DOI:
10.1039/C6QM00130K
(Research Article)
Mater. Chem. Front., 2017,
1, 499-506
Novel donor–acceptor type conjugated polymers based on quinoxalino[6,5-f]quinoxaline for photovoltaic applications†
Received
18th July 2016
, Accepted 17th August 2016
First published on 31st August 2016
Abstract
The field of polymer solar cells has undergone tremendous advancement in terms of power conversion efficiency in the past decade. However, there is still an urgent requirement to further enhance device performance for achieving large-scale commercialization. Here, we report the design and synthesis of three novel conjugated polymers alternatively copolymerized by a newly developed 2,3,8,9-tetrakis(3-(alkoxy)phenyl)-6,12-di(thiophen-2-yl)-2,3,8,9-tetrahydroquinoxalino[6,5-f]quinoxaline (DTNQx) acceptor unit and a conventional two-dimensional alkylthienyl-substituted benzodithiophene (BDT) unit. These polymers possess an identical main chain but different side chains. The structure–property relationship is systematically studied. All polymers exhibit an optical bandgap around 1.7 eV and a HOMO energy level around −5.16 eV. Compared with the quinoxaline unit, fused quinoxaline DTNQx leads to a downshifted HOMO energy level in the resulting polymers by 0.04 eV while keeping the bandgap unchanged. By changing the side chain length and by introducing branched side chains, polymer properties including absorption spectra and hole mobilities could be finely tuned. Under optimal conditions, polymer P2 shows the highest hole mobility of 2.7 × 10−4 cm2 V−1 s−1 and the best photovoltaic performance, with Jsc = 9.1 mA cm−2, Voc = 0.88 V, FF = 48%, and PCE = 3.8%.
Introduction
During the past decade, polymer solar cells (PSCs) have attracted extensive attention due to their light weight, low cost, and great potential for the fabrication of large-area flexible devices.1–10 Typically, PSCs adopt a bulk-heterojunction (BHJ) structure, where a nanoscale phase-separated blend of electron-donating conjugated polymers and electron-accepting fullerene derivatives (such as [6,6]-phenyl-C61-butyric acid methyl ester (PC61BM) or [6,6]-phenyl-C71-butyric acid methyl ester (PC71BM)) are used as the active layer. Significant improvements in power conversion efficiencies (PCEs) of PSCs have been achieved because of the invention of novel donor polymers, and the introduction of new interfacial materials and device structures. Recently, several groups reported high PCEs of 8–11% for single junction or tandem devices.11–21 However, PCEs need to be further improved prior to the success of OPV in real applications. One of the important methods to improve the PCEs is the development of new donor–acceptor (D–A) conjugated polymers with desired optoelectronic properties.22–27
Among many D–A conjugated polymers, quinoxaline-based copolymers have been successfully developed and applied in PSCs. The quinoxaline unit has an electron-deficient N-heterocycle, which can effectively tune the energy level and band gap of the resulting copolymers.28–34 In addition, as seen in most quinoxaline-based conjugated polymers, quinoxaline rings often link with two aromatic groups (typically benzene or thiophene) around the molecule core, which could not only red-shift the absorption spectra of the polymers but also provide abundant chemical modification sites to finely tune the solubility of the polymers. As a result, conjugated polymers with quinoxaline units as acceptor units often exhibit a narrow bandgap, a deep highest occupied molecular orbital (HOMO) energy level and good solubility. Several quinoxaline-based conjugated polymers have shown promising photovoltaic performances. For example, polymer TQ1 possesses a very low-lying HOMO energy level of −5.7 eV, a high open-circuit voltage (Voc) of 0.89 V, and a PCE of 6.0%.28 In another case, conjugated polymers based on quinoxaline and indacenodithiophen exhibited not only low-lying HOMO energy, but also high hole mobility up to 2.9 × 10−2 cm2 V−1 s−1.29 Recently, a fluorinated quinoxaline-based polymer PBDT-TFQ produced a prominent Jsc of 18.2 mA cm−2, which was among the highest values in PSCs.30
Recently, Huang and co-workers discovered that fusing some common acceptor units shoulder by shoulder can generate highly promising building blocks with large coplanar structures, and can thereby lead to state-of-the-art D–A conjugated polymers with superior photovoltaic properties than un-fused counterparts (Scheme 1).35–40 For example, the evolution from 2,1,3-benzothiadiazole (BT) to naphtho[1,2-c:5,6-c]bis(1,2,5-thiadiazole) (NT) brings a huge improvement of PCEs in solar cells.35 In the same vein, naphtho[1,2-c:5,6-c]bis(2-octyl-[1,2,3]triazole) (TZNT) shows better photovoltaic properties than benzo[d][1,2,3]triazole (BTA).41 The ascension of these performance comes mainly from two aspects: (1) fused molecules have a bigger conjugated plane, which enhances the interaction for efficient intermolecular packing in the resultant polymers; (2) the transformation of the molecule structure from axisymmetry to centrosymmetry well adjusts the polymer configuration, making molecules apt to a linear structure rather than a folded structure, which might be favorable for better molecular ordering.42–45 Although quinoxaline has been studied as a promising building block for photovoltaic applications for several years, no attempt has been made to modify it by the fusing method (shoulder by shoulder, as shown in Scheme 1). Therefore, the new building block (NQx) formed by fusing two quinoxaline units shoulder by shoulder will be highly useful for constructing state-of-the-art conjugated polymers for photovoltaic applications.
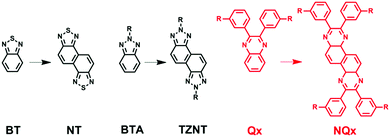 |
| | Scheme 1 Molecule structure from a single molecule to combined molecules. | |
In this manuscript, we report the design and synthesis of a new class of building block of 2,3,8,9-tetrakis(3-(alkoxy)phenyl)-6,12-di(thiophen-2-yl)-2,3,8,9-tetrahydroquinoxalino[6,5-f]quinoxaline (DTNQx), which contains a thiophene at each side of NQx for constructing well-performing D–A conjugated polymers. Three kinds of side chains were introduced onto DTNQx to optimize the solubility of the resulting polymers and BHJ morphology.43,47 Polymer P2 which contains ethylhexyl side chains shows an optimal film morphology and the highest hole mobility of 2.7 × 10−4 cm2 V−1 s−1. Champion photovoltaic performance was obtained from P2, with Jsc = 9.1 mA cm−2, Voc = 0.88 V, FF = 48% and PCE = 3.8%.
Experimental section
Materials
n-Butyllithium was purchased from Acros Organics. All other chemicals were purchased from Aldrich or Alfa Aesar and were used without further purification. The following compounds were synthesized according to modified literature procedures: diketone 1,34 3,7-dibromonaphtho[1,2-c:5,6-c]bis(1,2,5-thiadiazole),35 and 2,6-bis(trimethyltin)-4,8-bis(5-(2-ethylhexyl)thiophen-2-yl)benzo[1,2-b:4,5-b′]dithiophene.46
Synthesis of the monomers
5,10-Di(thiophen-2-yl)naphtho[1,2-c:5,6-c]bis(1,2,5-thiadiazole) (2).
To a flask, 2-(tributylstannyl)-thiophene (8.8 g, 18 mmol), 3,7-dibromonaphtho[1,2-c:5,6-c]bis(1,2,5-thiadiazole) (2.89 g, 7.2 mmol), and anhydrous DMF (30 mL) were added. The mixture was purged with argon for 15 min. Then, the catalyst tetrakis(triphenylphosphine)palladium(0) (Pd(PPh3)4) (160 mg) was added and the solution was purged with argon for an additional 15 min. The reaction mixture was heated to 120 °C and stirred for 8 h. Then, the reaction mixture was cooled to room temperature and precipitated in methanol. The crude product was collected by filtration, and then purified by chromatography using a mixture solvent of petroleum ether and CH2Cl2 (5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 in v:v) as the eluent to give compound 2 as a red solid (1.53 g, 34% yield).
:1 in v:v) as the eluent to give compound 2 as a red solid (1.53 g, 34% yield).
2,3,8,9-Tetrakis(3-(octyloxy)phenyl)-6,12-di(thiophen-2-yl)-2,3,8,9-tetrahydroquinoxalino[6,5-f]quinoxaline (3a).
To a flask, compound 2 (3.03 g, 10.3 mmol), zinc dust (8.43 g, 129 mmol), and acetic acid (100 mL) were added. The mixture was purged with argon for 10 min and then heated to reflux with stirring. After the red emulsion turned white-pink, the mixture was cooled to room temperature. It was found that the reduction product was extremely active and easy to oxidize when exposed to air, so the reaction mixture was directly used for the next step. 1,2-Bis-(3-octyloxyphenyl)-ethane-1,2-dione (1a) in 15 mL of CH2Cl2 was then added in one portion to the flask and the mixture was stirred for 1 h at room temperature. Then, the mixture was poured into water and extracted twice with CH2Cl2, and the combined organic layers were washed with water, dried with MgSO4, and the solvent was removed, which was subsequently subjected to column chromatography over silica gel using petroleum ether and ethyl acetate (10:1) as the eluent to afford compound 3a as an orange solid (40% yield). 1H NMR (CDCl3, 600 MHz) δ (ppm) = 9.89 (s, 2H), 8.13 (dd, J = 3.67, 0.92 Hz, 2H), 7.55 (dd, J = 5.13, 0.92 Hz, 2H), 7.42–7.45 (m, 2 H), 7.37 (d, J = 1.47 Hz, 2H), 7.30–7.33 (m, 6H), 7.28 (d, J = 7.89 Hz, 2H), 7.24 (dd, J = 5.14, 3.67 Hz, 2H), 7.00 (dt, J = 5.96, 2.89 Hz, 2H), 6.95–6.98 (m, 2H), 3.92–3.97 (m, 8H), 1.74–1.82 (m, 8H), 1.43–1.51 (m, 8H), 1.33–1.41 (m, 32H), 0.93–0.95 (m, 12H). 13C NMR (CDCl3, 600 MHz) δ (ppm) = 159.11, 159.03, 152.06, 151.40, 140.38, 139.83, 139.04, 138.30, 138.10, 130.96, 129.86, 129.37, 129.15, 128.85, 127.25, 126.67, 123.51, 122.93, 122.50, 116.56, 116.14, 115.74, 115.66, 68.16, 31.58, 29.16, 25.75, 22.67, 14.07.
2,3,8,9-Tetrakis(3-((2-ethylhexyl)oxy)phenyl)-6,12-di(thiophen-2-yl)-2,3,8,9-tetrahydroquinoxalino[6,5-f]quinoxaline (3b).
Compound 3b was prepared according to the same procedure as that for compound 3a. 1H NMR (CDCl3, 600 MHz) δ (ppm) = 9.89 (s, 2H), 8.13 (dd, J = 3.67, 0.92 Hz, 2H), 7.55 (dd, J = 5.13, 0.92 Hz, 2H), 7.42–7.45 (m, 2 H), 7.37 (d, J = 1.47 Hz, 2H), 7.30–7.33 (m, 6H), 7.28 (d, J = 7.89 Hz, 2H), 7.24 (dd, J = 5.14, 3.67 Hz, 2H), 7.00 (dt, J = 5.96, 2.89 Hz, 2H), 6.95–6.98 (m, 2H), 3.92–3.97 (m, 8H), 1.74–1.82 (m, 4H), 1.43–1.51 (m, 8H), 1.33–1.41 (m, 24H), 0.93–0.95 (m, 24H). δ (ppm) = 159.39, 159.31, 152.13, 151.43, 140.33, 139.79, 139.09, 138.32, 138.12, 130.97, 129.88, 129.33, 129.12, 128.83, 127.22, 126.68, 123.52, 122.83, 122.42, 116.64, 116.28, 115.74, 115.65, 70.69, 39.26, 30.57, 29.06, 23.89, 23.11, 14.14, 11.11.
2,3,8,9-Tetrakis(3-(hexyloxy)phenyl)-6,12-di(thiophen-2-yl)-2,3,8,9-tetrahydroquinoxalino[6,5-f]quinoxaline (3c).
Compound 3c was prepared according to the same procedure as that for compound 3a. 1H NMR (CDCl3, 600 MHz) δ (ppm) = 9.89 (s, 2H), 8.13 (dd, J = 3.67, 0.92 Hz, 2H), 7.55 (dd, J = 5.13, 0.92 Hz, 2H), 7.42–7.45 (m, 2 H), 7.37 (d, J = 1.47 Hz, 2H), 7.30–7.33 (m, 6H), 7.28 (d, J = 7.89 Hz, 2H), 7.24 (dd, J = 5.14, 3.67 Hz, 2H), 7.00 (dt, J = 5.96, 2.89 Hz, 2H), 6.95–6.98 (m, 2H), 3.92–3.97 (m, 8H), 1.74–1.82 (m, 8H), 1.43–1.51 (m, 8H), 1.33–1.41 (m, 16H), 0.93–0.95 (m, 12H). 13C NMR (CDCl3, 600 MHz) δ (ppm) = 159.11, 159.03, 152.06, 151.40, 140.38, 139.83, 139.04, 138.30, 138.10, 130.96, 129.86, 129.37, 129.15, 128.85, 127.25, 126.67, 123.51, 122.93, 122.50, 116.56, 116.14, 115.74, 115.66, 68.16, 31.58, 29.16, 25.75, 22.67, 14.07.
6,12-Bis(5-bromothiophen-2-yl)-2,3,8,9-tetrakis(3-(octyloxy)phenyl)-2,3,8,9-tetrahydroquinoxalino[6,5-f]quinoxaline (M1).
Compound 3a (0.68 g, 1.0 mmol) was dissolved in 200 mL of CCl3/CH3COOH (3:1 in v:v), and then NBS (0.374 g, 2.1 mmol) was added in portions at 0 °C. The mixture was stirred at 0 °C for 3 h. Then, the mixture was poured into water and extracted twice with CH2Cl2. The combined organic layers were dried over MgSO4, and the solvent was removed. The crude product was purified with column chromatography over silica gel using petroleum ether and CH2Cl2 (3:1) as the eluent to afford M1 as an orange-red solid (92% yield). 1H NMR (CDCl3, 600 MHz) δ (ppm) = 9.63 (s, 2H), 7.65 (d, J = 3.67 Hz, 2H), 7.56 (s, 2H), 7.30 (t, J = 7.62 Hz, 2H), 7.24 (s, 2H), 7.17–7.22 (m, 4H), 7.13–7.17 (m, 2H), 7.10 (d, J = 3.67 Hz, 2H), 6.99 (d, J = 8.07 Hz, 4H), 4.05 (t, J = 6.24 Hz, 4H), 3.91 (t, J = 6.42 Hz, 4H), 1.83 (dt, J = 14.12, 6.88 Hz, 4H), 1.76 (m, 4H), 1.44–1.57 (m, 8H), 1.33–1.41 (m, 32H), 0.94 (t, J = 6.42 Hz, 12H). 13C NMR (CDCl3, 600 MHz) δ (ppm) = 159.35, 159.06, 151.96, 151.54, 140.18, 139.52, 139.28, 137.82, 130.01, 129.38, 129.09, 128.97, 126.23, 123.13, 122.38, 117.25, 117.12, 116.15, 115.61, 115.22, 68.38, 68.20, 31.89, 29.50, 29.43, 29.35, 29.22, 26.25, 26.12, 22.72, 14.14.
6,12-Bis(5-bromothiophen-2-yl)-2,3,8,9-tetrakis(3-((2-ethylhexyl)oxy)phenyl)-2,3,8,9-tetrahydroquinoxalino[6,5-f]quinoxaline (M2).
Compound M2 was prepared according to the same procedure as that for compound M1. 1H NMR (CDCl3, 600 MHz) δ (ppm) = 9.63 (s, 2H), 7.65 (d, J = 3.67 Hz, 2H), 7.56 (s, 2H), 7.30 (t, J = 7.62 Hz, 2H), 7.24 (s, 2H), 7.17–7.22 (m, 4H), 7.13–7.17 (m, 2H), 7.10 (d, J = 3.67 Hz, 2H), 6.99 (d, J = 8.07 Hz, 4H), 4.05 (t, J = 6.24 Hz, 4H), 3.91 (t, J = 6.42 Hz, 4H), 1.83 (dt, J = 14.12, 6.88 Hz, 4H), 1.44–1.57 (m, 8H), 1.33–1.41 (m, 24H), 0.93–0.95 (m, 24H). 13C NMR (CDCl3, 600 MHz) δ (ppm) = 159.54, 159.32, 152.23, 151.74, 140.13, 139.76, 139.35, 138.07, 137.39, 130.18, 129.63, 129.37, 129.13, 129.05, 126.32, 122.96, 122.33, 121.98, 117.17, 116.95, 116.28, 115.74, 115.42, 70.74, 70.63, 39.47, 39.25, 30.64, 30.54, 29.21, 29.05, 23.97, 23.87, 23.12, 23.10, 14.14, 14.13, 11.28, 11.12.
6,12-Bis(5-bromothiophen-2-yl)-2,3,8,9-tetrakis(3-(hexyloxy)phenyl)-2,3,8,9-tetrahydroquinoxalino[6,5-f]quinoxaline (M3).
Compound M3 was prepared according to the same procedure as that for compound M1. 1H NMR (CDCl3, 600 MHz) δ (ppm) = 9.63 (s, 2H), 7.65 (d, J = 3.67 Hz, 2H), 7.56 (s, 2H), 7.30 (t, J = 7.62 Hz, 2H), 7.24 (s, 2H), 7.17–7.22 (m, 4H), 7.13–7.17 (m, 2H), 7.10 (d, J = 3.67 Hz, 2H), 6.99 (d, J = 8.07 Hz, 4H), 4.05 (t, J = 6.24 Hz, 4H), 3.91 (t, J = 6.42 Hz, 4H), 1.83 (dt, J = 14.12, 6.88 Hz, 4H), 1.76 (m, 4H), 1.44–1.57 (m, 8H), 1.33–1.41 (m, 16H), 0.94 (t, J = 6.42 Hz, 12H). 13C NMR (CDCl3, 600 MHz) δ (ppm) = 159.38, 159.07, 152.04, 151.65, 140.20, 139.61, 139.30, 137.95, 137.31, 130.10, 129.55, 129.41, 129.13, 129.01, 126.29, 123.09, 122.40, 121.89, 117.22, 117.12, 116.12, 115.69, 115.24, 68.35, 68.19, 31.69, 31.60, 29.36, 29.14, 25.89, 25.75, 22.67, 14.08.
Synthesis of the copolymers
Synthesis of PBDT-DTNQx-mOC8.
2,6-Bis(trimethyltin)-4,8-bis(5-(2-ethylhexyl)thiophen-2-yl)benzo[1,2-b:4,5-b′]dithiophene (90.4 mg, 0.10 mmol), compound M1 (137.1 mg, 0.10 mmol), Pd(PPh3)4 (5 mg), and toluene (4 ml) were added to a 35 ml reaction vial. The vial was purged with argon and subsequently sealed. The vial was heated in a microwave reactor at 170 °C for 45 min. Then, the reaction mixture was cooled to ambient temperature and precipitated into methanol. The solid was collected by filtration. After drying in the vacuum drying oven, the crude product was purified by Soxhlet extraction with methanol, acetone and hexane to remove oligomers and the residual catalyst. Finally, the polymer PBDT-DTNQx-mOC8 was obtained as a dark green solid (153.7 mg, 85% yield).
Synthesis of PBDT-DTNQx-mOEH.
The polymerization procedure for 2,6-bis(trimethyltin)-4,8-bis(5-(2-ethylhexyl)thiophen-2-yl)benzo[1,2-b:4,5-b′]dithiophene (90.4 mg, 0.10 mmol) and compound M2 (137.1 mg, 0.10 mmol) was the same as that of PBDT-DTNQx-mOC8. PBDT-DTNQx-mOEH was obtained as a dark-green solid (126.7 mg, 70%).
Synthesis of PBDT-DTNQx-mOC6.
The polymerization procedure for 2,6-bis(trimethyltin)-4,8-bis(5-(2-ethylhexyl)thiophen-2-yl)benzo[1,2-b:4,5-b′]dithiophene (90.4 mg, 0.10 mmol) and compound M3 (127.5 mg, 0.10 mmol) was the same as that of PBDT-DTNQx-mOC8. PBDT-DTNQx-mOC6 was obtained as a dark-green solid (161.0 mg, 89%).
Instruments and characterization
NMR data were collected on a Bruker AVANCE Digital 600 MHz NMR workstation. Gel-permeation chromatography (150 °C in 1,2,4-trichlorobenzene) was performed on a Polymer Laboratories PL220 Chromatograph with linear polystyrene as a reference sample. UV/vis absorption spectra were recorded on a HP 8453 spectrophotometer. The solutions that were used for the UV/vis spectroscopic measurements were dissolved in o-DCB and the films were drop-coated onto a quartz substrate. Differential scanning calorimetry measurements were performed on a Netzsch DSC 204 under a N2 flow at heating and cooling rates of 10 °C min−1 each. Thermogravimetric analyses were performed on a Netzsch TG 209 under a N2 flow at a heating rate of 10 °C min−1. Electrochemical cyclic voltammetry was performed on a CHI 600D electrochemical workstation at a scan rate of 50 mV s−1. The mobilities were determined by fitting the dark currents to the model of a single carrier space-charge-limited-current (SCLC), which is described by the equation J = (9/8)ε0εrμh((V2)/(d3)), where J is the current, μh is the zero-field mobility, ε0 is the permittivity of free space, εr is the relative permittivity of the material, d is the thickness of the polymer layer, and V is the effective voltage. The effective voltage was obtained by subtracting the built-in voltage (Vbi) and the voltage drop (Vs) from the series resistance of the whole device except for the active layer from the applied voltage (Vappl), V = Vappl − Vbi − Vs. Vs = 10 Ω (the resistance of MoO3) × J (current, A), and we assume the resistance of ITO/PEDOT:PSS and Al to be zero as they have high conductivity. The mobilities were extracted by modeling the dark currents in the SCLC region and calculated from the slope of the J1/2–V curves.
Fabrication and characterization of polymer solar cells
The ITO-coated glass substrates were cleaned by sonication in detergent deionized water, acetone, and 2-propanol and dried in a stream of N2. Then, the PFN (0.5 mg mL−1) was spin-cast from MeOH/acetic acid (100:1, v/v) onto an ITO substrate to form a thin film (thickness: 5 nm). Then, the bulk heterojunction composite (thickness: ∼100 nm) was spin-coated onto a PFN layer from a hot solution in o-DCB of the polymer (8 mg mL−1) and PC71BM. Molybdenum oxide (MoO3, ∼10 nm) was thermally deposited on top of the active layer through a shadow mask in a vacuum chamber, with an evaporation rate of 0.1 Å s−1 under a vacuum of about 3 × 10−4 Pa. The effective area of the device was 0.16 cm2, as determined by the shadow mask that was used during the deposition of the Al electrode. PCEs were measured under an AM1.5G XES-40S1 150 W AAA Class solar simulator. The power of sun simulation was calibrated before testing by using a standard silicon solar cell, thus giving a value of 100 mW cm−2 during the test. The current-density–voltage (J–V) characteristics were recorded on a Keithley 2400 source measure unit. The spectroscopic response was measured by using a commercial photo modulation spectroscopic setup (Oriel). A calibrated Si photodiode was used to determine the photosensitivity. The external quantum efficiencies of inverted PSCs were measured by using a commercial photo modulation spectroscopic setup (DSR100UV-B), including a xenon lamp, an optical chopper, a monochromator, and a lock-in amplifier that was operated by a PC (Zolix Instruments); a calibrated Si photodiode was used as a standard.
Results and discussion
Synthesis and characterization
The synthetic routes to the monomers and polymers are shown in Scheme 2. Three NQx-based monomers with octyl, ethylhexyl and hexyl side chains, respectively, in the meta position of the suspended benzene were synthesized. The benzil derivative (1) was synthesized according to modified literature procedures with good yield. Compound 2 was prepared by a common Stille cross-coupling reaction of 3,7-dibromonaphtho[1,2-c:5,6-c]bis(1,2,5-thiadiazole) with 2-(tributylstannyl)thiophene, and then reduced by zinc powder in acetic acid. It was found that the reduction product was extremely active and easy to oxidize when exposed to air, so the reaction mixture was directly used for the next step. The tetramine formed was condensed with benzil and compound 3 was formed, which was then dibrominated with N-bromosuccinimide to yield a dibromide monomer. Three polymers P1, P2 and P3 were then synthesized by Stille coupling copolymerization with 2,6-bis(trimethyltin)-4,8-bis(5-(2-ethylhexyl)thiophen-2-yl)benzo[1,2-b:4,5-b′]dithiophene and M1, M2, and M3, respectively, using tetrakis(triphenylphosphine)palladium (Pd(PPh3)4) as a catalyst. The resulting polymers were precipitated in MeOH and purified successively by Soxhlet extraction with MeOH, acetone, and n-hexane (24 h each) to remove any oligomers and the residual catalyst. The molecular weights of the polymers were evaluated by high temperature gel-permeation chromatography (GPC) at 150 °C using 1,2,4-trichlorobenzene as the eluent. P1 and P2 are readily soluble in common organic solvents including tetrahydrofuran (THF), chloroform, chlorobenzene, dichlorobenzene (o-DCB), etc., at room temperature; however, P3 shows worse solubility and is soluble only in warm chlorobenzene or o-dichlorobenzene (o-DCB). The number-average molecular weights (Mns) of polymers P1, P2 and P3 were determined to be 21.2, 30.4, and 28.3 kDa, respectively, with the corresponding polydispersity indices (PDIs) of 2.4, 2.4, and 2.7, respectively (Table 1). The relatively high PDI of P3 might be caused by the poor solubility. Differential scanning calorimetry (DSC) and thermogravimetric analysis were used to study the thermal properties of the polymers. No phase transition was observed up to 300 °C for all of the polymers, based on the DSC analysis. All of the polymers had decomposition temperatures (defined as the temperature at which 5% weight loss is observed, Td) over 300 °C under a nitrogen atmosphere as determined by TGA analysis, thus indicating their excellent thermal stabilities.
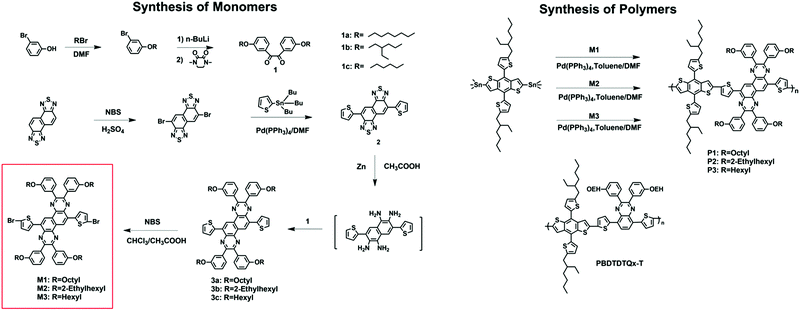 |
| | Scheme 2 Synthesis of the monomers and the polymers. | |
Table 1 Molecular weights, optical properties, and energy levels of P1, P2 and P3
| Copolymer |
M
nb [kDa] |
PDIb |
In solution |
As films |
E
optg [eV] |
E
HOMO [eV] |
E
LUMO [eV] |
|
λ
max [nm] |
λ
onset [nm] |
λ
max [nm] |
λ
onset [nm] |
|
The data of PBDTDTQx-T were acquired from a reported paper.
Determined by GPC at 150 °C using 1,2,4-trichlorobenzene as the eluent.
|
| P1 |
21.2 |
2.4 |
635 |
722 |
630 |
752 |
1.65 |
−5.16 |
−3.51 |
| P2 |
30.4 |
2.4 |
631 |
696 |
631 |
700 |
1.77 |
−5.16 |
−3.39 |
| P3 |
28.3 |
2.7 |
630 |
701 |
630 |
728 |
1.70 |
−5.16 |
−3.46 |
| PBDTDTQx-Ta |
49.5 |
1.6 |
605 |
— |
605 |
740 |
1.67 |
−5.12 |
−3.35 |
Optical and electrochemical properties
To investigate the photophysical properties of the polymers, the UV/vis absorption spectra of the polymers in solutions and in films were recorded (Fig. 1 and Table 1). All of the polymers exhibit two absorption bands both in solution and in thin films: short-wavelength peaks are located at about 300–450 nm, which originate from π–π* transitions, and long-wavelength peaks are located at about 600–700 nm, which could be ascribed to intramolecular charge transfer (ICT) between the donor and acceptor units. In solution, the three polymers showed almost identical absorption profiles, with a main peak located at around 630 nm and a shoulder peak at around 580 nm, which revealed the presence of a strong intermolecular interaction of polymer main chains in solution. Such an aggregation-induced absorption shoulder in solution was unusual for quinoxaline based polymers, and this could be explained by the strong π–π interaction induced by the enlarged planar aromatic structure of two fused quinoxaline rings. In addition to the absorption range, absorption coefficients of solutions were also characterized. It was found that all the polymers exhibit much similar solution absorption coefficients with the peak value in the range of 4.4 × 10−3 to 5.0 × 10−3 L mol−1 cm−1. When as solid films, all the polymers exhibit a wider absorption range, with the absorption onset red-shifted to 752, 700 and 728 nm for P1, P2 and P3, respectively. Interestingly, with a little change in the alkyl chain in the suspended benzene, P3 showed a much higher absorption coefficient than the other polymers over the whole visible-light region. As the solution absorption coefficients of these polymers are similar, the higher solid absorption coefficient might due to a closer packing of the polymer chain of P3 than the other two polymers in an aggregated state. This result is beneficial because a high absorption coefficient of a conjugated polymer means that it can harvest photons more efficiently and, therefore, achieve a large short-circuit current in PSCs. The optical bandgaps (Eoptg) of the polymers were determined from absorption onsets in thin films. The Eoptg of P1, P2 and P3 is around 1.70 eV (as seen in Table 1). The comparison between our new polymers and a reported quinoxaline-based polymer counterpart PBDTDTQx-T (Scheme 2) shows that they have a similar bandgap,46 suggesting that fused quinoxaline units have a similar electron withdrawing ability as a single quinoxaline unit.
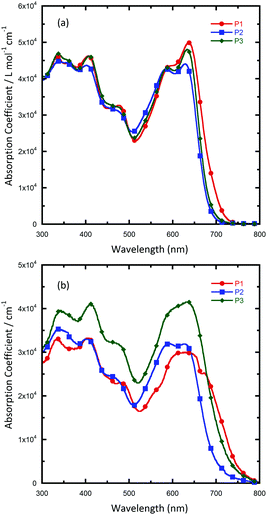 |
| | Fig. 1 UV/vis absorption spectra of the polymers: (a) in o-DCB; (b) as solid films. | |
Cyclic voltammetry (CV) measurements were performed to evaluate the electrochemical properties and energy levels of the conjugated polymers. Graphite, Pt wire, and Ag/AgCl were used as the working electrode, counter electrode, and reference electrode, respectively. All of the measurements were performed in a 0.1 M solution of tetrabutyl ammonium hexafluorophosphate in MeCN at a scan rate of 50 mV s−1 under an inert atmosphere. Fig. 2 shows the CV plots of P1, P2 and P3. It was found that the difference in alkyl chains had little effect on the energy level of polymers, and the HOMO levels of P1, P2 and P3 were all located at −5.16 eV, calculated using the equation HOMO = −e(Eox + 4.80). The LUMO levels were calculated from the HOMO levels and optical bandgaps according to the equation: LUMO = (HOMO + Eoptg) eV, which lie in the range of −3.39 to −3.51 eV. The electrochemical data are summarized in Table 1. It is found that the HOMO levels of DTNQx downshifted by 0.04 eV compared to the quinoxaline-based polymer PBDTDTQx-T. The low-lying HOMO levels can provide a higher Voc for DTNQx-based polymers in the resulting solar cells.
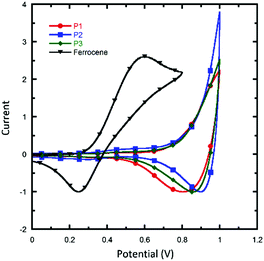 |
| | Fig. 2 Cyclic voltammograms of the polymer films in Bu4NPF6 solution (0.1 M in MeCN); scan rate: 50 mV s−1. | |
Photovoltaic properties
The PSC devices with an inverted structure of ITO/PFN/polymer:PC71BM/MoO3/Al were fabricated to investigate the photovoltaic properties of the polymers. A thin layer of PFN was applied on top of ITO to improve the electron selectivity at the cathode interface. The J–V characteristics of the PSCs acquired under AM1.5G with an illumination intensity of 100 mW cm−2 are shown in Fig. 4 and the corresponding device parameters (Jsc, Voc, FF, and PCE) are summarized in Table 2.
Table 2 Photovoltaic performance of devices measured under simulated light (AM 1.5G, 100 mW cm−2)
| Active layers |
J
sc (mA cm−2) |
V
oc (V) |
FF (%) |
PCE (%) |
|
The data of PBDTDTQx-T were acquired from a reported paper.
|
| P1:PC71BM |
4.9 |
0.80 |
43 |
1.7 |
| P2:PC71BM |
9.1 |
0.88 |
48 |
3.8 |
| P3:PC71BM |
8.0 |
0.79 |
42 |
2.7 |
| P2:PC71BM 2% DIO |
9.4 |
0.84 |
43 |
3.4 |
| PBDTDTQx-T:PC71BMa |
8.9 |
0.76 |
50 |
3.4 |
The polymers were tested in a 1:2 weight ratio against PC71BM. It is found that alkyl chains have a significant influence on the device performance of the polymers. The device based on P1 shows a PCE of 1.7%, with a Jsc of 4.9 mA cm−2, a Voc of 0.80 V and a FF of 43%. A slightly improved device performance was realized for P3, which exhibited a Voc of 0.79 V, a FF of 43%, and a dramatically improved Jsc of 8.0 mA cm−2, leading to a PCE of 2.7%. The best device performance with a PCE of 3.8% was achieved by P2, which showed a further improved Jsc of 9.1 mA cm−2, a Voc of 0.88 V and a FF of 48%. The higher photovoltaic performance of the P2 based device was mainly due to a higher Jsc value. To explain this difference, the mobility and surface morphology of these polymers were investigated and will be discussed later. 1,8-Diiodooctane (DIO) was then used as an additive during the PSC device fabrication process with an attempt to further optimize the PCE of P2 (ESI†). It was found that after adding DIO the Jsc was slightly improved from 9.1 mA cm−2 to 9.4 mA cm−2. However, the benefit was counteracted by the reduced Voc and FF, which ultimately led to a decreased PCE of 3.4%. These results may demonstrate that DIO was not suitable for this blend system. Since the HOMO energy levels of P1, P2 and P3 were downshifted by 0.04 eV compared to the quinoxaline-based polymer PBDTDTQx-T, we also investigated the influence of this energy level difference on the device performance. The PCE data of the PBDTDTQx-T:PC71BM based device with a weight ratio of 1:2 were collected from the related literature.46 Without any post-treatment process, the P2 based device, which exhibited the same main chain structure except for the quinoxaline unit compared with PBDTDTQx-T, showed an obviously higher Voc value of 0.12 V than the PBDTDTQx-T based device. This result clearly demonstrated that the deeper HOMO energy level of the DTNQx based polymer could lead to a higher Voc value and is beneficial for achieving a higher PCE. It was found that the FFs of P1, P2 and P3 were relatively low compared with the device based on PBDTDTQx-T. This might be attributed to the lower molecular weights and larger PDIs of P1, P2 and P3.
To study the spectroscopic response of the PSCs, EQE measurements were conducted (Fig. 3). The devices based on P2 showed the highest photoresponse over a broad range (380–700 nm) and the EQE peak value reached 45% at about 550 nm. The integrated current densities from the EQE spectra of all devices are in good agreement with the Jsc values obtained from J–V measurements.
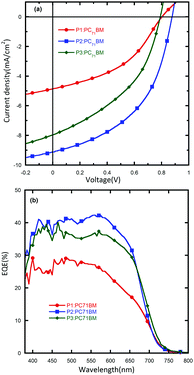 |
| | Fig. 3 (a) J–V curves and (b) EQE spectra of devices with the configuration: ITO/PFN/active-layer/MoO3/Al. | |
Mobility
The charge transport properties of the donor polymer in the BHJ film are important for determining the efficiency of the PSCs. In order to evaluate the hole mobility of the resultant polymers, hole-only devices with the configuration ITO/poly(3,4-ethylenedioxythiophene):poly-(styrenesulfonate) (PEDOT:PSS)/active layer/MoO3/Al were fabricated, in which the active layers were prepared under the optimal device conditions as discussed above. As shown in Fig. 4, the hole mobility of P1, P2 and P3 is 3.2 × 10−6, 2.7 × 10−4 and 8.9 × 10−5 cm2 V−1 s−1, respectively. The higher hole mobility of P2 indicates that ethylhexyl side chains could help improve the hole transport properties of the active layer, and explains the higher Jsc in the resulting PSCs to a certain degree. Although P3 exhibited the highest absorption coefficient in thin films, the low hole mobility limited carrier transport from active layer to electrode, thus partially counteracting the benefit of its high absorption coefficient.
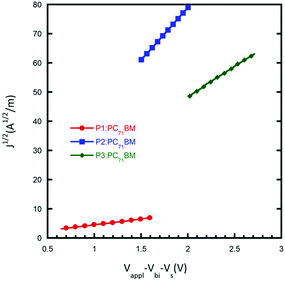 |
| | Fig. 4
J
1/2–V characteristics of devices with the configuration: ITO/PEDOT:PSS/active-layer/MoO3/Al. The solid lines represent the fit curves. | |
Morphology
AFM measurements were carried out to investigate the surface morphologies of the polymer/PC71BM blend films. The height images (5 × 5 μm2) are shown in Fig. 5, and the corresponding root mean square (RMS) roughnesses were observed to be 2.44, 2.48 and 1.04 nm for P1, P2 and P3, respectively. It was found that the difference in the alkyl side-chain type led to a huge difference in the film morphology. Compared with P1 and P3, polymer P2 exhibited a more obvious nanoscale phase separation and a larger domain size, which demonstrate a more continuous interpenetrating network in the blend film of P2. A good morphology is beneficial for enhancing the photovoltaic performance of the corresponding device due to the promotion of the exciton dissociation and charge transporting. Therefore, it could be concluded that the highest PCE of P2 can be ascribed to the optimal morphology as well as high hole mobility.
 |
| | Fig. 5 AFM height images of the polymer/PC71BM blend films: (a) P1; (b) P2; (c) P3. | |
Conclusions
In summary, three polymers based on a new DTNQx acceptor unit were designed and synthesized for constructing well-performing D–A conjugated polymers for use in PSCs. To investigate the relationship between the structure and the performance of the DTNQx-based polymers, three kinds of side chains were introduced. All polymers exhibit an optical bandgap around 1.7 eV and a HOMO energy level around −5.16 eV. Compared with the quinoxaline unit, fused quinoxaline DTNQx could downshift the HOMO levels of the resultant polymers by 0.04 eV while maintaining the bandgap unchanged. Through side chain engineering, polymer properties, such as absorption coefficient and hole mobility, are finely controlled. Polymer P2 which contains ethylhexyl side chains shows an optimal film morphology and the highest hole mobility of 2.7 × 10−4 cm2 V−1 s−1. As a result, P2 exhibits the best photovoltaic performance with Jsc = 9.1 mA cm−2, Voc = 0.88 V, FF = 48% and PCE = 3.8%. Our work demonstrates that the newly developed building block of DTNQx is a promising acceptor unit for constructing state-of-the-art conjugated polymers for use in PSCs.
Acknowledgements
The work was financially supported by the Ministry of Science and Technology (No. 2014CB643501), the Natural Science Foundation of China (No. 21125419 and 51361165301) and the Guangdong Natural Science Foundation (No. 2016A030310434).
Notes and references
- A. J. Heeger, Adv. Mater., 2014, 26, 10–28 CrossRef CAS PubMed.
- J. Chen and Y. Cao, Acc. Chem. Res., 2009, 42, 1709–1718 CrossRef CAS PubMed.
- F. C. Krebs, N. Espinosa, M. Hösel, R. R. Søndergaard and M. Jørgensen, Adv. Mater., 2014, 26, 29 CrossRef CAS PubMed.
- S. Gunes, H. Neugebauer and N. S. Sariciftci, Chem. Rev., 2007, 107, 1324 CrossRef PubMed.
- G. Yu, J. Gao, J. C. Hummelen, F. Wudl and A. J. Heeger, Science, 1995, 270, 1789 CAS.
- Y. Li, Acc. Chem. Res., 2012, 45, 723 CrossRef CAS PubMed.
- Z. He, H. Wu and Y. Cao, Adv. Mater., 2013, 26, 1006 CrossRef PubMed.
- G. Li, R. Zhu and Y. Yang, Nat. Photonics, 2012, 6, 153 CrossRef CAS.
- L. Lu, T. Zheng, Q. Wu, A. M. Schneider, D. Zhao and L. Yu, Chem. Rev., 2015, 115, 12666 CrossRef CAS PubMed.
- X. Zhan and D. Zhu, Polym. Chem., 2010, 1, 409 RSC.
- Z. He, C. Zhong, S. Su, M. Xu, H. Wu and Y. Cao, Nat. Photonics, 2012, 6, 591 Search PubMed.
- S. Liu, K. Zhang, J. Lu, J. Zhang, H.-L. Yip, F. Huang and Y. Cao, J. Am. Chem. Soc., 2013, 135, 15326 CrossRef CAS PubMed.
- J. You, L. Dou, K. Yoshimura, T. Kato, K. Ohya, T. Moriarty, K. Emery, C.-C. Chen, J. Gao, G. Li and Y. Yang, Nat. Commun., 2013, 4, 1446 CrossRef PubMed.
- C.-Z. Li, C.-Y. Chang, Y. Zang, H.-X. Ju, C.-C. Chueh, P.-W. Liang, N. Cho, D. S. Ginger and A. K. Y. Jen, Adv. Mater., 2014, 26, 6262 CrossRef CAS PubMed.
- S.-H. Liao, H.-J. Jhuo, Y.-S. Cheng and S.-A. Chen, Adv. Mater., 2013, 25, 4766 CrossRef CAS PubMed.
- C. Duan, K. Zhang, X. Guan, C. Zhong, H. Xie, F. Huang, J. Chen, J. Peng and Y. Cao, Chem. Sci., 2013, 4, 1298 RSC.
- Z. He, B. Xiao, F. Liu, H. Wu, Y. Yang, S. Xiao, C. Wang, T. P. Russell and Y. Cao, Nat. Photonics, 2015, 3, 174 CrossRef.
- Y. Liu, J. Zhao, Z. Li, C. Mu, W. Ma, H. Hu, K. Jiang, H. Lin, H. Ade and H. Yan, Nat. Commun., 2014, 5, 5293 CrossRef CAS PubMed.
- J. Zhao, Y. Li, G. Yang, K. Jiang, H. Lin, H. Ade, W. Ma and H. Yan, Nat. Energy, 2016, 1, 15027 CrossRef.
- J. Kim, J. B. Park, I. H. Jung, A. C. Grimsdale, S. C. Yoon, H. Yangd and D. Hwang, Energy Environ. Sci., 2015, 8, 2352 CAS.
- C. Duan, A. Furlan, J. J. van Franeker, R. E. M. Willems, M. M. Wienk and R. A. J. Janssen, Adv. Mater., 2015, 27, 4461 CrossRef CAS PubMed.
- R. Kroon, M. Lenes, J. C. Hummelen, P. W. M. Blom and B. de Boer, Polym. Rev., 2008, 48, 531 CrossRef CAS.
- C. Duan, F. Huang and Y. Cao, J. Mater. Chem., 2012, 22, 10416 RSC.
- L. Huo, S. Zhang, X. Guo, F. Xu, Y. Li and J. Hou, Angew. Chem., Int. Ed., 2011, 50, 9697 CrossRef CAS PubMed.
- E. Wang, Z. Ma, Z. Zhang, K. Vandewal, P. Henriksson, O. Inganäs, F. Zhang and M. R. Andersson, J. Am. Chem. Soc., 2011, 133, 14244 CrossRef CAS PubMed.
- L. Huo, J. Hou, S. Zhang, H.-Y. Chen and Y. Yang, Angew. Chem., Int. Ed., 2010, 49, 1500–1503 Search PubMed.
- E. Wang, W. Mammo and M. R. Andersson, Adv. Mater., 2014, 26, 1801–1826 CrossRef CAS PubMed.
- E. Wang, L. Hou, Z. Wang, S. Hellström, F. Zhang, O. Inganäs and M. R. Andersson, Adv. Mater., 2010, 22, 5240 CrossRef CAS PubMed.
- Y. Zhang, J. Zou, H.-L. Yip, K.-S. Chen, D. F. Zeigler, Y. Sun and A. K. Y. Jen, Chem. Mater., 2011, 23, 2289–2291 CrossRef CAS.
- H.-C. Chen, Y.-H. Chen, C.-C. Liu, Y.-C. Chien, S.-W. Chou and P.-T. Chou, Chem. Mater., 2012, 24, 4766 CrossRef CAS.
- X. Guo, M. Zhang, J. Tan, S. Zhang, L. Huo, W. Hu, Y. Li and J. Hou, Adv. Mater., 2012, 24, 6536 CrossRef CAS PubMed.
- C. L. Chochos, R. Singh, M. Kim, N. Gasparini, A. Katsouras, C. Kulshreshtha, V. G. Gregoriou, P. E. Keivanidis, T. Ameri, C. J. Brabec, K. Cho and A. Avgeropoulos, Adv. Funct. Mater., 2016, 26, 1840 CrossRef CAS.
- Q. Fan, H. Jiang, Y. Liu, W. Su, H. Tan, Y. Wang, R. Yang and W. Zhu, J. Mater. Chem. C, 2016, 4, 2606 RSC.
- J. Zhang, W. Cai, F. Huang, E. Wang, C. Zhong, S. Liu, M. Wang, C. Duan, T. Yang and Y. Cao, Macromolecules, 2011, 44, 894 CrossRef CAS.
- M. Wang, X. Hu, P. Liu, W. Li, X. Gong, F. Huang and Y. Cao, J. Am. Chem. Soc., 2011, 133, 9638 CrossRef CAS PubMed.
- T. Yang, M. Wang, C. Duan, X. Hu, L. Huang, J. Peng, F. Huang and X. Gong, Energy Environ. Sci., 2012, 5, 8208 CAS.
- X. Hu, C. Yi, M. Wang, C.-H. Hsu, S. Liu, K. Zhang, C. Zhong, F. Huang, X. Gong and Y. Cao, Adv. Energy Mater., 2014, 4, 1400378 CrossRef.
- L.-Q. Liu, G.-C. Zhang, P. Liu, J. Zhang, S. Dong, M. Wang, Y.-G. Ma, H.-L. Yip and F. Huang, Chem. – Asian J., 2014, 9, 2104 CrossRef CAS PubMed.
- P. Liu, S. Dong, F. Liu, X. Hu, L. Liu, Y. Jin, S. Liu, X. Gong, T. P. Russell, F. Huang and Y. Cao, Adv. Funct. Mater., 2015, 25, 6458–6469 CrossRef CAS.
- L. Liu, G. Zhang, B. He and F. Huang, Chin. J. Chem., 2015, 33, 902 CrossRef CAS.
- Y. Dong, X. Hu, C. Duan, P. Liu, S. Liu, L. Lan, D. Chen, L. Ying, S. Su, X. Gong, F. Huang and Y. Cao, Adv. Mater., 2013, 25, 3683 CrossRef CAS PubMed.
- I. Osaka, M. Shimawaki, H. Mori, I. Doi, E. Miyazaki, T. Koganezawa and K. Takimiya, J. Am. Chem. Soc., 2012, 134, 3498 CrossRef CAS PubMed.
- C. Duan, R. E. M. Willems, J. J. van Franeker, B. J. Bruijnaers, M. M. Wienk and R. A. J. Janssen, J. Mater. Chem. A, 2016, 4, 1855 CAS.
- V. Vohra, K. Kawashima, T. Kakara, T. Koganezawa, I. Osaka, K. Takimiya and H. Murata, Nat. Photonics, 2015, 9, 403 CrossRef CAS.
- K. Kawashima, Y. Tamai, H. Ohkita, I. Osaka and K. Takimiya, Nat. Commun., 2015, 6, 10085 CrossRef CAS PubMed.
- R. Duan, L. Ye, X. Guo, Y. Huang, P. Wang, S. Zhang, J. Zhang, L. Huo and J. Hou, Macromolecules, 2012, 45, 3032 CrossRef CAS.
- Z. Zhang and Y. Li, Sci. China: Chem., 2015, 58, 192 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6qm00130k |
|
| This journal is © the Partner Organisations 2017 |







