DOI:
10.1039/C6QM00133E
(Research Article)
Mater. Chem. Front., 2017,
1, 668-676
Pyrene-centered cyanophenyl end-capped starbursts: design, synthesis, stabilized blue electroluminescence and lasing properties†
Received
20th July 2016
, Accepted 16th September 2016
First published on 28th September 2016
Abstract
A novel class of monodisperse starburst molecules (T1–T3), which are comprised of a pyrene core, four short oligofluorene arms and cyanophenyl end-cappers, were designed and synthesized to explore the influence of electron-withdrawing moieties on their optoelectronic properties. The resulting materials demonstrated high fluorescence yields and excellent thermal stability with a high degradation temperature (Td) of over 400 °C. Non-doped electroluminescent devices with the configuration of ITO/PEDOT:PSS/T1–T3/TPBI/LiF/Al exhibited highly efficient and stabilized blue electroluminescence (EL). Among all these solution-processed devices, T1 possessed the best performance with a maximum luminous efficiency (LE) of 3.52 cd A−1 at 25 mA cm−2 and a maximum brightness of 9194 cd m−2 at 10 V, resulting from the improved electron affinity due to the large content of electron-withdrawing cyanophenyl moieties in the starbursts. Notably, the net gain coefficients reached 46–55 cm−1. Moreover, the amplified spontaneous emission (ASE) threshold value remained detectable upon thermal annealing temperatures up to 230 °C for T1 and 280 °C for T2–T3. 1D DFB lasing (50% fill factor, pumped source at 375 nm) was thus obtained, demonstrating a low lasing threshold. The results confirm that the incorporation of cyanophenyl moieties as end cappers can enhance the electrical properties of pyrene-centered starbursts without substantially sacrificing their great optical gain properties, which makes this series of materials very promising for electrically pumped organic lasers.
1. Introduction
Organic semiconductor emitters based on π-conjugated systems via solution processing have attracted increasing attention in the emerging field of organic electronics such as organic field-effect transistors (OFETs),1 organic light-emitting diodes (OLEDs),2–4 and organic lasers,5–7 mainly owing to their low-cost and large-area solution processing, broad photoluminescence (PL) spectra and promising optoelectronic characteristics.8 Despite achieving prominent performances in OLEDs, most organic emitters failed to fulfill the demands for organic lasers.9 In particular, it is extremely challenging for organic emitters to simultaneously achieve high carrier mobility, high luminescence efficiencies and low lasing thresholds with the aim to realize electrically pumped organic lasers.5,9 Great efforts in this area have thus been devoted to exploring robust organic gain media with highly efficient electroluminescence characteristics and strong optical gain.10–14
Owing to the distinct molecular structures, enhanced photoluminescence quantum yields (PLQY), stable thermal behaviors, excellent film-forming properties and good repeatability, starburst conjugated materials comprised of a central core with multiple conjugated arms15 have received considerable attention in the field of optoelectronic devices including OLEDs16–29 and organic lasers.30–36 They appear to be the most promising alternatives to conventional small molecules and polymers employed in optoelectronics. Based on our previous studies, we have developed several kinds of starburst molecules with prominent performances. For instance, monodisperse six-armed triazatruxenes22,23,37 and 2,3,7,8,12,13-hexaaryltruxenes are synthesized,35,38 which stand out as a key class of promising semiconductors in optoelectronics, demonstrating relatively stable thermal characteristics, high environmental stability, high solid-state PL quantum efficiencies, and low lasing thresholds.35–39 Nevertheless, despite the progresses made, the potential for the resulting conjugated starbursts as gain media has not been fully explored.
Pyrene derivatives have gained much interest due to their high photoluminescence efficiency,40 good charge carrier mobility and great hole-injection ability.41 Starburst molecules with pyrene units have been recently explored for organic electronic applications with promising luminescence properties.42–46 Despite all this, pyrene with characteristics like most of the p-type molecules is prone to possess lower electron affinity,1 reducing the current density and further hindering the potential application for electrically pumped organic lasers.5 It is well-known that the enhancement of the carrier injection and transport is crucial to balance the carrier injection. In order to accomplish this aim, the introduction of electron-withdrawing moieties into the architectures is an effective approach. Among the electron-withdrawing groups, the cyano-group is extensively employed because of its high electron affinity and excellent luminescence efficiency,47–49 which is highly desirable for OLED applications. For instance, several alternated polymers with cyanofluorene moieties were synthesized by Taranekar,50 demonstrating green to blue emission with relatively low turn-on voltages. Chen et al. developed a family of highly efficient blue-emitting molecules, which are composed of oligofluorene and electron-withdrawing cyanophenyl components.48,51 As revealed, the resulting novel molecules exhibited enhanced device performance due to the high electron affinity of the cyano-groups and thus improved electron injection and transport properties. We surmise that the integration of electron-withdrawing cyano-groups onto organic gain media would be interesting and may play a positive role in improving the electrical properties of the resultant emitters.
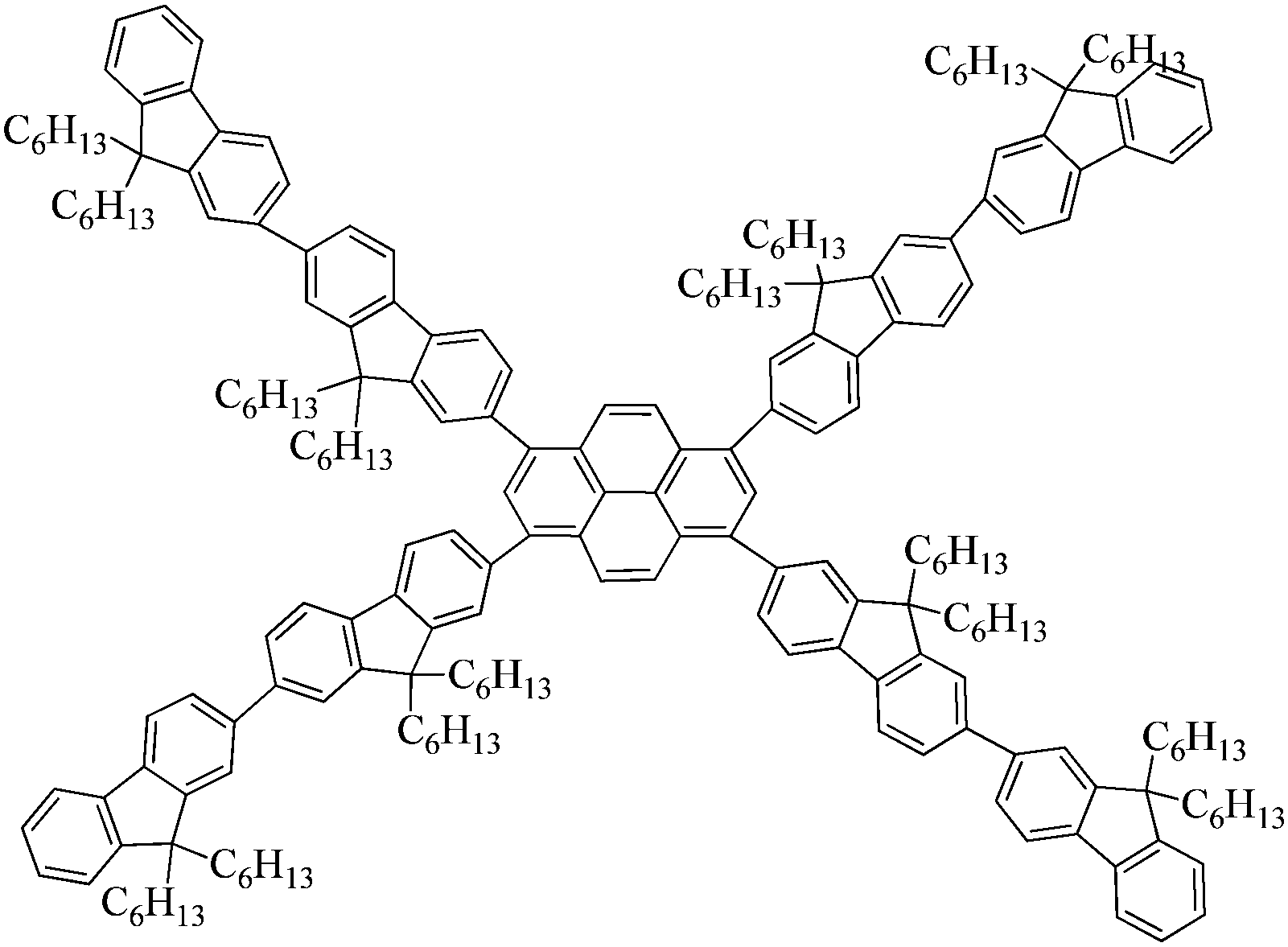
In this contribution, we designed and synthesized a novel family of pyrene-centered starbursts with four radial oligofluorene arms end-capped by cyanophenyl moieties (T1, T2 and T3), and explored the resulting starburst targets as efficient emitters for OLEDs and planar waveguide devices. Oligofluorenes were selected as the arms for their high quantum yields, good optical stability, and favorable film-forming ability.19–23,52,53 Cyanophenyl moieties were introduced as the end-cappers onto the pyrene-centered starburst molecular architectures because of their strong electron-withdrawing properties and good electron injection and transport properties. The relationships between the molecular structures and their photophysical, thermal, morphological, and electrochemical properties, as well as their electroluminescence (EL) characteristics, were systematically investigated. OLEDs based on T1 showed the best performance with a maximum luminous efficiency (LE) of 3.52 cd A−1 and a maximum brightness of 9194 cd m−2. Evidently, the electron affinity and mobility of these resulting materials could be greatly improved by introducing cyanophenyl moieties. Moreover, low thresholds of 2 μJ cm−2 (36 nJ pulse−1), 1.78 μJ cm−2 (32 nJ pulse−1), and 2.11 μJ cm−2 (38 nJ pulse−1) were recorded for T1–T343 and their corresponding ASE stabilities and lasing properties were investigated under an ambient atmosphere. The results confirm that incorporation of cyanophenyl moieties as end-cappers can enhance the electrical properties of pyrene-centered starbursts without substantially sacrificing their great optical gain properties, which results in this series of materials holding great promise for electrically pumped organic lasers.
2. Results and discussion
2.1 Synthesis and characterization
Synthetic routes of T1–T3 and the oligofluorene arms are depicted in Scheme 1. Oligofluorene boronic pinacol esters of different chain lengths (n = 1, 2, and 3) were prepared according to the procedures reported in our previous work.38 Monodisperse starburst molecules were synthesized in a rapid and facile way in minutes using a well-developed microwave-assisted multiple Suzuki coupling methodology.21,38 Detailed synthetic procedures for all intermediates and the final targets T1–T3 are provided in the ESI.† The well-defined structures and chemical purities have been adequately confirmed using 1H and 13C NMR spectroscopy, MALDI-TOF mass spectrometry and elemental analysis, which matched very well with the expected structures. The MALDI-TOF spectra, 1H NMR and 13C NMR spectra of the final targets T1–T3 are given in the ESI† (Fig. S1–S9).
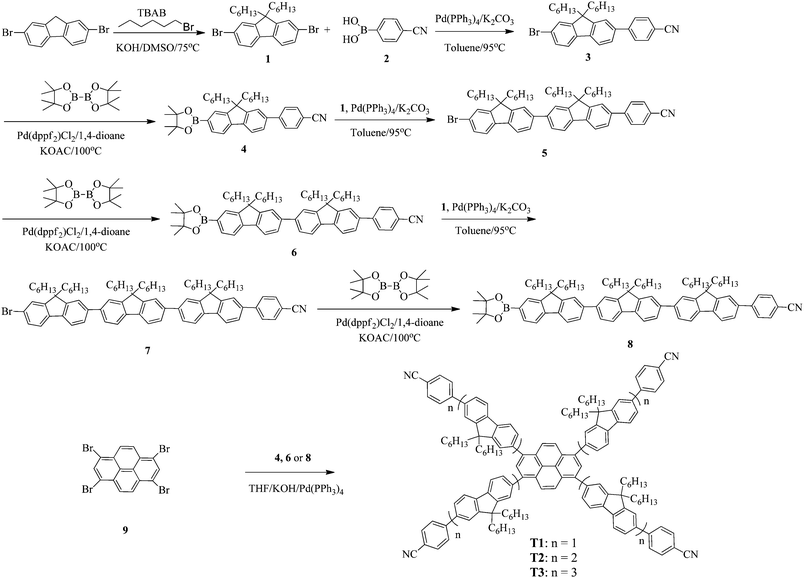 |
| | Scheme 1 The synthetic routes toward T1–T3. | |
2.2 Photophysical properties
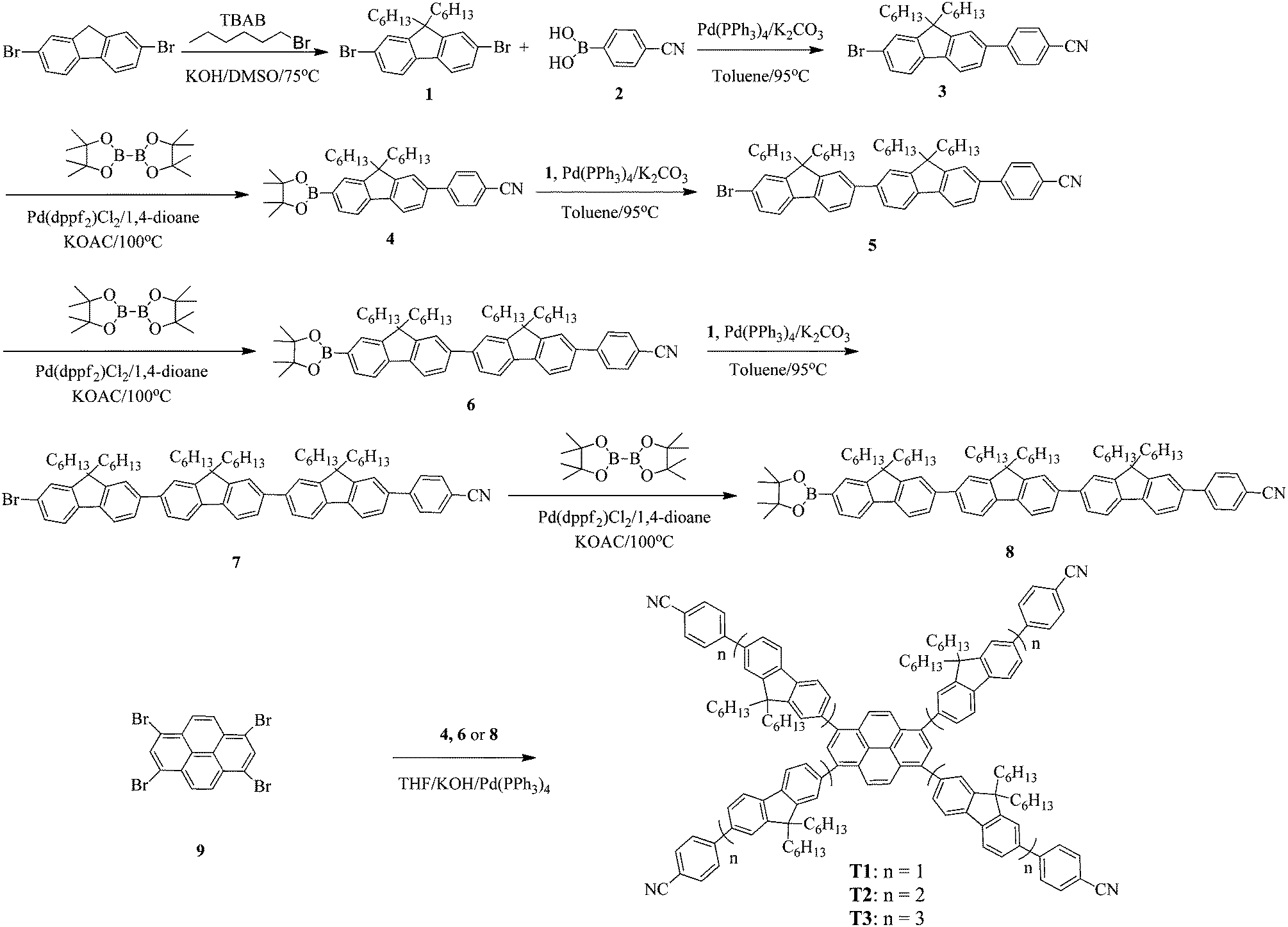
The absorption and photoluminescence (PL) characteristics of T1–T3 were demonstrated both in dilute solutions and thin films (Fig. 1). T1 showed two distinctive absorption peaks located at 405 and 348 nm, respectively, while T2 had one main absorption peak at 374 nm with a shoulder around 412 nm, and T3 showed only one main absorption peak at 374 nm. The main absorption peak in the dilute solution for T1–T3 was gradually red-shifted with the increase of the arm length. However, in the long wavelength region, the relative absorption intensity decreased when the arm length increased. More interestingly, the onset absorption edges of T1–T3 were almost the same, which were located at around 460 nm, indicating that the effective conjugation saturated quickly with the increase of the arm length.39 The absorption spectra of T1 in the films exhibited nearly identical peaks compared to those recorded from dilute solution, except for the relative intensity. Nevertheless, the absorption spectra of T2 and T3 were a little altered, demonstrating one main peak at 365 nm (T2) and 373 nm (T3), respectively, with a shoulder around 420 nm for T2. In light of the PL spectra in dilute solutions (Fig. 1a), the emission peak was almost consistent. In the film state, the PL spectra of T2 and T3 peaked at 478 nm and 475 nm, respectively, which were slightly blue-shifted compared with that of T1 (484 nm). According to the result, the effective conjugation could quickly saturate by increasing the arm length. Furthermore, approximately similar photoluminescence quantum yields (PLQYs) of 0.41 for T1, 0.48 for T2 and 0.44 for T3 were recorded, respectively, which were measured on the quartz plates using an integrating sphere, indicating that the PLQYs were almost independent of the lengths of oligofluorene arms for this set of materials. The key optical data are summarized in Table 1.
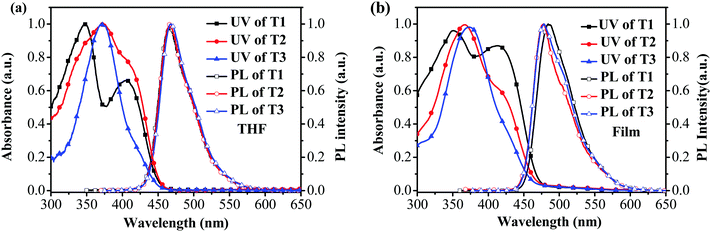 |
| | Fig. 1 UV-vis absorption and PL spectra of T1–T3 in (a) THF at 1 × 10−6 M; and (b) thin films. | |
Table 1 Thermal and photophysical data of T1–T3
| |
T
d (°C) |
λ
abs
(nm) |
λ
abs
(nm) |
λ
em,max
(nm) |
λ
em,max
(nm) |
Φ
PL
(%) |
|
Measured in toluene.
Measured in films.
|
|
T1
|
424 |
348, 405 |
350, 413 |
465 |
484 |
41 |
|
T2
|
439 |
374, 412 |
365, 420 |
466 |
478 |
48 |
|
T3
|
436 |
374 |
373 |
468 |
475 |
44 |
2.3 Thermal and morphological properties
The thermal properties of the final starburst targets were evaluated by means of thermo-gravimetric analysis (TGA) and differential scanning calorimetry (DSC). The samples were heated to 600 °C at a heating rate of 10 °C min−1 under a dry nitrogen atmosphere. As shown in Fig. S10a (ESI†), T1–T3 showed high decomposition temperatures (Td, which correspond to 5% weight loss) ranging between 424 and 439 °C, which was around 50 °C higher than that of molecules without cyanophenyl end-cappers,39 indicating the good thermal stability of these compounds. DSC measurements showed that these materials exhibited amorphous morphologies with no obvious crystallization or melting processes during the repeated scan cycles. The glass transition temperatures (Tg) for T1 were observed at 80 °C, whereas no evident Tg for T2 and T3 was found within the heating scans from room temperature to 220 °C (Fig. S10a, ESI† inset), mainly resulting from enhanced steric hindrance with increasing arm length. Their wide-angle X-ray diffraction (WAXD) patterns (Fig. S10b, ESI†) demonstrated weak and broad halos, further confirming the amorphous morphologies of these materials.
2.4 Electroluminescence properties
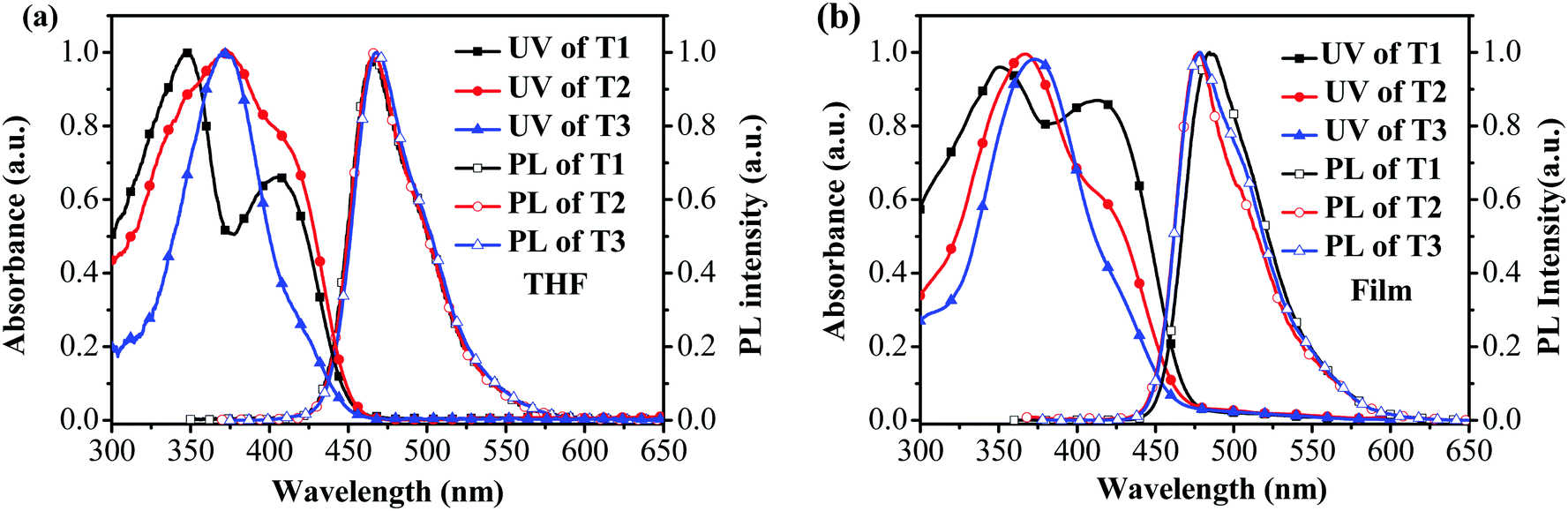
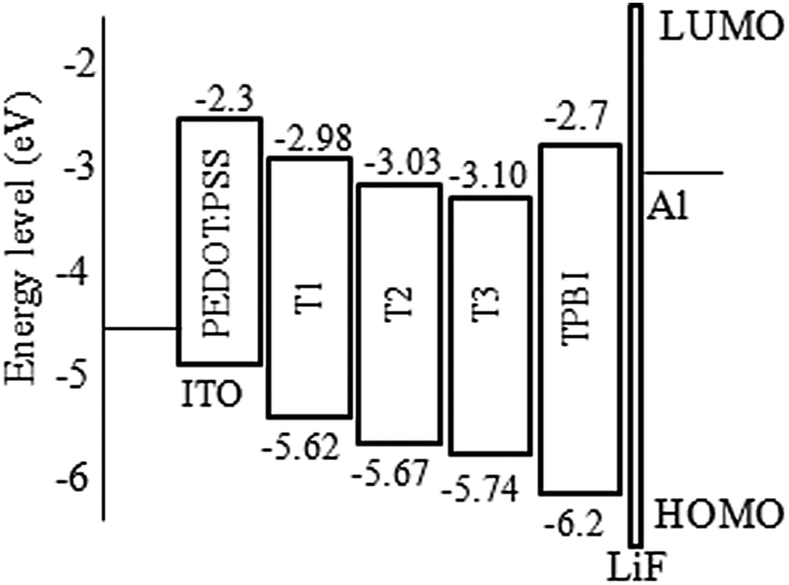
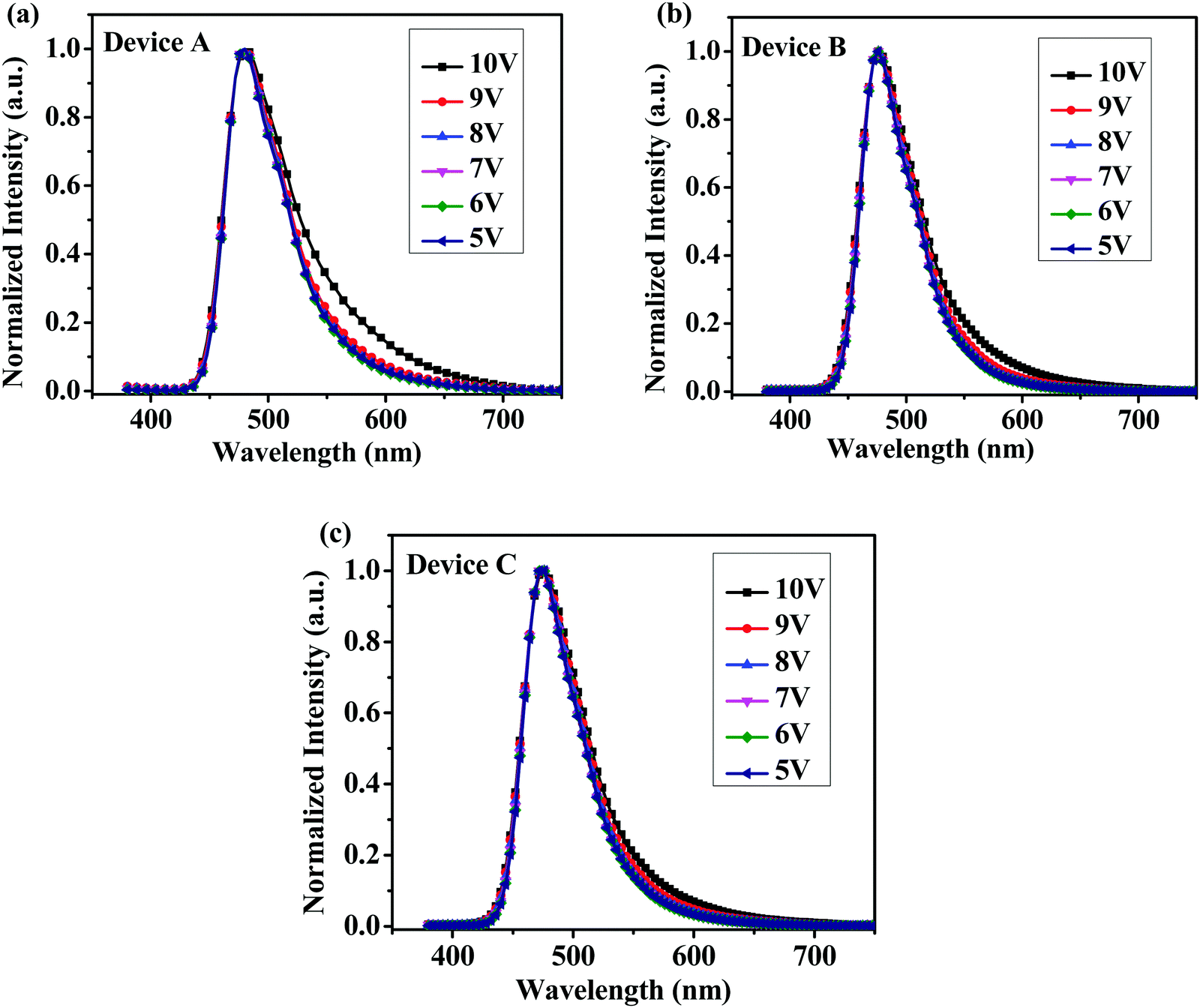
From cyclic voltammetry (CV) studies (Fig. S11, ESI†), the corresponding energy level diagram of T1–T3 is depicted in Fig. 2. Single-emissive layer devices were fabricated with the configuration of ITO/PEDOT: PSS(40 nm)/T1–T3 (80 nm)/TPBI (40 nm)/LiF (0.5 nm)/Al (100 nm) (emitter: T1, device A; T2, device B; T3, device C), in which the emission layer was spin-coated from chloroform solution on top of PEDOT:PSS. PEDOT:PSS and LiF were used as the hole- and electron-injecting layers, respectively, and TPBI was inserted between the EML and LiF as an electron-transporting layer. Fig. 3 shows the EL spectra of device A (a), device B (b), and device C (c) at various driving voltages. The central peaks were recorded at 480 nm, 476 nm and 476 nm, respectively, for the EL devices based on T1, T2, and T3. With increasing driving voltages from 5 to 10 V, the EL spectra of all these devices remained nearly unchanged, suggesting remarkable voltage-independent and stable EL spectra. Obviously, these devices had striking blue EL color stability at various driving voltages. The excellent color stability can be attributed to the non-coplanar conformation, which may suppress close packing of the molecules in the solid state and limit crystallization and transition-induced deterioration, and thus help to improve the morphological stability during device operation.
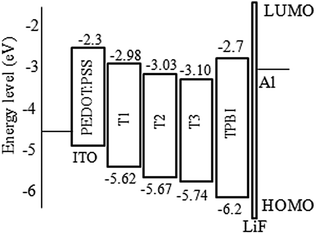 |
| | Fig. 2 Energy level diagrams for T1–T3. | |
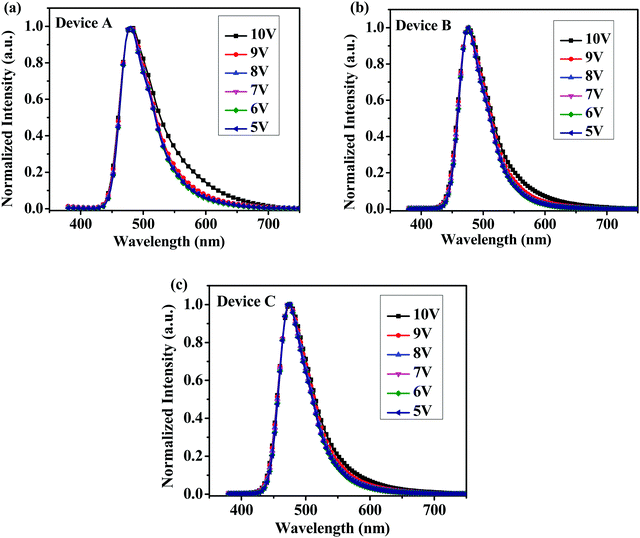 |
| | Fig. 3 Normalized EL spectra of (a) device A, (b) device B and (c) device C with increasing driving voltages. | |
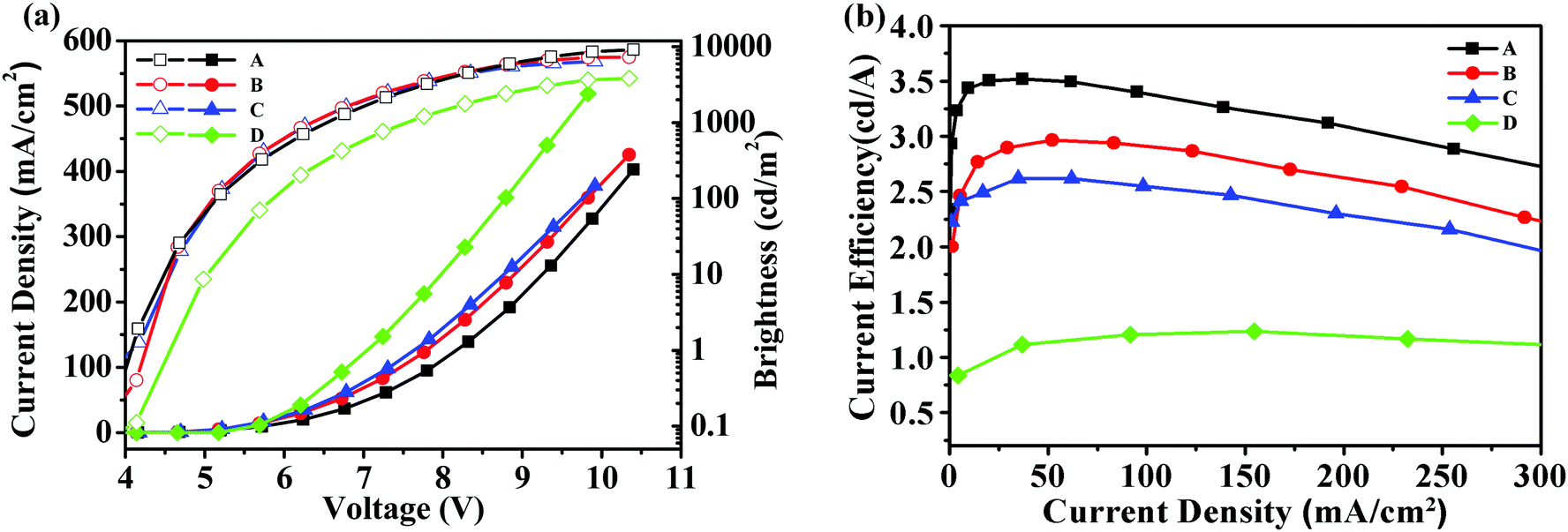
Fig. 4 showed the brightness-voltage characteristics of the devices and the corresponding data are summarized in Table 2. All the devices displayed a low turn-on voltage of around 4 V, which may result from the high HOMO levels of these starbursts that matched well with the HOMO level of the adjacent PEDOT:PSS/ITO anode, facilitating the hole injection of the device. The best EL performance was achieved for device A with a maximum brightness of 9194 cd m−2 and a maximum current efficiency of 3.52 cd A−1. For device B, the maximum brightness was 7253 cd m−2, and the maximum current efficiency reached 2.97 cd A−1. Device C had a maximum brightness of 6401 cd m−2, and a maximum current efficiency of 2.62 cd A−1.
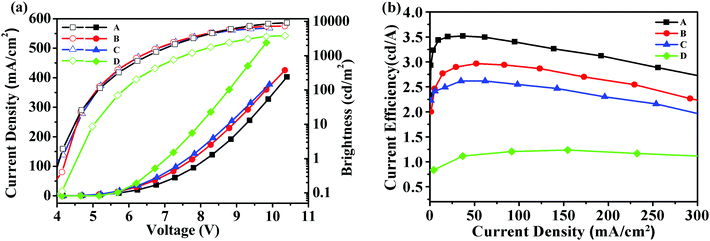 |
| | Fig. 4 (a) Current density–voltage–brightness (J–V–L) characteristics for devices A–D; (b) current efficiency–current density characteristics for devices A–D. | |
Table 2 EL characteristics of T1–T3 OLED devices
| Device |
V
on (V) |
L
max (cd m−2) |
LEmax (cd A−1) |
CIE (x, y) at 9 V |
λ
EL, nm (film) |
λ
PL, nm (film) |
|
V
on: turn-on voltage; Lmax: maximum luminance; LEmax: maximum current efficiency; CIE (x, y): Commission International de 1 Eclairage coordinates, measured at 9 V. |
| A |
4.05 |
9194 |
3.52 |
(0.18, 0.35) |
480 |
484 |
| B |
4.27 |
7253 |
2.97 |
(0.15, 0.29) |
476 |
478 |
| C |
4.07 |
6401 |
2.62 |
(0.16, 0.28) |
476 |
475 |
According to the results, device A exhibited a better EL performance, which could be elucidated as follows. (1) In Fig. 2, the LUMO level of T1 (−2.98 eV) is 0.05 eV and 0.12 eV lower than that of T2 and T3, respectively. Thus, device A had a lower electron injection barrier between TPBI (LUMO: −2.7 eV) and the EML than device B and device C. Therefore, a more balanced charge flux could be anticipated in device A. (2) In terms of hole injection, the hole injection barriers in devices A, B and C were 0.46 eV, 0.50 eV and 0.59 eV, respectively, in which device A was the lowest. (3) T1 as the active layer of device A had the best film-forming ability in the solid state among the three compounds. (4) The shorter oligofluorene arm length for T1 may also contribute to enable more efficient exciton migration and energy transfer between the pyrene center and the cyanophenyl end-cappers within the intramolecular conjugated system.
To understand the impact of cyanophenyl end-cappers, a pyrene-centered starburst oligofluorene analogue without cyanophenyl end-cappers (M2, seen in Scheme 2, its synthetic route and related characterization are reported in our previous studies39) was studied as the reference compound. With the same configuration as device B, a control device D using M2 as the EML was fabricated. For device D, the maximum brightness was 3697 cd m−2 and the maximum current efficiency approached 1.67 cd A−1, which were much lower than that of device B. The results indicate that introducing a cyanophenyl group into the oligomers can significantly improve the device performance without altering the EL spectra substantially. Obviously, the superior brightness and current efficiency of device B over device D should be ascribed to the enhanced electron injection and transport in the EML of T2, due to the introduction of the electron-withdrawing cyanophenyl groups.
 |
| | Scheme 2 Structure of M2. | |
2.5 Amplified spontaneous emission (ASE) stability and lasing properties
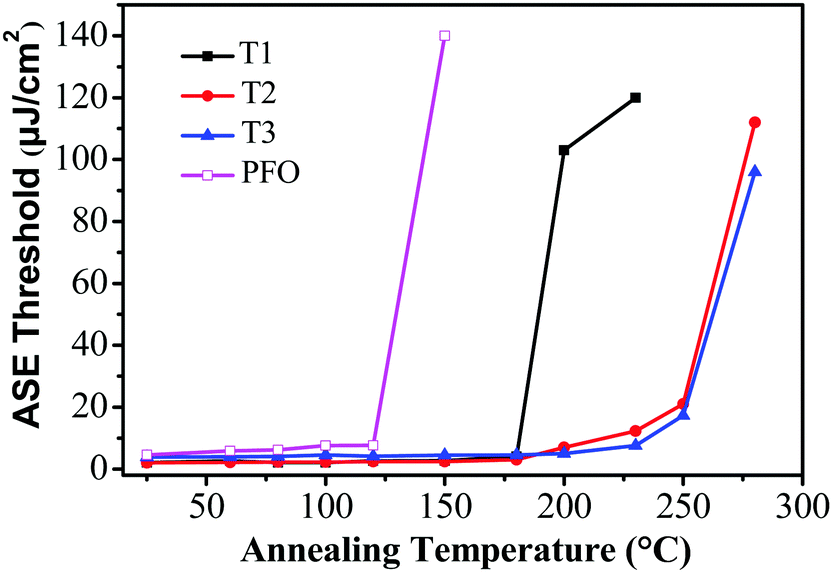
For potential application as gain media for electrically pumped organic semiconductor lasers, high stability is one of the most important prerequisites. To evaluate the photostability of the thin-film samples, the ASE threshold values of T1–T3 were recorded as a function of annealing temperature, which were kept in air for 10 min at each annealing temperature during the test. Remarkably, the ASE thresholds remained almost the same from room temperature up to 180 °C for T1 and 200 °C for T2 and T3 (Fig. 5). The ASE can still be detectable even when the annealing temperature was increased to 230 °C for T1 and 280 °C for T2 and T3. Fig. 5 illustrates that the ASE threshold of T1 remained almost the same from room temperature up to 180 °C and subsequently increased by 51-fold (from 2 μJ cm−2 to 102 μJ cm−2) upon heating at 200 °C for 10 min. However, the ASE was not detectable upon annealing above 230 °C, implying a significant change in the morphologies and structures during the process. In contrast, the ASE spectra of T2 and T3 remained almost unchanged until the annealing temperature was increased to 230 °C, suggesting that no drastic changes occurred in the molecular structures or electronic properties over this temperature range. Meanwhile, the ASE threshold increased by 11-fold (from 1.78 μJ cm−2 to 21 μJ cm−2) for T2 and 8-fold (from 2.11 μJ cm−2 to 17.3 μJ cm−2) for T3 at 250 °C. By comparison, for the linear polyfluorene anologue (PFO) thin films, the ASE threshold increased by 17-fold (from 8 μJ cm−2 to 139 μJ cm−2) upon heating at 150 °C for 10 min and the ASE was not detectable upon annealing above 150 °C. For the starburst analogue M2 film sample, the ASE threshold increased by 10-fold when heating up to 170 °C and the ASE was not detectable upon annealing above 170 °C as revealed in our previous studies.36 The results implied that both the PFO and M2 film samples were liable to experience some drastic morphological changes that caused a large optical loss during thermal annealing.36 The excellent morphological and optical stability of T2 and T3 under the harsh annealing conditions is thus quite impressive. This is believed to be crucially important for electrically-pumped lasers in the future, which may undergo a large amount of heat generated by the nonradiative losses resulting from many routes under high current density.
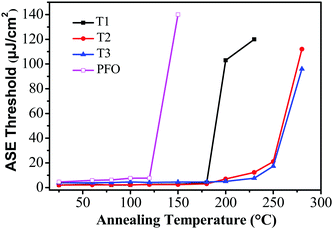 |
| | Fig. 5 Evolution of the ASE thresholds for T1 (solid squares), T2 (solid circles), T3 (solid triangles) and PFO (open squares) slab waveguides as a function of annealing temperatures. | |
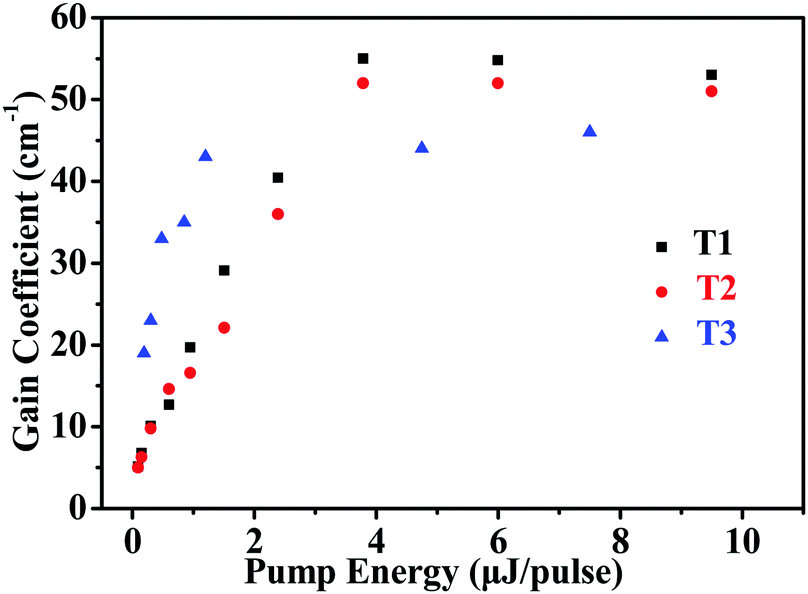
The net gains for T1–T3 was determined using the variable stripe length (VSL) technique.54,55Fig. 6 shows the net gain coefficients as a function of pump energy for T1–T3. According to the results, the net gain increased approximately linearly up to a saturation value. The corresponding maximum net gain coefficients are g = 55 cm−1 (pump energy E = 3.8 μJ pulse−1), g = 52 cm−1 (pump energy E = 6.0 μJ pulse−1) and g = 46 cm−1 (pump energy E = 7.5 μJ pulse−1) for T1–T3, respectively.
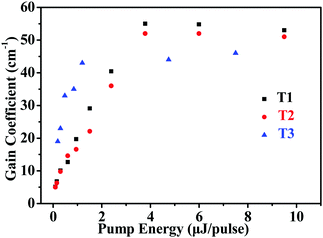 |
| | Fig. 6 Net gain coefficients as a function of pump energy for T1–T3. | |
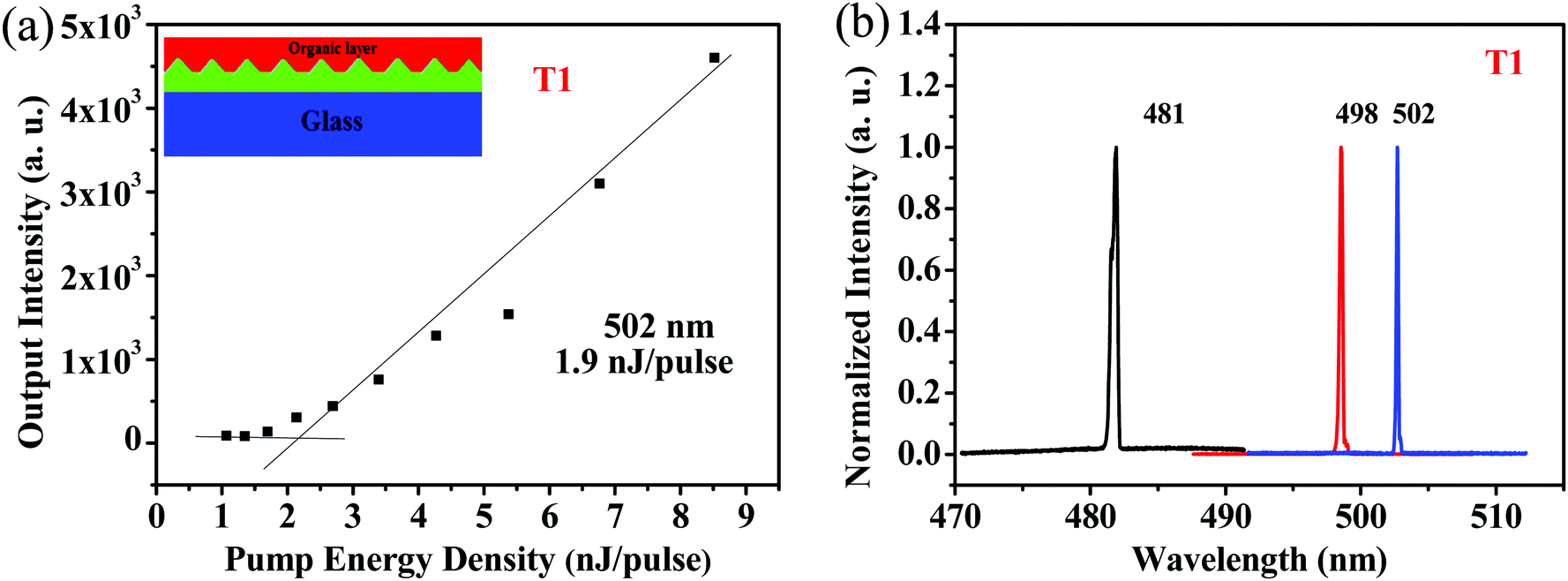
Because of its good film-forming properties and superior ASE performance, T1 was chosen to further explore lasing properties, and one-dimensional distributed-feedback (DFB) gratings were fabricated via nanoimprint lithography (NIL). The 2nd order gratings (50% fill factor) consisted of arrays of lines with a periodicity of 280 nm, 290 nm, and 300 nm, respectively. T1 was spin-coated on the DFB gratings to form laser devices, which were optically pumped at a wavelength of 375 nm with Q-switched at room temperature. The lasing output intensity from the organic lasers detected using a spectrometer upon various pump energies was recorded to determine their lasing thresholds. As shown in Fig. 7a, with increasing excitation fluence, a change in the slope of the output intensity was observed for the device, which was denoted as the threshold of the DFB lasers. The minimum lasing threshold measured for the T1 (180 nm) laser was 158.3 kW cm−2 (1.9 nJ pulse−1, 791 nJ cm−2) at 502 nm with a grating period of 300 nm. Above the lasing thresholds, the surface-emission spectra based on the DFB resonators for the device narrowed into one peak with a FWHM of around 0.3 nm. Following the principle of tuning the lasing peaks by means of regulating the periods of DFB gratings, wavelength-tunable DFB lasers can be achieved. As shown in Fig. 7b, by simply modulating various grating periods such as 280 nm, 290 nm and 300 nm, lasing spectra for T1 were obtained with a spectral window of 21 nm (from 481 nm to 502 nm).
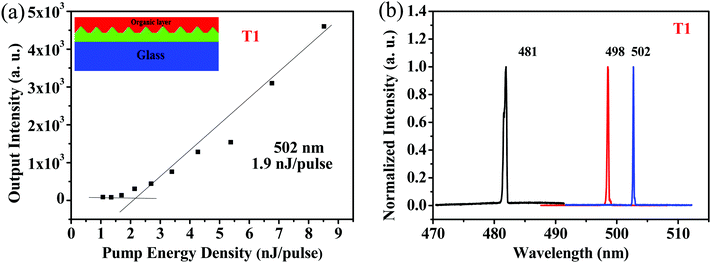 |
| | Fig. 7 (a) Output intensity versus the pump intensity for T1. Inset shows the schematic illustration of the DFB laser device. (b) Lasing spectra based on various DFB resonators for T1 films at different periods: 180 nm with Λ = 280 nm for λ = 481 nm; 180 nm with Λ = 290 nm for λ = 498 nm; 180 nm with Λ = 300 nm for λ = 502 nm. | |
3. Conclusion
In summary, we have developed a series of starburst oligofluorenes (T1–T3) based on a rigid planar pyrene core with four short oligofluorene arms and cyanophenyl end groups. All of the starbursts showed excellent thermal stability, pronounced PL efficiency and good solution processability. By means of solution processing, all the single-emissive-layer devices based on these materials exhibited efficient EL performances. Among these, device A based on T1 achieved a maximum current efficiency of 3.52 cd A−1 and a maximum brightness (Lmax) of 9194 cd m−2 because of its improved electron affinity. The lower ASE threshold of 2 μJ cm−2 (36 nJ pulse−1) and the largest net gain coefficient (g = 55 cm−1) was also recorded for T1 due to its excellent optical performance. A tunable spectral window of 21 nm for T1 (from 481 nm to 502 nm) was achieved by adjusting the grating periods. In addition, detailed investigation on the gain properties of T1 in the thin films indicated its great optical gain properties. 1D DFB lasing (50% fill factor, pumped source at 375 nm) demonstrated a lasing threshold of 158.3 kW cm−2 (1.9 nJ pulse−1, 791 nJ cm−2) at 502 nm with a grating period of 300 nm. The results suggest that well-designed pyrene-centered starbursts with oligofluorene arms end-capped by cyanophenyl moieties are prominent materials for the application of optically pumped organic lasers and will open up bright prospects for electrically pumped organic lasers.
4. Experimental section
General methods
Commercially available chemicals were used as received. Microwave-assisted reactions were performed using a CEM discovery single-mode focused microwave system. All reactions were monitored using thin layer chromatography (TLC) with silica gel 60 F254 (Merck, 0.2 mm). Column chromatography was carried out on silica gel (300–400 mesh). 1H NMR and 13C NMR spectra were recorded on a Bruker 400 plus at 295 K. The MALDI-TOF mass spectroscopy measurements were carried out using a Bruker mass spectrometer. Differential scanning calorimetry (DSC) and thermo-gravimetric analysis (TGA) were carried out on Shimadzu DSC-60A and DTG-60H equipment, respectively. UV-vis absorption spectra were recorded on a Shimadzu UV-3600 spectrophotometer. Fluorescence spectra were measured using a Shimadzu RF-5301PC spectrofluorimeter with a xenon lamp as the light source.
OLED fabrication and measurements
The hole-injection material poly(3,4-ethylenedioxy-thiophene): polystyrenesulfone acid (PEDOT:PSS) and the electron-transporting material 1,3,5-tris(1-phenyl-1H-benzimidazol-2-yl)benzene (TPBI) were commercially available. Commercial ITO-coated glass with a sheet resistance of 10 Ω square−1 was used as the starting substrate. Before device fabrication, the ITO/glass substrates were pre-cleaned carefully and treated with oxygen plasma for 4 min. PEDOT:PSS was spin-coated to smooth the ITO surface and promote hole injection, and then the emissive layer was spin-coated from the chloroform solution. The samples were annealed at 80 °C for 30 min to remove the residual solvent. The thickness of the emissive layer (EML) was about 80 nm. Finally, an electron-transporting layer of TPBI (40 nm) and a cathode composed of lithium fluoride (LiF, 0.5 nm) and aluminum (Al, 100 nm) were sequentially deposited onto the substrate via vacuum deposition at 10−6 Pa. The luminance–current–voltage characteristics of the devices were measured using a combination of a Keithley source-meter (model 2602) and a calibrated luminance meter. Electroluminescence (EL) spectra and Commission International de l’Eclairage (CIE) coordinates were obtained using a spectra-scan PR655 spectrophotometer. The determination of film thicknesses was conducted on a Bruker DektakXT Stylus Profiling System. All measurements were carried out at room temperature under ambient conditions.
Organic laser fabrication and measurements
For lasing measurements, the samples (150–200 nm thickness), prepared by spin-coating the solutions in chloroform (25 mg mL−1) onto pre-cleaned Spectrosil substrates, were optically pumped with a Q-switched, neodymium ion doped yttrium aluminium garnate [Nd3+:YAG] laser pumped, type-II β-BaB2O4[BBO], optical parametric oscillator that delivered 5 ns pulses at a repetition rate of 10 Hz. Calibrated neutral density filters were inserted into the beam path to adjust the pulse energy incident on the sample. An adjustable slit and a cylindrical lens were utilized to create a narrow excitation strip (4.1 mm × 440 μm) vertically placed at the edge of the sample film. The edge emission from the samples was collected using a fiber-coupled grating spectrometer (Andor Co.) equipped with a charge coupled device (CCD) detector (Newton Co.). One-dimensional (1D) distributed-feedback (DFB) gratings (50% fill factor, 30 nm modulation depth) were fabricated by nanoimprint lithography (NIL) with periods of 280 nm, 290 nm and 300 nm. The gain media were spin-coated on DFB gratings to form DFB lasers, which were optically pumped at a wavelength of 375 nm at room temperature. The lasing output intensity upon various pump energies from the organic lasers was detected using a spectrometer.
Acknowledgements
We acknowledge financial support of the National Key Basic Research Program of China (973 Program, 2014CB648300), the National Natural Science Foundation of China (21422402, 20904024, 61136003, 21674050), the Natural Science Foundation of Jiangsu Province (BK20140060, BK20130037, BK20140865, BM2012010), Program for Jiangsu Specially-Appointed Professors (RK030STP15001), Program for New Century Excellent Talents in University (NCET-13-0872), Specialized Research Fund for the Doctoral Program of Higher Education (20133223110008, and 20113223110005), the Synergetic Innovation Center for Organic Electronics and Information Displays, the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD), the NUPT “1311 Project”, the Six Talent Plan (2012XCL035), the 333 Project (BRA2015374) and the Qing Lan Project of Jiangsu Province.
References
- G. Horowitz, Adv. Mater., 1998, 10(5), 365–377 CrossRef CAS.
- A. Kraft, A. C. Grimsdale and A. B. Holmes, Angew. Chem., Int. Ed., 1998, 37(4), 402–428 CrossRef.
- G. Gustafsson, Y. Cao, G. M. Treacy, F. Klavetter, N. Colaneri and A. J. Heeger, Nature, 1992, 357(6378), 477–479 CrossRef CAS.
- S. R. Forrest, Nature, 2004, 428(6986), 911–918 CrossRef CAS PubMed.
- I. D. W. Samuel and G. A. Turnbull, Chem. Rev., 2007, 107(4), 1272–1295 CrossRef CAS PubMed.
- J. R. Lawrence, G. A. Turnbull, I. D. W. Samuel, G. J. Richards and P. L. Burn, Opt. Lett., 2004, 29(8), 869–871 CrossRef CAS PubMed.
- N. Tessler, Adv. Mater., 1999, 11(5), 363–370 CrossRef CAS.
- L. H. Xie, C. R. Yin, W. Y. Lai, Q. L. Fan and W. Huang, Prog. Polym. Sci., 2012, 37(9), 1192–1264 CrossRef CAS.
- I. D. W. Samuel, E. B. Namdas and G. A. Turnbull, Nat. Photonics, 2009, 3(10), 546–549 CrossRef CAS.
- D. Amarasinghe, A. Ruseckas, A. E. Vasdekis, G. A. Turnbull and I. D. W. Samuel, Adv. Mater., 2009, 21(1), 107–110 CrossRef CAS.
- B. K. Yap, R. Xia, M. Campoy-Quiles, P. N. Stavrinou and D. D. C. Bradley, Nat. Mater., 2008, 7(5), 376–380 CrossRef CAS PubMed.
- E. B. Namdas, M. Tong, P. Ledochowitsch, S. R. Mednick, J. D. Yuen, D. Moses and A. J. Heeger, Adv. Mater., 2009, 21(7), 799–802 CrossRef CAS.
- X. F. Jiang, Y. F. Xiao, C. L. Zou, L. He, C. H. Dong, B. B. Li, Y. Li, F. W. Sun, L. Yang and Q. Gong, Adv. Mater., 2012, 24(35), OP260–OP264 CrossRef CAS PubMed.
- M. M. Mroz, G. Sforazzini, Y. Zhong, K. S. Wong, H. L. Anderson, G. Lanzani and J. Cabanillas-Gonzalez, Adv. Mater., 2013, 25(31), 4347–4351 CrossRef CAS PubMed.
- A. L. Kanibolotsky, I. F. Perepichka and P. J. Skabara, Chem. Soc. Rev., 2010, 39(7), 2695–2728 RSC.
- W. D. Xu, W. Y. Lai, Q. Hu, X. Y. Teng, X. W. Zhang and W. Huang, Polym. Chem., 2014, 5(8), 2942–2950 RSC.
- Q. D. Liu, J. P. Lu, J. F. Ding, M. Day, Y. Tao, P. Barrios, J. Stupak, K. Chan, J. J. Li and Y. Chi, Adv. Funct. Mater., 2007, 17(6), 1028–1036 CrossRef CAS.
- H. Huang, Q. A. Fu, S. Q. Zhuang, Y. K. Liu, L. Wang, J. S. Chen, D. G. Ma and C. L. Yang, J. Phys. Chem. C, 2011, 115(11), 4872–4878 CAS.
- B. S. Li, J. Li, Y. Q. Fu and Z. S. Bo, J. Am. Chem. Soc., 2004, 126(11), 3430–3431 CrossRef CAS PubMed.
- A. L. Kanibolotsky, R. Berridge, P. J. Skabara, I. F. Perepichka, D. D. C. Bradley and M. Koeberg, J. Am. Chem. Soc., 2004, 126(42), 13695–13702 CrossRef CAS PubMed.
- W. Y. Lai, Q. Q. Chen, Q. Y. He, Q. L. Fan and W. Huang, Chem. Commun., 2006,(18), 1959–1961 RSC.
- W. Y. Lai, Q. Y. He, R. Zhu, Q. Q. Chen and W. Huang, Adv. Funct. Mater., 2008, 18(2), 265–276 CrossRef CAS.
- W. Y. Lai, R. Zhu, Q. L. Fan, L. T. Hou, Y. Cao and W. Huang, Macromolecules, 2006, 39(11), 3707–3709 CrossRef CAS.
- L. Chen, P. Li, Y. Cheng, Z. Xie, L. Wang, X. Jing and F. Wang, Adv. Mater., 2011, 23(26), 2986–2990 CrossRef CAS PubMed.
- Y. Zou, J. Zou, T. Ye, H. Li, C. Yang, H. Wu, D. Ma, J. Qin and Y. Cao, Adv. Funct. Mater., 2013, 23(14), 1781–1788 CrossRef CAS.
- C. Liu, Y. H. Li, Y. F. Li, C. L. Yang, H. B. Wu, J. G. Qin and Y. Cao, Chem. Mater., 2013, 25(16), 3320–3327 CrossRef CAS.
- C. Liu, Y. H. Li, Y. Y. Zhang, C. L. Yang, H. B. Wu, J. G. Qin and Y. Cao, Chem. – Eur. J., 2012, 18(22), 6928–6934 CrossRef CAS PubMed.
- H. Huang, Q. Fu, S. Q. Zhuang, Y. K. Liu, L. Wang, J. S. Chen, D. G. Ma and C. L. Yang, J. Phys. Chem. C, 2011, 115(11), 4872–4878 CAS.
- X. H. Zhou, J. C. Yan and J. Pei, Org. Lett., 2003, 5(19), 3543–3546 CrossRef CAS PubMed.
- C. R. Belton, A. L. Kanibolotsky, J. Kirkpatrick, C. Orofino, S. E. T. Elmasly, P. N. Stavrinou, P. J. Skabara and D. D. C. Bradley, Adv. Funct. Mater., 2013, 23(22), 2792–2804 CrossRef CAS.
- B. Guilhabert, N. Laurand, J. Herrnsdorf, Y. Chen, A. R. Mackintosh, A. L. Kanibolotsky, E. Gu, P. J. Skabara, R. A. Pethrick and M. D. Dawson, J. Opt., 2010, 12(3), 035503 CrossRef.
- P. W. Wang, Y. J. Liu, C. Devadoss, P. Bharathi and J. S. Moore, Adv. Mater., 1996, 8(3), 237–241 CrossRef CAS.
- J. C. Ribierre, G. Tsiminis, S. Richardson, G. A. Turnbull, I. D. W. Samuel, H. S. Barcena and P. L. Burn, Appl. Phys. Lett., 2007, 91(8), 081108 CrossRef.
- G. Tsiminis, N. A. Montgomery, A. L. Kanibolotsky, A. Ruseckas, I. F. Perepichka, P. J. Skabara, G. A. Turnbull and I. D. W. Samuel, Semicond. Sci. Technol., 2012, 27(9), 094005 CrossRef.
- W. Y. Lai, R. Xia, Q. Y. He, P. A. Levermore, W. Huang and D. D. C. Bradley, Adv. Mater., 2009, 21(3), 355–360 CrossRef CAS.
- R. Xia, W. Y. Lai, P. A. Levermore, W. Huang and D. D. C. Bradley, Adv. Funct. Mater., 2009, 19(17), 2844–2850 CrossRef CAS.
- G. L. Feng, W. Y. Lai, S. J. Ji and W. Huang, Tetrahedron Lett., 2006, 47(39), 7089–7092 CrossRef CAS.
- W. Y. Lai, R. Xia, D. D. C. Bradley and W. Huang, Chem. – Eur. J., 2010, 16(28), 8471–8479 CrossRef CAS PubMed.
- F. Liu, W. Y. Lai, C. Tang, H. B. Wu, Q. Q. Chen, B. Peng, W. Wei, W. Huang and Y. Cao, Macromol. Rapid Commun., 2008, 29(8), 659–664 CrossRef CAS.
- P. C. Bevilacqua, R. Kierzek, K. A. Johnson and D. H. Turner, Science, 1992, 258(5086), 1355–1357 CAS.
- W. L. Jia, T. McCormick, Q. D. Liu, H. Fukutani, M. Motala, R. Y. Wang, Y. Tao and S. N. Wang, J. Mater. Chem., 2004, 14(22), 3344–3350 RSC.
- W. D. Xu, J. P. Yi, W. Y. Lai, L. Zhao, Q. Zhang, W. B. Hu, X. W. Zhang, Y. Jiang, L. Liu and W. Huang, Adv. Funct. Mater., 2015, 25(29), 4617–4625 CrossRef CAS.
- Q. Zhang, Y. Zhang, W. D. Xu, X. C. Li, J. G. Liu, X. R. Guo, R. D. Xia and W. Huang, Opt. Express, 2015, 23(11), A465–A470 CrossRef PubMed.
- M. Gingras, V. Placide, J. M. Raimundo, G. Bergamini, P. Ceroni and V. Balzani, Chem. – Eur. J., 2008, 14(33), 10357–10363 CrossRef CAS PubMed.
- Y. J. Pu, M. Higashidate, K. I. Nakayama and J. Kido, J. Mater. Chem., 2008, 18(35), 4183–4188 RSC.
- T. M. Figueira-Duarte, S. C. Simon, M. Wagner, S. I. Drtezhinin, K. A. Zachariasse and K. Müllen, Angew. Chem., Int. Ed., 2008, 47(52), 10175–10178 CrossRef CAS PubMed.
- S. Cheylan, H. J. Bolink, A. Fraleoni-Morgera, C. J. Puigdollers, C. Voz, I. Mencarelli, L. Setti, R. Alcubilla and G. Badenes, Org. Electron., 2007, 8(6), 641–647 CrossRef CAS.
- C. G. Zhen, Z. K. Chen, Q. D. Liu, Y. F. Dai, R. Y. C. Shin, S. Y. Chang and J. Kieffer, Adv. Mater., 2009, 21(23), 2425–2429 CrossRef CAS.
- A. Pucci, T. Biver, G. Ruggeri, L. I. Meza and Y. Pang, Polymer, 2005, 46(25), 11198–11205 CrossRef CAS.
- P. Taranekar, M. Abdulbaki, R. Krishnamoorti, S. Phanichphant, P. Waenkaew, D. Patton, T. Fulghum and R. Advincula, Macromolecules, 2006, 39(11), 3848–3854 CrossRef CAS.
- M. Zhu and C. Yang, Chem. Soc. Rev., 2013, 42(12), 4963–4976 RSC.
- J. Pei, J. L. Wang, X. Y. Cao, X. H. Zhou and W. B. Zhang, J. Am. Chem. Soc., 2003, 125(33), 9944–9945 CrossRef CAS PubMed.
- Q. D. Liu, J. Lu, J. Ding, M. Day, Y. Tao, P. Barrios, J. Stupak, K. Chan, J. Li and Y. Chi, Adv. Funct. Mater., 2007, 17(6), 1028–1036 CrossRef CAS.
- M. D. McGehee, R. Gupta, S. Veenstra, E. K. Miller, M. A. Diaz-Garcia and A. J. Heeger, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 73(17), 2453–2455 Search PubMed.
- K. L. Shaklee and J. E. Rowe, Appl. Opt., 1970, 9(3), 627–632 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Details of 1H NMR spectra, 13C NMR spectra and MADIL-TOF spectra; TGA thermograms, DSC traces, XRD patterns, CV curves for T1, T2 and T3. See DOI: 10.1039/c6qm00133e |
| ‡ These authors contributed equally to this work. |
|
| This journal is © the Partner Organisations 2017 |