Park, H. Lee and J. W. Choi, Adv. Mater., 2013, 25, 1571–1576 CrossRef CAS PubMed
- T. W. Kwon, Y. K. Jeong, I. Lee, T. S. Kim, J. W. Choi and A. Coskun, Adv. Mater., 2014, 26, 7979–7985 CrossRef CAS PubMed
- Y. K. Jeong, T. W. Kwon, I. Lee, T. S. Kim, A. Coskun and J. W. Choi, Energy Environ. Sci., 2015, 8, 1224–1230 CAS
- Y. K. Jeong, T. W. Kwon, I. Lee, T. S. Kim, A. Coskun and J. W. Choi, Nano Lett., 2014, 14, 864–870 CrossRef CAS PubMed
- M. Murase, N. Yabuuchi, Z. J. Han, J. Y. Son, Y. T. Cui, H. Oji and S. Komaba, ChemSusChem, 2012, 5, 2307–2311 CrossRef CAS PubMed
- J. Liu, Q. Zhang, Z. Y. Wu, J. H. Wu, J. T. Li, L. Huang and S. G. Sun, Chem. Commun., 2014, 50, 6386–6389 RSC
- B. Jin, H. B. Gu and K. W. Kim, J. Solid State Electrochem., 2008, 12, 105–111 CrossRef CAS
- Z. Chen, C. Wang, J. Lopez, Z. Lu, Y. Cui and Z. Bao, Adv. Energy Mater., 2015, 5, 1401826 CrossRef
- D. Molina Piper, T. Evans, K. Leung, T. Watkins, J. Olson, S. C. Kim, S. S. Han, V. Bhat, K. H. Oh, D. A. Buttry and S. H. Lee, Nat. Commun., 2015, 6, 6230 CrossRef PubMed
- M. Q. Li, M. Z. Qu, X. Y. He and Z. L. Yu, Electrochim. Acta, 2009, 54, 4506–4513 CrossRef CAS
- F. H. Du, K. X. Wang and J. S. Chen, J. Mater. Chem. A, 2016, 4, 32–50 CAS
- J. C. Araque, J. J. Hettige and C. J. Margulis, J. Phys. Chem. B, 2015, 119, 12727–12740 CrossRef CAS PubMed
- M. Ulldemolins, F. Le Cras, B. Pecquenard, V. P. Phan, L. Martin and H. Martinez, J. Power Sources, 2012, 206, 245–252 CrossRef CAS
- D. Aurbach, B. Markovsky, M. D. Levi, E. Levi, A. Schechter, M. Moshkovich and Y. Cohen, J. Power Sources, 1999, 81, 95–111 CrossRef
- D. P. Abraham, M. M. Furczon, S. H. Kang, D. W. Dees and A. N. Jansen, J. Power Sources, 2008, 180, 612–620 CrossRef CAS
- C. Xu, F. Lindgren, B. Philippe, M. Gorgoi, F. Bjorefors, K. Edstrom and T. Gustafsson, Chem. Mater., 2015, 27, 2591–2599 CrossRef CAS
- I. A. Shkrob, J. F. Wishart and D. P. Abraham, J. Phys. Chem. C, 2015, 119, 14954–14964 CAS
- I. A. Profatilova, C. Stock, A. Schmitz, S. Passerini and M. Winter, J. Power Sources, 2013, 222, 140–149 CrossRef CAS
- C. C. Nguyen and B. L. Lucht, J. Electrochem. Soc., 2014, 161, A1933–A1938 CrossRef
- K. Leung, S. B. Rempe, M. E. Foster, Y. Ma, J. M. M. del la Hoz, N. Sai and P. B. Balbuena, J. Electrochem. Soc., 2014, 161, A213–A221 CrossRef CAS
- S. Dalavi, P. Guduru and B. L. Lucht, J. Electrochem. Soc., 2012, 159, A642–A646 CrossRef CAS
- X. Chen, X. Li, D. Mei, J. Feng, M. Y. Hu, J. Hu, M. Engelhard, J. Zheng, W. Xu and J. Xiao, ChemSusChem, 2014, 7, 549–554 CrossRef CAS PubMed
- H. Ota, Y. Sakata, A. Inoue and S. Yamaguchi, J. Electrochem. Soc., 2004, 151, A1659–A1669 CrossRef CAS
- L. B. Chen, K. Wang, X. H. Xie and J. Y. Xie, J. Power Sources, 2007, 174, 538–543 CrossRef CAS
- L. B. Chen, K. Wang, X. H. Xie and J. Y. Xie, Electrochem. Solid-State Lett., 2006, 9, A512–A515 CrossRef CAS
- D. Aurbach, K. Gamolsky, B. Markovsky, Y. Gofer, M. Schmidt and U. Heider, Electrochim. Acta, 2002, 47, 1423–1439 CrossRef CAS
- Y. Xiao, D. Hao, H. X. Chen, Z. L. Gong and Y. Yang, ACS Appl. Mater. Interfaces, 2013, 5, 1681–1687 CAS
- Y. Li, G. J. Xu, Y. F. Yao, L. G. Xue, S. Zhang, Y. Lu, O. Toprakci and X. W. Zhang, J. Solid State Electrochem., 2013, 17, 1393–1399 CrossRef CAS
- G. B. Han, M. H. Ryou, K. Y. Cho, Y. M. Lee and J. K. Park, J. Power Sources, 2010, 195, 3709–3714 CrossRef CAS
- G. B. Han, J. N. Lee, J. W. Choi and J. K. Park, Electrochim. Acta, 2011, 56, 8997–9003 CrossRef CAS
- N. Liu, L. Hu, M. T. McDowell, A. Jackson and Y. Cui, ACS Nano, 2011, 5, 6487–6493 CrossRef CAS PubMed
- M. W. Forney, M. J. Ganter, J. W. Staub, R. D. Ridgley and B. J. Landi, Nano Lett., 2013, 13, 4158–4163 CrossRef CAS PubMed
- J. Zhao, Z. D. Lu, H. T. Wang, W. Liu, H. W. Lee, K. Yan, D. Zhuo, D. C. Lin, N. Liu and Y. Cui, J. Am. Chem. Soc., 2015, 137, 8372–8375 CrossRef CAS PubMed
- J. Zhao, H. W. Lee, J. Sun, K. Yan, Y. Y. Liu, W. Liu, Z. D. Lu, D. C. Lin, G. M. Zhou and Y. Cui, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 7408–7413 CrossRef CAS PubMed
- J. Y. Zhang, C. Q. Zhang, S. M. Wu, X. Zhang, C. B. Li, C. L. Xue and B. W. Cheng, Nanoscale Res. Lett., 2015, 10, 395 CrossRef PubMed
- N. H. Yang, Y. S. Wu, J. Chou, H. C. Wu and N. L. Wu, J. Power Sources, 2015, 296, 314–317 CrossRef CAS
- M. Marinaro, M. Weinberger and M. Wohlfahrt-Mehrens, Electrochim. Acta, 2016, 206, 99–107 CrossRef CAS
- H. J. Kim, S. Choi, S. J. Lee, M. W. Seo, J. G. Lee, E. Deniz, Y. J. Lee, E. K. Kim and J. W. Choi, Nano Lett., 2015, 16, 282–288 CrossRef PubMed
- S. Chang, J. Moon, K. Cho and M. Cho, Comput. Mater. Sci., 2015, 98, 99–104 CrossRef CAS
- J. Wu, P. Liu, Y. Hu and H. Li, Energy Storage Sci. Technol., 2016, 5, 443–453 Search PubMed
- B. Liu, Q. Xu, G. Chu, H. Lu, Y. Yin, F. Luo, J. Zheng, Y. Guo and H. Li, Energy Storage Sci. Technol., 2016, 5, 418–421 Search PubMed
- F. Luo, B. Liu, J. Zheng, G. Chu, K. Zhong, H. Li, X. Huang and L. Chen, J. Electrochem. Soc., 2015, 162, A2509–A2528 CrossRef CAS
- K. S. Eom, T. Joshi, A. Bordes, I. Do and T. F. Fuller, J. Power Sources, 2014, 249, 118–124 CrossRef CAS
- N. Dupré, P. Moreau, E. De Vito, L. Quazuguel, M. Boniface, A. Bordes, C. Rudisch, P. Bayle-Guillemaud and D. Guyomard, Chem. Mater., 2016, 28, 2557–2572 CrossRef PubMed
- M.-S. Balogun, W. Qiu, Y. Luo, H. Meng, W. Mai, A. Onasanya, T. K. Olaniyi and Y. Tong, Nano Res., 2016, 9, 2823–2851 CrossRef CAS
- V. Aravindan, Y. S. Lee and S. Madhavi, Adv. Energy Mater., 2015, 5, 1402225 CrossRef
- C. Chae, H. J. Noh, J. K. Lee, B. Scrosati and Y. K. Sun, Adv. Funct. Mater., 2014, 24, 3036–3042 CrossRef CAS
- D.-T. Nguyen, J. Kang, K.-M. Nam, Y. Paik and S.-W. Song, J. Power Sources, 2016, 303, 150–158 CrossRef CAS
- K. Kierzek and J. Machnikowski, Electrochim. Acta, 2016, 192, 475–481 CrossRef CAS
- N. Delpuech, N. Dupre, P. Moreau, J. S. Bridel, J. Gaubicher, B. Lestriez and D. Guyomard, ChemSusChem, 2016, 9, 841–848 CrossRef CAS PubMed
- S. D. Beattie, M. J. Loveridge, M. J. Lain, S. Ferrari, B. J. Polzin, R. Bhagat and R. Dashwood, J. Power Sources, 2016, 302, 426–430 CrossRef CAS
- R. V. Salvatierra, A. R. O. Raji, S. K. Lee, Y. Ji, L. Li and J. M. Tour, Adv. Energy Mater., 2016, 1600918 CrossRef
- A. Krause, S. Dörfler, M. Piwko, F. M. Wisser, T. Jaumann, E. Ahrens, L. Giebeler, H. Althues, S. Schädlich and J. Grothe, Sci. Rep., 2016, 6, 27982 CrossRef CAS PubMed
- J. Yang, S. Muhammad, M. R. Jo, H. Kim, K. Song, D. A. Agyeman, Y.-I. Kim, W.-S. Yoon and Y.-M. Kang, Chem. Soc. Rev., 2016, 45, 5717–5770 RSC
- M. T. McDowell, S. W. Lee, C. Wang, W. D. Nix and Y. Cui, Adv. Mater., 2012, 24, 6034–6041 CrossRef CAS PubMed
- X. H. Liu and J. Y. Huang, Energy Environ. Sci., 2011, 4, 3844–3860 CAS
- P. Limthongkul, Y. I. Jang, N. J. Dudney and Y. M. Chiang, Acta Mater., 2003, 51, 1103–1113 CrossRef CAS
- J. Li and J. R. Dahn, J. Electrochem. Soc., 2007, 154, A156–A161 CrossRef CAS
- H. Li, X. J. Huang, L. Q. Chen, G. W. Zhou, Z. Zhang, D. P. Yu, Y. J. Mo and N. Pei, Solid State Ionics, 2000, 135, 181–191 CrossRef CAS
- B. Key, M. Morcrette, J. M. Tarascon and C. P. Grey, J. Am. Chem. Soc., 2011, 133, 503–512 CrossRef CAS PubMed
- A. Bordes, E. De Vito, C. Haon, A. Boulineau, A. Montani and P. Marcus, Chem. Mater., 2016, 28, 1566–1573 CrossRef CAS
- Y. He, L. Zhong, F. Fan, C. Wang, T. Zhu and S. X. Mao, Nat. Nanotechnol., 2016, 11, 866–871 CrossRef CAS PubMed
- J. Yin, M. Wada, K. Yamamoto, Y. Kitano, S. Tanase and T. Sakai, J. Electrochem. Soc., 2006, 153, A472–A477 CrossRef CAS
- H. Xia, S. Tang and L. Lu, Mater. Res. Bull., 2007, 42, 1301–1309 CrossRef CAS
- J. W. Wang, Y. He, F. Fan, X. H. Liu, S. Xia, Y. Liu, C. T. Harris, H. Li, J. Y. Huang, S. X. Mao and T. Zhu, Nano Lett., 2013, 13, 709–715 CrossRef CAS PubMed
- E. Pollak, G. Salitra, V. Baranchugov and D. Aurbach, J. Phys. Chem. C, 2007, 111, 11437–11444 CAS
- A. Netz, R. A. Huggins and W. Weppner, J. Power Sources, 2003, 119, 95–100 CrossRef
- M. T. McDowell, S. W. Lee, J. T. Harris, B. A. Korgel, C. Wang, W. D. Nix and Y. Cui, Nano Lett., 2013, 13, 758–764 CrossRef CAS PubMed
- H. Jung, M. Park, Y.-G. Yoon, G.-B. Kim and S.-K. Joo, J. Power Sources, 2003, 115, 346–351 CrossRef CAS
- H. Jung, M. Park, S. H. Han, H. Lim and S.-K. Joo, Solid State Commun., 2003, 125, 387–390 CrossRef CAS
- Y. He, X. Q. Yu, Y. H. Wang, H. Li and X. J. Huang, Adv. Mater., 2011, 23, 4938–4941 CrossRef CAS PubMed
- L. Chen, J. Xie, H. Yu and T. Wang, J. Appl. Electrochem., 2009, 39, 1157–1162 CrossRef CAS
- K. Schulmeister and W. Mader, J. Non-Cryst. Solids, 2003, 320, 143–150 CrossRef CAS
- A. Hirata, S. Kohara, T. Asada, M. Arao, C. Yogi, H. Imai, Y. W. Tan, T. Fujita and M. W. Chen, Nat. Commun., 2016, 7, 11591 CrossRef CAS PubMed
- C. J. Wen and R. A. Huggins, J. Solid State Chem., 1981, 37, 271–278 CrossRef CAS
- R. A. Sharma and R. N. Seefurth, J. Electrochem. Soc., 1976, 123, 1763–1768 CrossRef CAS
- F. F. Fan, S. Huang, H. Yang, M. Raju, D. Datta, V. B. Shenoy, A. C. T. van Duin, S. L. Zhang and T. Zhu, Modell. Simul. Mater. Sci. Eng., 2013, 21, 074002 CrossRef
- A. M. Wilson, B. M. Way, J. R. Dahn and T. van Buuren, J. Appl. Phys., 1995, 77, 2363 CrossRef CAS
- J.-H. Kim, C.-M. Park, H. Kim, Y.-J. Kim and H.-J. Sohn, J. Electroanal. Chem., 2011, 661, 245–249 CrossRef CAS
- I. Ryu, J. W. Choi, Y. Cui and W. D. Nix, J. Mech. Phys. Solids, 2011, 59, 1717–1730 CrossRef CAS
- X. H. Liu, L. Zhong, S. Huang, S. X. Mao, T. Zhu and J. Y. Huang, ACS Nano, 2012, 6, 1522–1531 CrossRef CAS PubMed
- X. H. Liu, H. Zheng, L. Zhong, S. Huang, K. Karki, L. Q. Zhang, Y. Liu, A. Kushima, W. T. Liang, J. W. Wang, J. H. Cho, E. Epstein, S. A. Dayeh, S. T. Picraux, T. Zhu, J. Li, J. P. Sullivan, J. Cumings, C. Wang, S. X. Mao, Z. Z. Ye, S. Zhang and J. Y. Huang, Nano Lett., 2011, 11, 3312–3318 CrossRef CAS PubMed
- X. D. Wu, Z. X. Wang, L. Q. Chen and X. J. Huang, Electrochem. Commun., 2003, 5, 935–939 CrossRef CAS
- P. Verma, P. Maire and P. Novak, Electrochim. Acta, 2010, 55, 6332–6341 CrossRef CAS
- A. Tokranov, R. Kumar, C. Z. Li, S. Minne, X. C. Xiao and B. W. Sheldon, Adv. Energy Mater., 2016, 6, 1502302 CrossRef
- A. L. Michan, G. Divitini, A. J. Pell, M. Leskes, C. Ducati and C. P. Grey, J. Am. Chem. Soc., 2016, 138, 7918–7931 CrossRef CAS PubMed
- G. Li, H. Li, Y. Mo, L. Chen and X. Huang, J. Power Sources, 2002, 104, 190–194 CrossRef CAS
- S. P. Kim, A. C. T. van Duin and V. B. Shenoy, J. Power Sources, 2011, 196, 8590–8597 CrossRef CAS
- C. K. Chan, R. Ruffo, S. S. Hong and Y. Cui, J. Power Sources, 2009, 189, 1132–1140 CrossRef CAS
- J. Yu, J. Yang, X. Feng, H. Jia, J. Wang and W. Lu, Ind. Eng. Chem. Res., 2014, 53, 12697–12704 CrossRef CAS
- A. M. Wilson and J. R. Dahn, J. Electrochem. Soc., 1995, 142, 326–332 CrossRef CAS
- M.-S. Wang and L.-Z. Fan, J. Power Sources, 2013, 244, 570–574 CrossRef CAS
- J. Park, G.-P. Kim, I. Nam, S. Park and J. Yi, Nanotechnology, 2012, 24, 025602 CrossRef PubMed
- S. H. Ng, J. Z. Wang, D. Wexler, K. Konstantinov, Z. P. Guo and H. K. Liu, Angew. Chem., Int. Ed., 2006, 45, 6896–6899 CrossRef CAS PubMed
- A. Magasinski, P. Dixon, B. Hertzberg, A. Kvit, J. Ayala and G. Yushin, Nat. Mater., 2010, 9, 353–358 CrossRef CAS PubMed
- X.-y. Zhou, J.-j. Tang, J. Yang, J. Xie and L.-l. Ma, Electrochim. Acta, 2013, 87, 663–668 CrossRef CAS
- H. Tao, L.-Z. Fan, W.-L. Song, M. Wu, X. He and X. Qu, Nanoscale, 2014, 6, 3138–3142 RSC
- L. Su, Z. Zhou and M. Ren, Chem. Commun., 2010, 46, 2590–2592 RSC
- L. Su, J. Xie, Y. Xu, L. Wang, Y. Wang and M. Ren, Phys. Chem. Chem. Phys., 2015, 17, 17562–17565 RSC
- Y. Park, N. S. Choi, S. Park, S. H. Woo, S. Sim, B. Y. Jang, S. M. Oh, S. Park, J. Cho and K. T. Lee, Adv. Energy Mater., 2013, 3, 206–212 CrossRef CAS
- L. Pan, H. Wang, D. Gao, S. Chen, L. Tan and L. Li, Chem. Commun., 2014, 50, 5878–5880 RSC
- X. Li, P. Meduri, X. Chen, W. Qi, M. H. Engelhard, W. Xu, F. Ding, J. Xiao, W. Wang and C. Wang, J. Mater. Chem., 2012, 22, 11014–11017 RSC
- H.-H. Li, J.-W. Wang, X.-L. Wu, H.-Z. Sun, F.-M. Yang, K. Wang, L.-L. Zhang, C.-Y. Fan and J.-P. Zhang, RSC Adv., 2014, 4, 36218–36225 RSC
- H. Li, X. J. Huang, L. Q. Chen, Z. G. Wu and Y. Liang, Electrochem. Solid-State Lett., 1999, 2, 547–549 CrossRef CAS
- S. Chen, M. L. Gordin, R. Yi, G. Howlett, H. Sohn and D. Wang, Phys. Chem. Chem. Phys., 2012, 14, 12741–12745 RSC
- R. D. Cakan, M.-M. Titirici, M. Antonietti, G. Cui, J. Maier and Y.-S. Hu, Chem. Commun., 2008, 3759–3761 RSC
- J. Bae, Colloid Polym. Sci., 2011, 289, 1233–1241 CAS
- H. Kim, M. Seo, M. H. Park and J. Cho, Angew. Chem., Int. Ed., 2010, 49, 2146–2149 CrossRef CAS PubMed
- X. S. Zhou, Y. X. Yin, L. J. Wan and Y. G. Guo, Adv. Energy Mater., 2012, 2, 1086–1090 CrossRef CAS
- J. K. Lee, K. B. Smith, C. M. Hayner and H. H. Kung, Chem. Commun., 2010, 46, 2025–2027 RSC
- Y. Yao, M. T. McDowell, I. Ryu, H. Wu, N. Liu, L. Hu, W. D. Nix and Y. Cui, Nano Lett., 2011, 11, 2949–2954 CrossRef CAS PubMed
- M. S. Wang, Y. Song, W. L. Song and L. Z. Fan, ChemElectroChem, 2014, 1, 2124–2130 CrossRef CAS
- W.-h. Lee, D.-Y. Kang, J. S. Kim, J. K. Lee and J. H. Moon, RSC Adv., 2015, 5, 17424–17428 RSC
- P. Gao, J. Fu, J. Yang, R. Lv, J. Wang, Y. Nuli and X. Tang, Phys. Chem. Chem. Phys., 2009, 11, 11101–11105 RSC
- N. Liu, H. Wu, M. T. McDowell, Y. Yao, C. Wang and Y. Cui, Nano Lett., 2012, 12, 3315–3321 CrossRef CAS PubMed
- Z. Wen, J. Stark, R. Saha, J. Parker and P. A. Kohl, J. Phys. Chem. C, 2013, 117, 8604–8610 CAS
- R. Teki, M. K. Datta, R. Krishnan, T. C. Parker, T. M. Lu, P. N. Kumta and N. Koratkar, Small, 2009, 5, 2236–2242 CrossRef CAS PubMed
- R. Ruffo, S. S. Hong, C. K. Chan, R. A. Huggins and Y. Cui, J. Phys. Chem. C, 2009, 113, 11390–11398 CAS
- K.-Q. Peng, X. Wang, L. Li, Y. Hu and S.-T. Lee, Nano Today, 2013, 8, 75–97 CrossRef CAS
- K. Peng, J. Jie, W. Zhang and S.-T. Lee, Appl. Phys. Lett., 2008, 93, 033105 CrossRef
- H. T. Nguyen, F. Yao, M. R. Zamfir, C. Biswas, K. P. So, Y. H. Lee, S. M. Kim, S. N. Cha, J. M. Kim and D. Pribat, Adv. Energy Mater., 2011, 1, 1154–1161 CrossRef CAS
- M. T. McDowell, S. W. Lee, I. Ryu, H. Wu, W. D. Nix, J. W. Choi and Y. Cui, Nano Lett., 2011, 11, 4018–4025 CrossRef CAS PubMed
- X. H. Liu, L. Q. Zhang, L. Zhong, Y. Liu, H. Zheng, J. W. Wang, J. H. Cho, S. A. Dayeh, S. T. Picraux, J. P. Sullivan, S. X. Mao, Z. Z. Ye and J. Y. Huang, Nano Lett., 2011, 11, 2251–2258 CrossRef CAS PubMed
- A. M. Chockla, K. C. Klavetter, C. B. Mullins and B. A. Korgel, Chem. Mater., 2012, 24, 3738–3745 CrossRef CAS
- C. K. Chan, R. Ruffo, S. S. Hong, R. A. Huggins and Y. Cui, J. Power Sources, 2009, 189, 34–39 CrossRef CAS
- C. K. Chan, H. L. Peng, G. Liu, K. McIlwrath, X. F. Zhang, R. A. Huggins and Y. Cui, Nat. Nanotechnol., 2008, 3, 31–35 CrossRef CAS PubMed
- Y. Zhou, X. Jiang, L. Chen, J. Yue, H. Xu, J. Yang and Y. Qian, Electrochim. Acta, 2014, 127, 252–258 CrossRef CAS
- S. Soleimani-Amiri, S. A. S. Tali, S. Azimi, Z. Sanaee and S. Mohajerzadeh, Appl. Phys. Lett., 2014, 105, 193903 CrossRef
- S. H. Nguyen, J. C. Lim and J. K. Lee, Electrochim. Acta, 2012, 74, 53–58 CrossRef CAS
- N. S. Hieu, J. C. Lim and J. K. Lee, Microelectron. Eng., 2012, 89, 138–140 CrossRef CAS
- F. F. Cao, J. W. Deng, S. Xin, H. X. Ji, O. G. Schmidt, L. J. Wan and Y. G. Guo, Adv. Mater., 2011, 23, 4415–4420 CrossRef CAS PubMed
- J. K. Yoo, J. Kim, Y. S. Jung and K. Kang, Adv. Mater., 2012, 24, 5452–5456 CrossRef CAS PubMed
- H. Wu, G. Chan, J. W. Choi, Y. Yao, M. T. McDowell, S. W. Lee, A. Jackson, Y. Yang, L. Hu and Y. Cui, Nat. Nanotechnol., 2012, 7, 310–315 CrossRef CAS PubMed
- M.-H. Park, M. G. Kim, J. Joo, K. Kim, J. Kim, S. Ahn, Y. Cui and J. Cho, Nano Lett., 2009, 9, 3844–3847 CrossRef CAS PubMed
- X. Yu, F. Xue, H. Huang, C. Liu, J. Yu, Y. Sun, X. Dong, G. Cao and Y. Jung, Nanoscale, 2014, 6, 6860–6865 RSC
- Y. H. Wang, Y. P. Liu, J. Y. Zheng, H. Zheng, Z. X. Mei, X. L. Du and H. Li, Nanotechnology, 2013, 24, 424011 CrossRef PubMed
- C. Pereira-Nabais, J. Światowska, M. Rosso, F. o. Ozanam, A. Seyeux, A. l. Gohier, P. Tran-Van, M. Cassir and P. Marcus, ACS Appl. Mater. Interfaces, 2014, 6, 13023–13033 CAS
- M. Park, G. Wang, H. Liu and S. Dou, Electrochim. Acta, 2006, 51, 5246–5249 CrossRef CAS
- Z. Lu, J. Zhu, D. Sim, W. Zhou, W. Shi, H. H. Hng and Q. Yan, Chem. Mater., 2011, 23, 5293–5295 CrossRef CAS
- J. C. Li, A. K. Dozier, Y. C. Li, F. Q. Yang and Y. T. Cheng, J. Electrochem. Soc., 2011, 158, A689–A694 CrossRef CAS
- H. Al-Taay, M. Mahdi, D. Parlevliet, Z. Hassan and P. Jennings, Superlattices Microstruct., 2014, 68, 90–100 CrossRef CAS
- A. Xing, S. Tian, H. Tang, D. Losic and Z. Bao, RSC Adv., 2013, 3, 10145–10149 RSC
- D. P. Wong, R. Suriyaprabha, R. Yuvakumar, V. Rajendran, Y.-T. Chen, B.-J. Hwang, L.-C. Chen and K.-H. Chen, J. Mater. Chem. A, 2014, 2, 13437–13441 CAS
- M.-S. Wang, L.-Z. Fan, M. Huang, J. Li and X. Qu, J. Power Sources, 2012, 219, 29–35 CrossRef CAS
- L. Shen, X. Guo, X. Fang, Z. Wang and L. Chen, J. Power Sources, 2012, 213, 229–232 CrossRef CAS
- Y. Liu, B. Chen, F. Cao, H. L. Chan, X. Zhao and J. Yuan, J. Mater. Chem., 2011, 21, 17083–17086 RSC
- N. Liu, K. Huo, M. T. McDowell, J. Zhao and Y. Cui, Sci. Rep., 2013, 3, 1919 Search PubMed
- H. Kim, B. Han, J. Choo and J. Cho, Angew. Chem., Int. Ed., 2008, 120, 10305–10308 CrossRef
- H. Jia, P. Gao, J. Yang, J. Wang, Y. Nuli and Z. Yang, Adv. Energy Mater., 2011, 1, 1036–1039 CrossRef CAS
- M. Ge, J. Rong, X. Fang, A. Zhang, Y. Lu and C. Zhou, Nano Res., 2013, 6, 174–181 CrossRef CAS
- M. Ge, Y. Lu, P. Ercius, J. Rong, X. Fang, M. Mecklenburg and C. Zhou, Nano Lett., 2013, 14, 261–268 CrossRef PubMed
- P. Gao, H. Jia, J. Yang, Y. Nuli, J. Wang and J. Chen, Phys. Chem. Chem. Phys., 2011, 13, 20108–20111 RSC
- A. Esmanski and G. A. Ozin, Adv. Funct. Mater., 2009, 19, 1999–2010 CrossRef CAS
- J. Cho, J. Mater. Chem., 2010, 20, 4009–4014 RSC
- D. Chen, X. Mei, G. Ji, M. Lu, J. Xie, J. Lu and J. Y. Lee, Angew. Chem., Int. Ed., 2012, 51, 2409–2413 CrossRef CAS PubMed
- B. M. Bang, J. I. Lee, H. Kim, J. Cho and S. Park, Adv. Energy Mater., 2012, 2, 878–883 CrossRef CAS
- D. A. Agyeman, K. Song, G.-H. Lee, M. Park and Y.-M. Kang, Adv. Energy Mater., 2016, 1600904, DOI:10.1002/aenm.201600904
- X. Feng, J. Yang, Y. Bie, J. Wang, Y. Nuli and W. Lu, Nanoscale, 2014, 6, 12532–12539 RSC
- X. Gao, J. Li, Y. Xie, D. Guan and C. Yuan, ACS Appl. Mater. Interfaces, 2015, 7, 7855–7862 CAS
- D. S. Jung, T. H. Hwang, S. B. Park and J. W. Choi, Nano Lett., 2013, 13, 2092–2097 CrossRef CAS PubMed
- H. Kim, B. Han, J. Choo and J. Cho, Angew. Chem., Int. Ed., 2008, 47, 10151–10154 CrossRef CAS PubMed
- J. I. Lee, N. S. Choi and S. Park, Energy Environ. Sci., 2012, 5, 7878–7882 CAS
- D. C. Lin, Z. D. Lu, P. C. Hsu, H. R. Lee, N. Liu, J. Zhao, H. T. Wang, C. Liu and Y. Cui, Energy Environ. Sci., 2015, 8, 2371–2376 CAS
- B. Liu, P. Soares, C. Checkles, Y. Zhao and G. Yu, Nano Lett., 2013, 13, 3414–3419 CrossRef CAS PubMed
- J. Liu, P. Kopold, P. A. van Aken, J. Maier and Y. Yu, Angew. Chem., Int. Ed., 2015, 54, 9632–9636 CrossRef CAS PubMed
- N. Liu, Z. Lu, J. Zhao, M. T. McDowell, H. W. Lee, W. Zhao and Y. Cui, Nat. Nanotechnol., 2014, 9, 187–192 CrossRef CAS PubMed
- A. Magasinski, P. Dixon, B. Hertzberg, A. Kvit, J. Ayala and G. Yushin, Nat. Mater., 2010, 9, 353–358 CrossRef CAS PubMed
- C. M. Xiao, N. Du, X. X. Shi, H. Zhang and D. R. Yang, J. Mater. Chem. A, 2014, 2, 20494–20499 CAS
- R. Yi, F. Dai, M. L. Gordin, S. R. Chen and D. H. Wang, Adv. Energy Mater., 2013, 3, 295–300 CrossRef CAS
- Q. Yun, X. Qin, Y.-B. He, W. Lv, Y. V. Kaneti, B. Li, Q.-H. Yang and F. Kang, Electrochim. Acta, 2016, 211, 982–988 CrossRef CAS
- Q. B. Yun, X. Y. Qin, W. Lv, Y. B. He, B. H. Li, F. Y. Kang and Q. H. Yang, Carbon, 2015, 93, 59–67 CrossRef CAS
- Z. Zhang, Y. Wang, W. Ren, Q. Tan, Y. Chen, H. Li, Z. Zhong and F. Su, Angew. Chem., Int. Ed., 2014, 53, 5165–5169 CAS
- Y. C. Zhang, Y. You, S. Xin, Y. X. Yin, J. Zhang, P. Wang, X. S. Zheng, F. F. Cao and Y. G. Guo, Nano Energy, 2016, 25, 120–127 CrossRef CAS
- K. Evanoff, A. Magasinski, J. B. Yang and G. Yushin, Adv. Energy Mater., 2011, 1, 495–498 CrossRef CAS
- B. Fuchsbichler, C. Stangl, H. Kren, F. Uhlig and S. Koller, J. Power Sources, 2011, 196, 2889–2892 CrossRef CAS
- S. Jeong, J. P. Lee, M. Ko, G. Kim, S. Park and J. Cho, Nano Lett., 2013, 13, 3403–3407 CrossRef CAS PubMed
- Y. N. Jo, Y. Kim, J. S. Kim, J. H. Song, K. J. Kim, C. Y. Kwag, D. J. Lee, C. W. Park and Y. J. Kim, J. Power Sources, 2010, 195, 6031–6036 CrossRef CAS
- S. W. Kim, J. H. Yun, B. Son, Y. G. Lee, K. M. Kim, Y. M. Lee and K. Y. Cho, Adv. Mater., 2014, 26, 2977–2982 CrossRef CAS PubMed
- S. Y. Kim, J. Lee, B. H. Kim, Y. J. Kim, K. S. Yang and M. S. Park, ACS Appl. Mater. Interfaces, 2016, 8, 12109–12117 CAS
- M. Ko, S. Chae, J. Ma, N. Kim, H.-W. Lee, Y. Cui and J. Cho, Nat. Energy, 2016, 1, 16113 CrossRef CAS
- J. H. Lee, W. J. Kim, J. Y. Kim, S. H. Lim and S. M. Lee, J. Power Sources, 2008, 176, 353–358 CrossRef CAS
- F. S. Li, Y. S. Wu, J. Chou and N. L. Wu, Chem. Commun., 2015, 51, 8429–8431 RSC
- M. Li, X. Hou, Y. Sha, J. Wang, S. Hu, X. Liu and Z. Shao, J. Power Sources, 2014, 248, 721–728 CrossRef CAS
- S. Li, X. Qin, H. Zhang, J. Wu, Y.-B. He, B. Li and F. Kang, Electrochem. Commun., 2014, 49, 98–102 CrossRef CAS
- X. Li, M. Gu, S. Hu, R. Kennard, P. Yan, X. Chen, C. Wang, M. J. Sailor, J. G. Zhang and J. Liu, Nat. Commun., 2014, 5, 4105 CAS
- W. F. Ren, Z. L. Zhang, Y. H. Wang, Q. Q. Tan, Z. Y. Zhong and F. B. Su, J. Mater. Chem. A, 2015, 3, 5859–5865 CAS
- H. Wang, J. Xie, S. Zhang, G. Cao and X. Zhao, RSC Adv., 2016, 6, 69882–69888 RSC
- Y.-X. Wang, S.-L. Chou, J. H. Kim, H.-K. Liu and S.-X. Dou, Electrochim. Acta, 2013, 93, 213–221 CrossRef CAS
- Q. Xu, J. Y. Li, Y. X. Yin, Y. M. Kong, Y. G. Guo and L. J. Wan, Chem. – Asian J., 2016, 11, 1205–1209 CrossRef CAS PubMed
- C.-H. Yim, F. M. Courtel and Y. Abu-Lebdeh, J. Mater. Chem. A, 2013, 1, 8234 CAS
- J. Yu, H. H. Zhan, Y. H. Wang, Z. L. I. Zhang, H. Chen, H. Li, Z. Y. Zhong and F. B. Su, J. Power Sources, 2013, 228, 112–119 CrossRef CAS
- X. Y. Zhu, H. Chen, Y. H. Wang, L. H. Xia, Q. Q. Tan, H. Li, Z. Y. Zhong, F. B. Su and X. S. Zhao, J. Mater. Chem. A, 2013, 1, 4483–4489 CAS
- Q. Xu, J. Y. Li, J. K. Sun, Y. X. Yin, L. J. Wan and Y. G. Guo, Adv. Energy Mater., 2016, 7, 1601481 CrossRef
- H. Zhao, Q. Yang, N. Yuca, M. Ling, K. Higa, V. S. Battaglia, D. Y. Parkinson, V. Srinivasan and G. Liu, Nano Lett., 2016, 16, 4686–4690 CrossRef CAS PubMed
- S. S. Suh, W. Y. Yoon, D. H. Kim, S. U. Kwon, J. H. Kim, Y. U. Kim, C. U. Jeong, Y. Y. Chan, S. H. Kang and J. K. Lee, Electrochim. Acta, 2014, 148, 111–117 CrossRef CAS
- M. Miyachi, H. Yamamoto, H. Kawai, T. Ohta and M. Shirakata, J. Electrochem. Soc., 2005, 152, A2089–A2091 CrossRef CAS
- X. Liu, H. L. Zhao, J. Y. Xie, P. P. Lv, K. Wang and J. J. Cui, Prog. Chem., 2015, 27, 336–348 Search PubMed
- X. S. Zhou, Y. X. Yin, L. J. Wan and Y. G. Guo, Chem. Commun., 2012, 48, 2198–2200 RSC
- X. S. Zhou, L. J. Wan and Y. G. Guo, Small, 2013, 9, 2684–2688 CrossRef CAS PubMed
- X. S. Zhou and Y. G. Guo, J. Mater. Chem. A, 2013, 1, 9019–9023 CAS
- X. S. Zhou, A. M. Cao, L. J. Wan and Y. G. Guo, Nano Res., 2012, 5, 845–853 CrossRef CAS
- Y. X. Yin, S. Xin, L. J. Wan, C. J. Li and Y. G. Guo, J. Phys. Chem. C, 2011, 115, 14148–14154 CAS
- Q. Xu, J. Y. Li, Y. X. Yin, Y. M. Kong, Y. G. Guo and L. J. Wan, Chem. – Asian J., 2016, 11, 1205–1209 CrossRef CAS PubMed
- S. Xin, Y. G. Guo and L. J. Wan, Acc. Chem. Res., 2012, 45, 1759–1769 CrossRef CAS PubMed
- X. L. Wu, Y. G. Guo and L. J. Wan, Chem. – Asian J., 2013, 8, 1948–1958 CrossRef CAS PubMed
- H. Takezawa, K. Iwamoto, S. Ito and H. Yoshizawa, J. Power Sources, 2013, 244, 149–157 CrossRef CAS
- Y. Ren, J. Ding, N. Yuan, S. Jia, M. Qu and Z. Yu, J. Solid State Electrochem., 2012, 16, 1453–1460 CrossRef CAS
- J. Wang, N. Yang, H. Tang, Z. Dong, Q. Jin, M. Yang, D. Kisailus, H. Zhao, Z. Tang and D. Wang, Angew. Chem., 2013, 125, 6545–6548 CrossRef
- J. Qi, X. Lai, J. Wang, H. Tang, H. Ren, Y. Yang, Q. Jin, L. Zhang, R. Yu and G. Ma, Chem. Soc. Rev., 2015, 44, 6749–6773 RSC
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6qm00302h |
| This journal is © the Partner Organisations 2017 |