DOI:
10.1039/C6QM00120C
(Research Article)
Mater. Chem. Front., 2017,
1, 646-653
Fabrication of circular polarized luminescent helical fibers from chiral phenanthro[9,10]imidazole derivatives†
Received
12th July 2016
, Accepted 25th October 2016
First published on 11th November 2016
Abstract
This work provides a simple but efficient way of constructing violet fluorescent helical nanofibers by the self-assembly of chiral π–π conjugated molecules, phenanthro[9,10-d]imidazole (PIM) derivatives. PIM derivatives are well-known functional molecules, but the construction of PIM into functional architectures has not been carried out to date. By introducing L- and D-aniline pendants into PIM derivatives, PIM-D-Ala and PIM-L-Ala are synthesized, which not only have the properties of aggregation induced circular dichroism (AICD) and circular polarized luminescence (CPL), but also have the capacity to self-assemble into helical fibers. They have important applications in the increasing demand for the miniatured optics and electronic devices.
Introduction
π–π conjugated molecules are important functional molecules due to their applications in organic molecular electronic devices, such as organic solar cells, field effect transistors and light-emitting diodes.1 The construction of π–π conjugated molecules into functional architectures by self-assembly may further broaden their application in nano/micro science and technologies. In nature, chirality is an important inducer to guide the construction of novel architectures of biomolecules, such as the duplex of DNA and α-helix of protein. If π–π conjugated molecules are endowed with chirality, novel architectures and properties can also be generated.2 Two strategies are usually utilized to introduce chirality to π–π conjugated molecules, either by chemically modifying the π-conjugated chromophores with chiral pendants or by adding chiral dopants to π-conjugated chromophores.3 Either way could efficiently generate spatial hindrance, guide the helical association of the chromophores and endow the helical assemblies with circularly polarized luminescence (CPL) as well.4 The novel architectures with CPL properties are of considerable interest due to their potential application as materials for nano/micro optics and electronics. CPL reflects the intrinsic chiral feature of the architectures formed by the conjugated molecules. The degree of CPL is characterized by the emission dissymmetry factor (gem) of photoluminescence, which is defined as gem = 2(IL − IR)/(IL + IR), where IL and IR are the intensities of luminescence of the left- and right-handed emissions, respectively.5 For most of the organic molecules carrying chiral chromophores, the reported gem values barely exceed 0.01 in solution and they even get worse at the film state due to aggregation caused quenching (ACQ).4 Novel nano/micro architectures which have CPL properties are seldom explored and systematical works in this approach remain to be carried out. The rational design of functional π-conjugated chromophores and the hybridization of them with suitable chiral inducers are still a challenging task. We have made some exploration in this approach and successfully utilized silole and tetraphenylethylene (TPE) as the luminescent scaffolds and amino acid pendants as the inducers to guide the construction of helical luminescent fibers that have excellent CPL properties.6 Through this work, we are further broadening the components of π-conjugated chromophores and exploring the possibility of constructing violet luminescent helical fibers based on the self-assembly of chiral PIM derivatives.
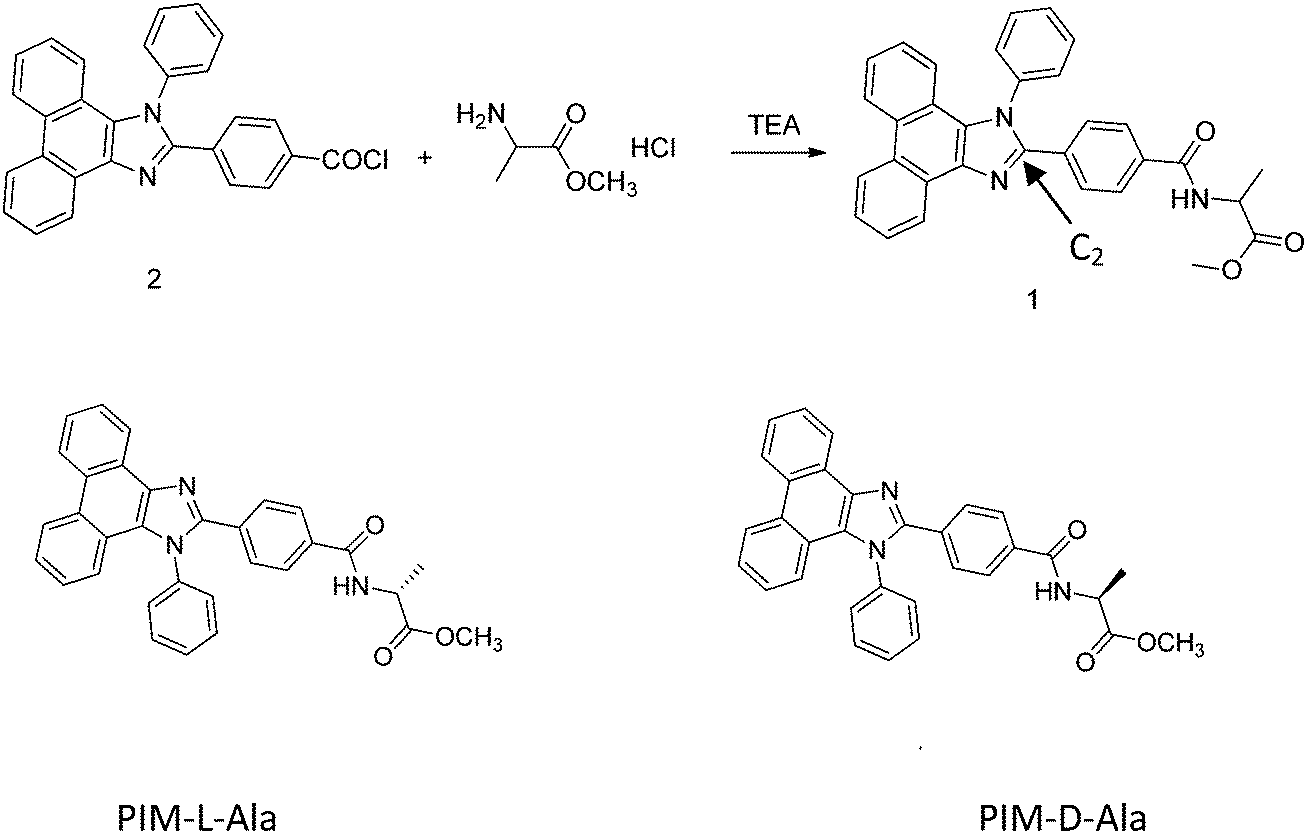
PIM derivatives emit violet light and have a high luminescence efficiency, an intrinsic wide band gap and good thermal stability.7 PIM derivatives are the ideal luminescent scaffolds of CPL materials and the incorporation of chiral motifs into PIM scaffolds may endow the molecules with excellent CPL properties and self-assembling capacities as well. In this work, we report the fabrication of helical luminescent fibers with CPL properties via the self-assembly of chiral PIM derivatives. We chose the 1,2-diphenyl-phenanthro[9,10-d]imidazole derivative as the scaffold, modified it with D- and L-aniline, and synthesized two types of chiral PIM molecules, respectively (PIM-D-Ala and PIM-L-Ala, Scheme 1). The position of the chiral motif is critical for controlling the chirality, self-assembling behavior and optical properties of the chiral PIM derivatives. Systematic research studies have shown that the photoluminescence and electroluminescence of PIM derivatives mainly come from the contribution of imidazole. In particular, the groups connecting to the C2 atom of imidazole have a pivotal impact on the optical properties of the molecule.7d Thus C2 is chosen as the location where the amino acid attachment should happen. The incorporation of chiral attachment introduces an asymmetric force field to the imidazole scaffold to induce the PIM scaffold to helically rotate and adopt helical packing, which is stabilized by the judicious patterns of the noncovalent interactions of the chiral attachments, such as hydrogen bonds and van der Waals interactions. The helical conformation is expected to amplify in the higher order architectures of PIM–Ala as helical fibers or ribbons.8 With this rational design, we provide a simple and efficient way to fabricate helical fluorescent nano-/micro fibers with CPL properties which have important applications in optoelectronic nanodevices.
 |
| | Scheme 1 Chemical structures of PIM containing alanine attachment with (L-) and (D-) configurations. | |
Results and discussion
Synthesis of PIM-D-Ala and PIM-L-Ala
PIM-D-Ala and PIM-L-Ala (Scheme 1) were synthesized by modifying the phenyl group of the C2 atom of imidazole of PIM with D- and L-amino acid attachment. The PIM intermediate 2 was prepared from a one-pot cyclization and substitution reaction9 (the synthetic route of PPI-Ala is provided in Scheme S1, ESI†).
Aggregation induced CD and CPL
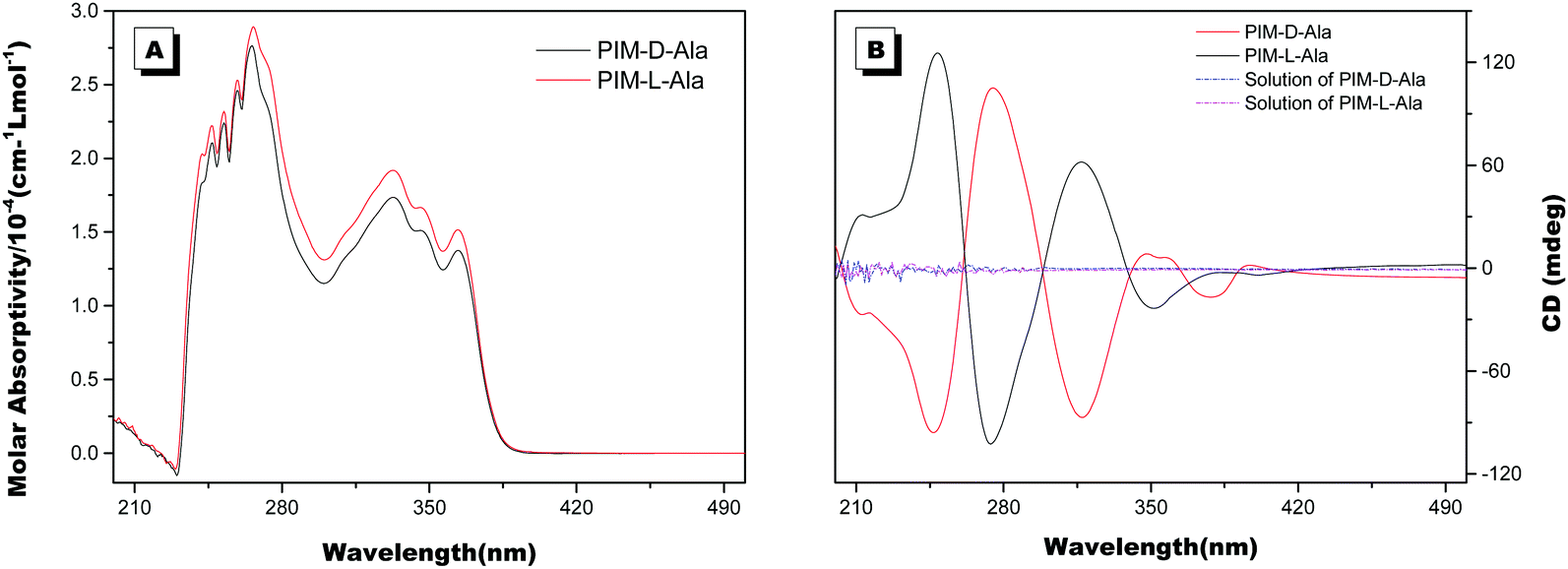
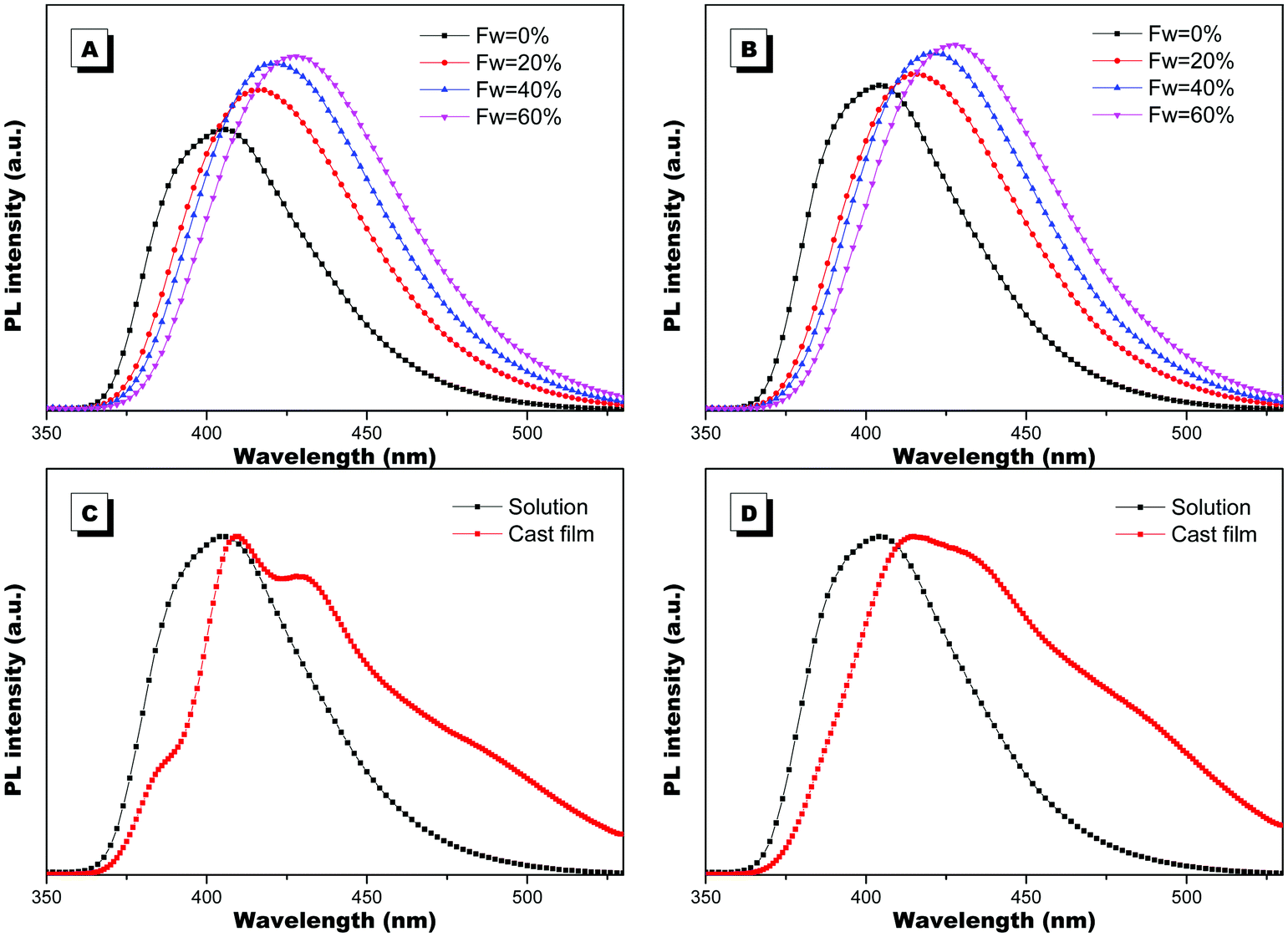
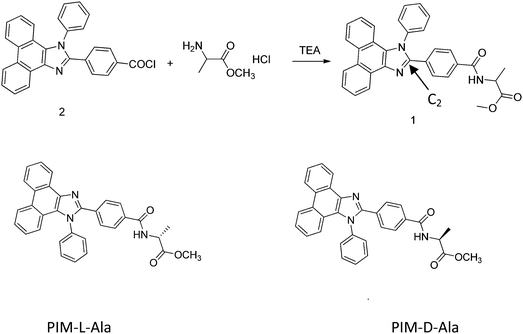
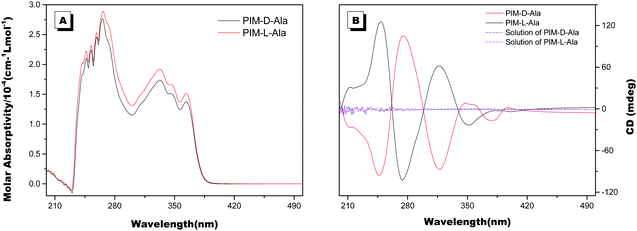
The optical properties of PIM–Ala were explored through UV, CD, PL and CPL measurements. As shown in Fig. 1A, PIM-D-Ala and PIM-L-Ala exhibited similar absorption peaks at a wavelength of 270 nm and a band between 320 and 360 nm.10 The absorption peaks at a wavelength of 270 nm is attributed to the π–π* electronic transition of the isolated benzene ring connected to the imidazole. While the absorption band at 320–370 nm is attributed to the π–π* electronic transition of the conjugated phenanthro[9,10-d]imidazole. Both forms of molecules are CD silent in the solution (Fig. 1B) and suspension (not shown), but they have significant absorption at the film state. They showed a mirror bisignate Cotton effect (containing both positive and negative components) at a wavelength of 270, 320 and 360 nm at the film state, showing typical aggregation-induced circular dichroism (AICD).6 Since Ala attachments have no CD absorption at a wavelength longer than 270 nm,11 these peaks at the longer wavelength are assigned to the absorption of phenanthro[9,10-d]imidazole, suggesting that the helicity of the amino acid attachment has been successfully transferred to the conjugating system of phenanthro[9,10-d]imidazole. The mirror Cotton effects at the solid state imply that the helical sense of the molecule mainly depends on the configuration of the chiral inducer, Ala.12 Hydrogen bonds and van der Waals interactions help to guide the transfer of chirality from the amino acid attachments to the conjugated chromophores. The photoluminescence of the two forms of molecules is investigated in solution, in suspension and at the film state, respectively. As illustrated in Fig. 2, both forms of molecules give similar intensive fluorescence at a wavelength of 390 nm, which corresponds to the emission of imidazole. Compared with the reported emission of the PIM scaffold in solution,10 the fluorescence peak of PIM–Ala has a slight red shift due to the enhanced conjugation and strong hydrogen bonding which are caused by the amino acid attachments. More obvious red shifts appear in the suspension and the red shift becomes most intensive at 50% water content. Similarly, both forms of molecules also exhibit ∼10 nm red shift at the film state (Fig. 2C and D). The red shift is usually a sign of the occurrence of aggregation, suggesting that aggregates have been formed in the suspension and at the film state. Along with the obvious red shift, a slight enhancement in fluorescence emission also occurs in the suspension and the emission is most intensive at 50% water content, suggesting that both types of the molecules exhibit aggregation induced enhanced emission (AIEE).13 The photoluminescence of PIM–Ala is mainly from the contribution of imidazole and the phenyl group, which is directly connected to the C2 carbon atom of it. The phenyl group has a propeller arrangement with a nearly 90° dihedral angle with the imidazole plane,14 which can efficiently prevent the imidazole planes from stacking in a face to face manner and avoid the disturbance of ACQ upon aggregation. Meanwhile, the free rotation of the phenyl group is also restricted and therefore the related nonradioactive energy loss is inhibited. The collaborative effect of the above two factors determines that both forms of the molecules have intensive photoluminescence in solution and AIEE properties at the film state as well.
 |
| | Fig. 1 (A) The UV and (B) CD spectra of PIM-D-Ala and PIM-L-Ala measured in their THF solution and from the cast film. The concentration for UV and CD measurements: 1 × 10−4 M, and the concentration for preparing the cast film: 1.0 × 10−3 M. | |
 |
| | Fig. 2 PL spectra of PIM-D-Ala (A) and PIM-L-Ala (B) in THF/water mixtures with different water fractions (fw). PL spectra of PIM-D-Ala (C) and PIM-L-Ala (D) in THF solution and at the film state. Concentration: 1.0 × 10−5 M; the concentration for preparing the cast film: 1.0 × 10−3 M, λex: 270 nm. | |
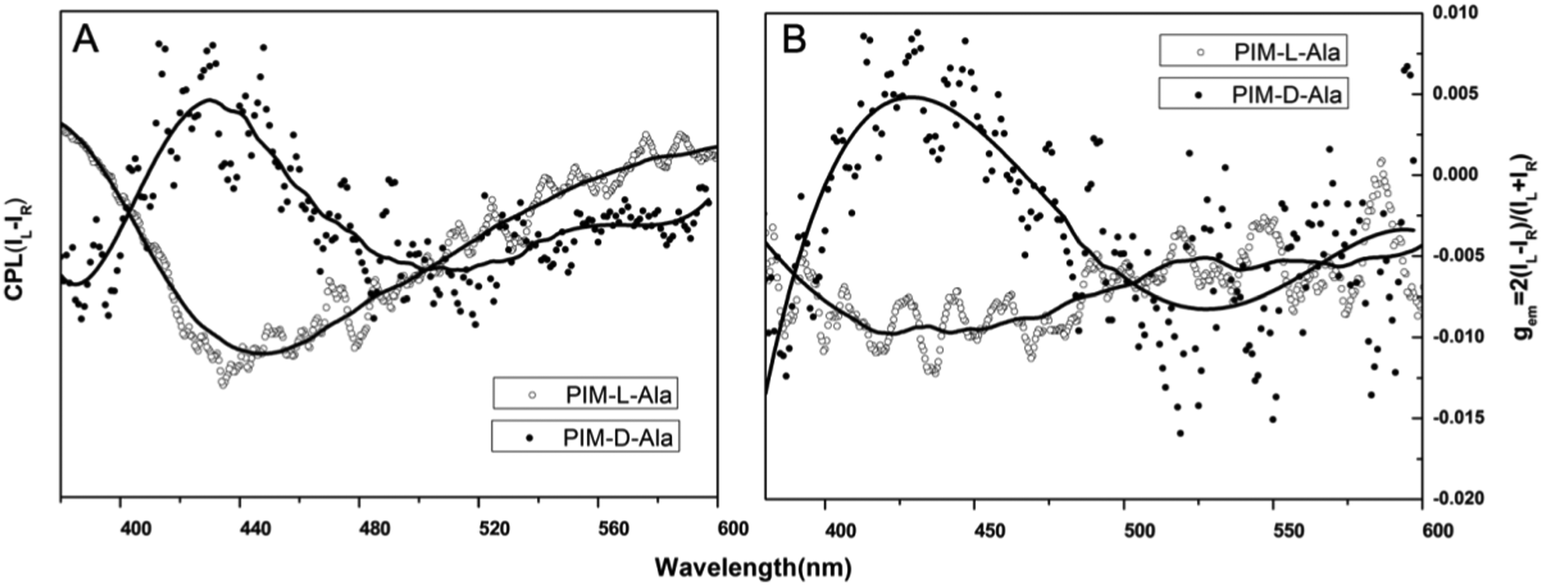
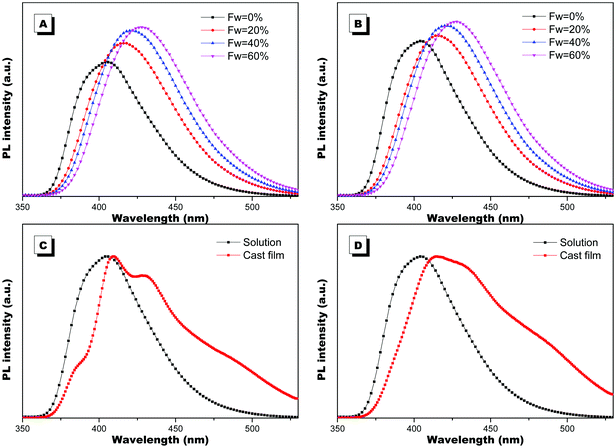
When the luminophores or the ensemble of luminophores of a molecule is chiral, it exhibits CPL properties.4 We explored the CPL properties of the two forms of molecules. Both forms of molecules give no CPL signal in solution, but exhibit intensive CPL signals at the film state, which is similar to the CD signal, suggesting that the CPL properties are only generated in the aggregation process. The CPL signals of the two forms of molecules have mirror signs, indicating that the CPL properties are mainly determined by the chiral inducer. PIM-L-Ala gives a maximum asymmetric factor gem of 0.01 and PIM-D-Ala has a maximum asymmetric factor gem of −0.005.4 Considering that the typical gem values for organic photoluminescent molecules are in the range of 10−2–10−6, and they even get worse at the film state because of the ACQ phenomenon, PIM-L-Ala and PIM-D-Ala both have excellent CPL performances (Fig. 3).
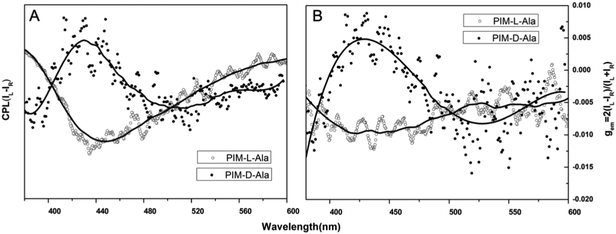 |
| | Fig. 3 Plots of CPL (A) and CPL dissymmetry factor gem (B) of PIM-D-Ala and PIM-L-Ala of the cast film on quartz prepared by the evaporation of their THF solution (concentration: 1.0 × 10−4 M, λex: ∼320 nm). | |
Thermal properties and hydrogen bonds of PIM–Ala
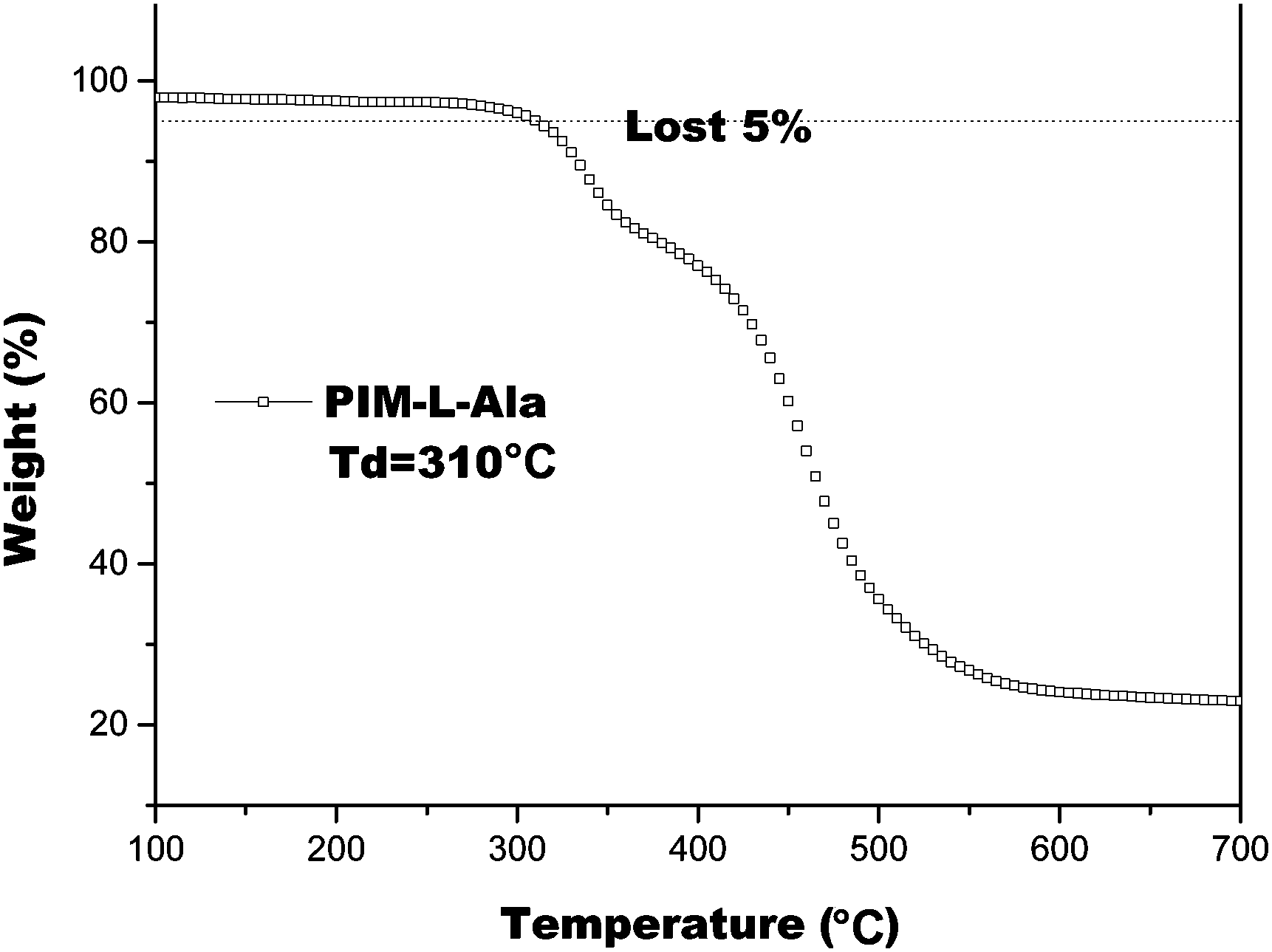
We also studied the thermal properties of PIM(Ala) by carrying out differential scanning calorimetry (DSC) and thermal gravimetric analysis (TGA) under a nitrogen atmosphere. The melting point (Tm) of PIM–Ala is higher than 200 °C (Fig. S9, ESI†), which is likely due to the intermolecular hydrogen bonds and C–H⋯π interactions that induce the condensed molecular packing of the molecules. The TGA thermogram shows that the decomposition temperature (Td) of PIM-L-Ala with a 5% weight loss is 310 °C. The high Tm and Td values indicate that the compound is thermally stable and an ideal candidate for electronic devices (Fig. 4).
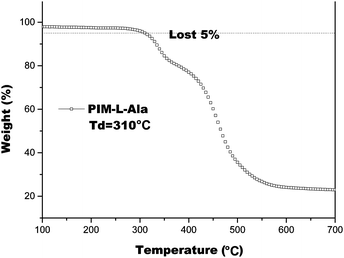 |
| | Fig. 4 TGA thermogram of PIM–Ala recorded under nitrogen at a heating rate of 20 °C min−1. | |
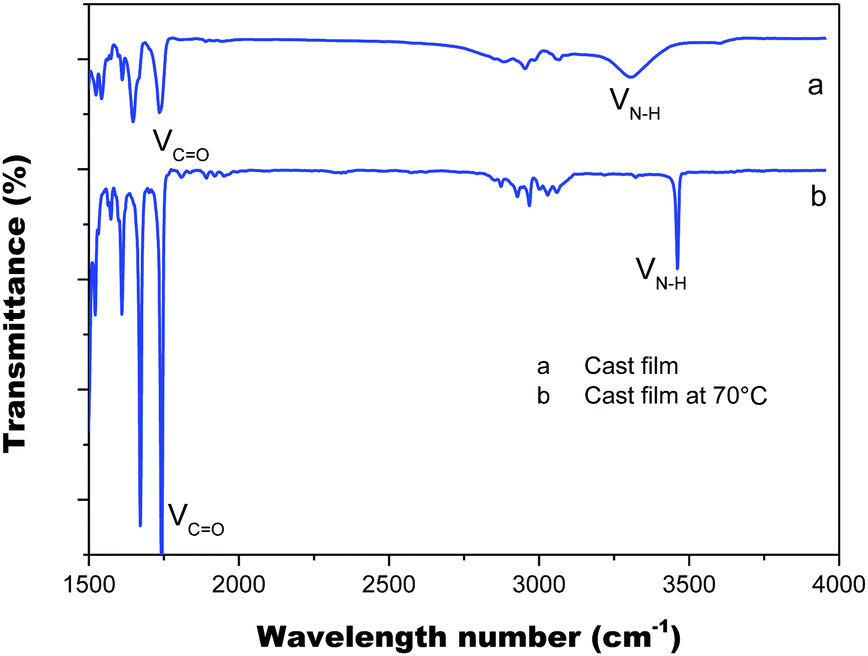
The intermolecular hydrogen bond of the amino acid attachments is critical for the excellent thermal properties of the molecules. It also plays an important role in transferring the chirality of the amino acids to the PIM scaffold and in stabilizing it. We thus also recorded the IR spectra of the cast film of PIM–Ala to study the intermolecular hydrogen bonds of the amino acid attachments. As shown in Fig. 5, under ambient conditions the cast film has an absorption at 1750 cm−1, which corresponds to the stretching mode of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, while there is also a broad absorption band at 3250–3400 cm−1 with medium intensity, which corresponds to the associated N–H stretching mode. When the cast film is heated to 70 °C, the broad band at 3250–3400 cm−1 vanished; instead a sharp peak appears at 3520 cm−1, which is assigned to the stretching mode of the unassociated –NH group. Meanwhile, the absorption intensity of CO also becomes much sharper. The characteristic broad band supports the formation of intermolecular hydrogen bonds via NH⋯NH and the hydrogen bonds are destroyed after thermal treatment. The considerable variation of the intensity of CO suggests a transition from associated to the unassociated state, implying that CO might also join in the formation of hydrogen bonds. Thus it is clear that the introduction of amino acids successfully brings in hydrogen bonds between molecules, which enhances the intermolecular interactions and helps to stabilize the helical conformation and helical assemblies of PIM–Ala.
O, while there is also a broad absorption band at 3250–3400 cm−1 with medium intensity, which corresponds to the associated N–H stretching mode. When the cast film is heated to 70 °C, the broad band at 3250–3400 cm−1 vanished; instead a sharp peak appears at 3520 cm−1, which is assigned to the stretching mode of the unassociated –NH group. Meanwhile, the absorption intensity of CO also becomes much sharper. The characteristic broad band supports the formation of intermolecular hydrogen bonds via NH⋯NH and the hydrogen bonds are destroyed after thermal treatment. The considerable variation of the intensity of CO suggests a transition from associated to the unassociated state, implying that CO might also join in the formation of hydrogen bonds. Thus it is clear that the introduction of amino acids successfully brings in hydrogen bonds between molecules, which enhances the intermolecular interactions and helps to stabilize the helical conformation and helical assemblies of PIM–Ala.
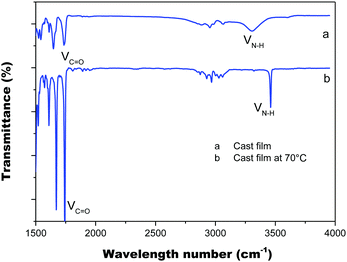 |
| | Fig. 5 FT-IR spectra of the PIM–Ala cast film from THF solution (0.5 mg mL−1) under ambient conditions (a) and after thermal treatment at 70 °C (b). | |
Helical luminescent fibers
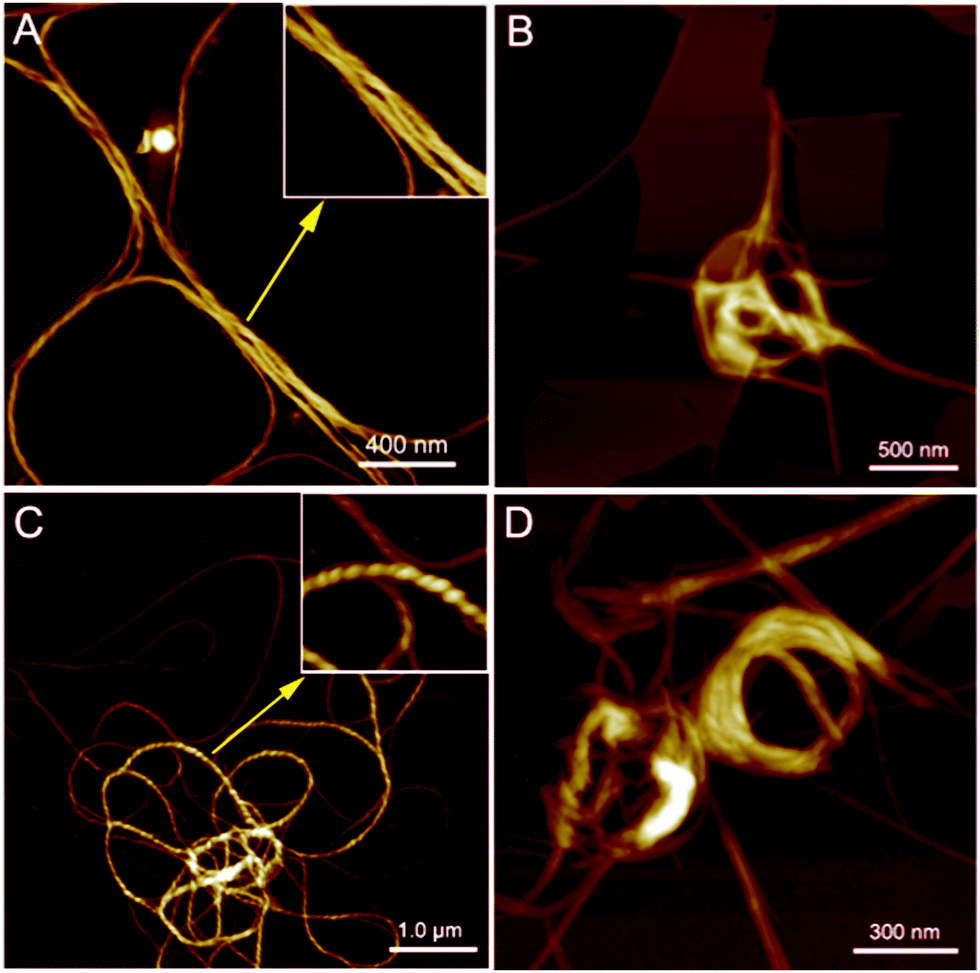
PIM–Ala exhibited significant AIEE, AICD and CPL properties at the aggregation state and therefore the aggregates they form are critical for understanding their optical properties. We then further scrutinize the aggregates formed by the molecules. As illustrated in Fig. 6A and C, helical fibers are formed by both forms of PIM–Ala upon the evaporation of their THF solution. The high resolution image inserted in Fig. 6A manifests the characteristic left-handedness of the helical fibers formed by PIM-D-Ala. Compared with the uniform fibers formed by PIM-D-Ala, PIM-L-Ala also forms left-handed fibers, but the fibers rotate more tightly and they are at multiple association levels. Accordingly, they exhibit a broad distribution of dimensions, with the thin fibers having their helical pitches beyond the observation of AFM and thick fibers having their characteristic left-handedness clearly shown in the inserted image in Fig. 6C. The helical pitches vary in a wide range from several tens of nanometers to several hundreds of nanometers. Both forms of PIM–Ala give the same handedness of supramolecular helical fibers. It is in sharp contrast to their mirror absorption in CD and CPL spectra. This divergence is due to the different levels of chirality reflected by the AFM, CD and CPL spectra. The resolution of AFM is good enough to reveal the supramolecular chirality, but it is unable to discern the molecular chirality of the molecule. So AFM only reveals a corner of the whole tip of the chiral assemblies. The CD and CPL spectra reflect the total enantiomeric excess of chirality, which includes the contribution from the molecular and supramolecular levels as well.15 The two forms of PIM–Ala have mirror CD and CPL spectra, but the same handed helical fibers, suggesting that each form of PIM–Ala forms more than one kind of chirality. A certain percentage of opposite chirality exists in the different levels of assemblies of each form of the molecule.
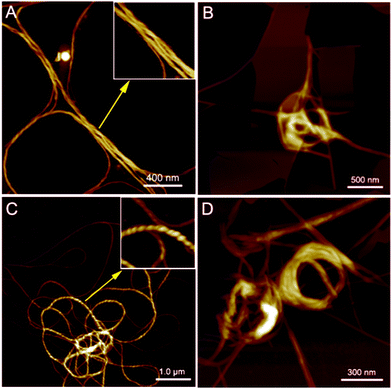 |
| | Fig. 6 AFM images of the helical assemblies formed by PIM-D-Ala (A) and PIM-L-Ala (C) upon the evaporation of their THF solution. Aggregates formed by PIM-D-Ala (B) and PIM-L-Ala (D) upon the evaporation of the THF/water mixture. The inserted images in A and C are the high-resolution images zoomed in from the corresponding labeled areas. Concentration of THF solution: 1.0 × 10−5 M. The water content is 50%. | |
In the presence of a poor solvent, both forms of the molecule adopt a similar transition trend in that they both take a more tight association to assemble into a loop-like morphology. They still keep the left-handedness, as can be seen from Fig. 6B and D. Besides the helical fibers, both forms of PIM also form thin films upon the evaporation of THF/water (shown in Fig. 6B and D). Images of the combined morphologies show that helical fibers usually occur at the edge of the films. It shows an important clue that the elementary fibers are wrapped up from the thin films. The formation of the film is similar to that of β-sheets formed by proteins,16 which are mainly stabilized by intermolecular hydrogen bonds between amino acids. The film gets elongated axially through a head-to-tail hydrogen bond and gets extended laterally in a side by side hydrogen bond via NH⋯NH. For PIM–Ala, the incorporation of Ala attachments allows the formation of intermolecular hydrogen bonds, which join the molecules in a similar fashion to those in proteins. Due to the existence of multiple chiral centers in the film, dislocations will occur when more molecules join in the packing, which are further amplified as a significant twist or even wrapped up to form helical fibers.
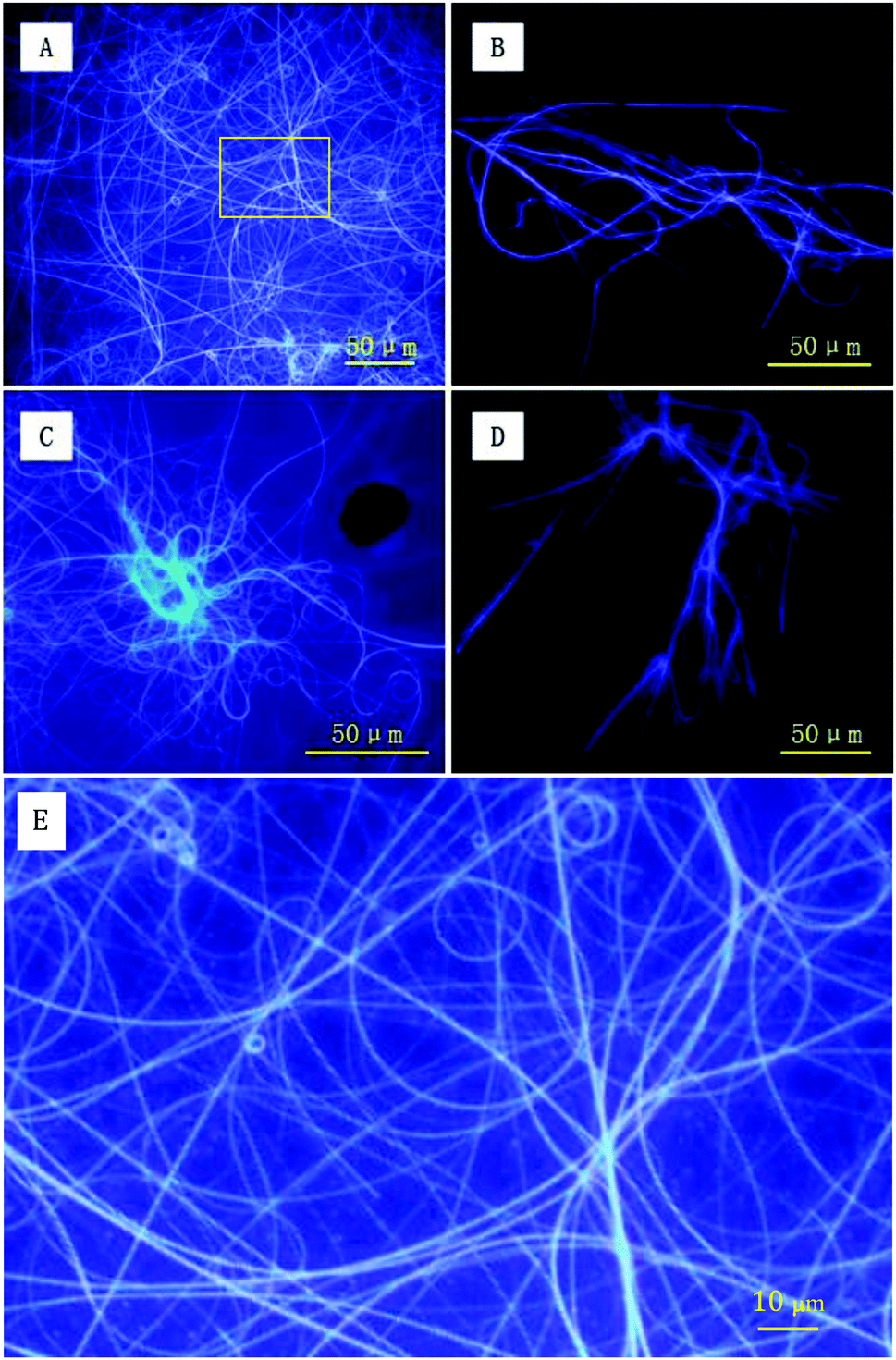
To directly capture the image of luminescent helical fibers, the assemblies were also explored using fluorescence microscopy. As shown in Fig. 7A and C, both forms of PIM–Ala form violet luminescent fibers upon the evaporation of THF. The helical fibers are several hundreds of microns in length and the violet luminescence of the fibers is consistent with the emission wavelength of the PL and CPL of the molecules, thus clearly showing the correlation between the PL and CPL properties. From the high resolution image in Fig. 7E, the helical fibers show a broad distribution of helical pitches varying from 2 μm to 5 μm. The helical fibers also show different handedness, which is complementary to nanofibers with dominant left-handedness revealed by AFM at the nanoscale. They cooperatively confirm our previous analysis that each form of PIM–Ala gives helical fibers with both handedness, but different dominant ones. The resolution of AFM allows it to discern the helical pitches of the elementary helical fibers, which are beyond the resolution of the fluorescence microscope. The fluorescence microscope can only reveal the helical pitches of fibers that are several microns long and at a much higher order of assembly. Thus the helical fibers observed under the fluorescence microscope are much longer and thicker than the elementary ones observed under the AFM microscope. Comparing the individual handedness of the helical fibers revealed by AFM and fluorescence microscopy, CD reflects statistical information of the excessive handedness of the chiral assemblies on all levels of molecular architectures, including single molecular chirality and higher orders of supramolecular assemblies as well. For the two forms of PIM–Ala, they self-assembled into helical fibers with both handedness, but with a different dominant handedness.
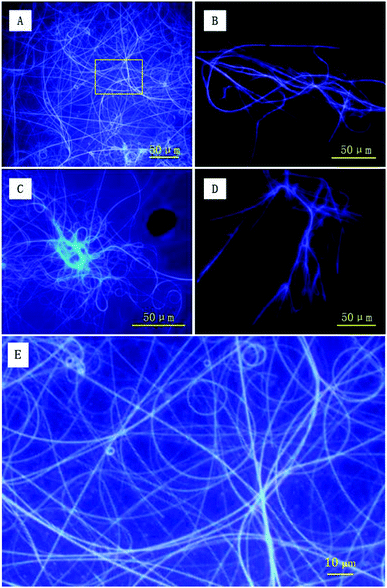 |
| | Fig. 7 Fluorescence images of PIM-D-Ala (A) and PIM-L-Ala (C) upon the evaporation of THF solution. Fluorescence images of PIM-D-Ala (B) and PIM-L-Ala (D) upon the evaporation of their THF/water solution. (E) Zoomed-in image of the labeled area in image A. Concentration: 1.0 × 10−3 M; water content: 80%. | |
AFM and fluorescence microscopy have different sample requirements and fibers of different dimensions are thus formed because of the different sample preparation methods required. For imaging samples using a fluorescence microscope, more molecules are required for sample preparation. The solution volume for deposition is generally several times that used for AFM sample preparation, and the concentration is around 10 times that used for AFM sample preparation. Accordingly, the assembled fibers for fluorescence imaging are formed with more molecules and are associated at higher levels of assembly. Therefore, they get much longer and thicker than that revealed by AFM. In terms of AFM imaging, it is only suitable for scanning very smooth samples by using a radium tiny tip of several nanometers. Because of the sample requirement of smoothness of AFM imaging, fluorescence samples are too rough to be scanned using AFM. It can be seen that the two imaging tools characterize the different levels of the assembled fibers formed by PIM–Ala. They cooperatively provide complementary information of the supramolecular assembling behaviors of the molecules, from the construction of elementary fibers at the nanoscale to supramolecular fibers at the microscale. The formation of different levels of fibers offers the possibility that the helical fibers can be tuned from the nanoscale to the microscale and this flexibility also makes this material have more broad applications in the field of optics and electronics.
Conclusion
In summary, we have provided a simple but effective strategy to fabricate helical luminescent fibers with CPL properties via self-assembly of violet-emitting phenanthroimidazole carrying chiral amino acid motifs. The chiral amino acid attachments are pivotal in transferring the chirality of amino acid attachment to the phenanthroimidazole conjugated scaffold and directing the self-assembling process of the molecules. The helical fibers can be tuned from the nanoscale to the microscale. The efficient fabrication of chiral nano/micro architectures offers many opportunities to develop novel functional materials which have versatile optical and electronic properties with promising applications in the nano/micro field. It is an area that requires further efforts to explore with respect to realizing the precise self-assembling control and function optimization.
Experimental section
Instruments and methods
1H and 13C NMR spectra were recorded on a VNMRS 400 (Varian, USA) NMR spectrometer, using CDCl3 as the solvent and tetramethylsilane (TMS) as the internal references. High-resolution mass spectra (HR-MS) were recorded using a Thermo Finnigan MAT TSQ 7000 spectrometer (USA). The mass spectrometer system was operated in the MALDI-TOF mode. UV spectra were recorded using a UV-2600 spectrometer (Shimadzu, Japan). CD spectra were recorded using a Chirascan spectrometer (Applied Photophysics, England). Fluorescence spectra were recorded using a F-7000 fluorescence spectrometer (Hitachi, Japan). CPL spectra were recorded using a CPL-200 instrument (JASCO, Japan) at room temperature. AFM images were captured using a Multimode VIII AFM instrument (Bruker, USA). DSC measurements were carried out on a NETZSCH DSC 200F3 instrument. TGA analysis was performed on a NETZSCH SA409PC thermogravimeter. IR spectra were recorded on a Spectrum Two FT-IR spectrophotometer (Perkin-Elmer, USA). Fluorescence images were captured using the fluorescence microscope DHG-9070A (Olympus, Japan).
Materials
Synthesis of 4-(1-phenyl-1H-phenanthro[9,10-d]imidazol-2-yl) benzoic acid (3).
The product was prepared by refluxing 9,10-phenanthrenequinone (2.08 g, 10 mmol), 4-carboxybenzaldehyde (1.50 g, 10 mmol), benzenamine (1.1 mL, 12 mmol), and ammonium acetate (9.41 g, 122 mmol) in glacial acetic acid (50 mL) for 24 h under an atmosphere of nitrogen. After cooling to room temperature, a pale yellow mixture was obtained and poured into a methanol solution under stirring. The separated solid was filtered off, washed with methanol, and dried to give a pale yellow solid. The solid was purified using column chromatography (CH2Cl2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :ethanol, 20:1) on silica gel. A white powder was finally obtained after it was stirred in refluxing ethanol, filtered, and dried in a vacuum. Yield: 3.2 g (77%). 1H NMR (DMSO-d6, 400 MHz), δ (TMS, ppm): 13.03 (s, 1H), 8.84 (d, J = 13.1 Hz, 2H), 8.69 (s, 1H), 8.20–7.41 (m, 12H), 7.29 (s, 1H), 7.06 (s, 1H). 13C NMR (DMSO-d6, 100 MHz), δ (TMS, ppm): 167.38, 150.05, 138.54, 137.28, 134.76, 132.4, 131.46, 131.24, 129.49, 129.30, 128.71, 128.38, 128.26, 127.26, 126.50, 126.06, 125.11, 124.28, 122.82, 122.64, 120.89, 60.35. HERMS (MALDI-TOF), m/z 415.14, (M+, calcd 414.45), anal. calcd for C28H18N2O2: C, 81.14; H, 4.38; N, 6.76; O, 7.72.
:ethanol, 20:1) on silica gel. A white powder was finally obtained after it was stirred in refluxing ethanol, filtered, and dried in a vacuum. Yield: 3.2 g (77%). 1H NMR (DMSO-d6, 400 MHz), δ (TMS, ppm): 13.03 (s, 1H), 8.84 (d, J = 13.1 Hz, 2H), 8.69 (s, 1H), 8.20–7.41 (m, 12H), 7.29 (s, 1H), 7.06 (s, 1H). 13C NMR (DMSO-d6, 100 MHz), δ (TMS, ppm): 167.38, 150.05, 138.54, 137.28, 134.76, 132.4, 131.46, 131.24, 129.49, 129.30, 128.71, 128.38, 128.26, 127.26, 126.50, 126.06, 125.11, 124.28, 122.82, 122.64, 120.89, 60.35. HERMS (MALDI-TOF), m/z 415.14, (M+, calcd 414.45), anal. calcd for C28H18N2O2: C, 81.14; H, 4.38; N, 6.76; O, 7.72.
Synthesis of 4-(1-phenyl-1H-phenanthro[9,10-d]imidazol-2-yl)benzoyl chloride (2).
To 2.07 g (5 mmol) of 4-(1-phenyl-1H-phenanthro[9,10-d] imidazol-2-yl)benzoic acid (3) was added 25 mL (50 mmol) of 2 M SOCl2 in CH2Cl2. After heating at reflux for 2 h, the clear solution was cooled and the solvent was removed under a reduced pressure. The remaining solid residue was dried in a vacuum to give 2.16 g of the crude acid chloride. 1H NMR (CDCl3, 400 MHz), δ (TMS, ppm): 8.86 (d, J = 7.8 Hz, 1H), 8.71 (dd, J = 26.2, 8.3 Hz, 2H), 7.93 (dd, J = 31.5, 20.0 Hz, 2H), 7.69 (ddt, J = 21.3, 14.1, 7.1 Hz, 7H), 7.57–7.41 (m, 3H), 7.26 (dd, J = 9.8, 5.1 Hz, 1H), 7.15 (d, J = 8.2 Hz, 1H). 13C NMR (CDCl3, 100 MHz), δ (TMS, ppm): 167.79, 148.27, 138.30, 137.63, 136.77, 132.60, 131.13, 130.44, 129.80, 129.45, 128.88, 128.47, 127.49, 126.64, 126.30, 126.08, 125.50, 124.18, 123.16, 122.72, 120.98, 29.68. HERMS (MALDI-TOF), m/z 433.90, (M+, calcd 432.10). Anal. calcd for C28H17ClN2O: C, 77.69; H, 3.96; N, 6.47; O, 3.70.
Synthesis of N-(3-oxobutan-2-yl)-4-(1-phenyl-1H-phenanthro[9,10-d]imidazol-2-yl)benzamide (1).
Into a 100 mL round-bottom flask were added 186 mg (1.22 mmol) of L-alanine methyl ester hydrochloride and 500 mg (1.11 mmol) of 4-(1-phenyl-1H-phenanthro[9,10-d]imidazol-2-yl) benzoyl chloride under nitrogen. A mixture of DCM (20 mL) and TEA (10 mL) was injected into the flask. The mixture was refluxed for 4 hours. After cooling, the mixture was then diluted with 100 mL of DCM, and the resultant solution was washed twice with dilute HCl solution and once with water. The organic layer was dried over 5 g of magnesium sulfate. After filtration of the solid and removal of the solvent, the crude product was purified on a silica gel column using a mixture of CHCl3/acetone (20:1 by volume) as the eluent. Evaporation of the solvents gave 0.45 g of the product as a white solid. 1H NMR (300 MHz; DMSO-d6; Me4Si) δ H [ppm]: 1.40–1.42 (d, 3H), 3.65 (s, 3H), 4.5 (m,1H) 7.10–7.12 (d, 1H), 7.33 (t, 1H), 7.54 (t, 1H), 7.65–7.78 (m, 9H), 7.85–7.87 (d, 2H) 8.76 (dd, J = 15.2, 8.2 Hz, 3H). 13C NMR (100 MHz, DMSO) δ: 173.08, 165.59, 149.63, 137.98, 136.59, 133.59, 132.92, 130.40, 129.10, 128.89, 128.64, 127.99, 127.74, 127.53, 127.27, 126.67, 126.60, 125.89, 124.50, 122.37, 122.06, 120.26, 51.91, 48.33, 40.07, 39.86, 39.65, 39.44, 39.23, 39.02, 38.82, 16.66. HERMS (MALDI-TOF), m/z 499.19, (M+, calcd 499.56). Anal. calcd for C32H25N3O3: C, 76.94; H, 5.04; N, 8.41; O, 9.61.
Acknowledgements
This work was partially supported by the National Natural Science Foundation of China (21574085), the Innovation Research foundation of Shenzhen (JCYJ20140509172609153), the National Natural Science Foundation of Guangdong Province (2016A030312002), the National Basic Research Program of China (973 program, 2013CB834701) and the Innovation and Technology Commission (ITC-CNERC14SC01).
Notes and references
-
(a) G. M. Whitesides and B. Grzybowski, Science, 2002, 295, 2418 CrossRef CAS PubMed;
(b) K. Tahara, H. Yamaga, E. Ghijsens, K. Inukai, J. Adisoejoso, M. O. Blunt, S. De Feyter and Y. Tobe, Nat. Chem., 2011, 3, 714 CrossRef CAS PubMed;
(c) T. Chen, W.-H. Yang, D. Wang and L.-J. Wan, Nat. Commun., 2013, 4, 1389 CrossRef PubMed;
(d) S. J. George, R. de Bruijin, Z. Tomovic, B. Van Averbeke, D. Beljionne, R. Lazzaroni, A. P. H. J. Schenning and E. W. Meijer, J. Am. Chem. Soc., 2012, 134, 17789 CrossRef CAS PubMed;
(e) N. Suzuki, M. Fujiki, R. Kimpinde-Kalunga and J. R. Koe, J. Am. Chem. Soc., 2013, 135, 13079 CrossRef PubMed;
(f) J. Yang, J. Huang, Q. Li and Z. Li, J. Mater. Chem. C, 2016, 4, 2663 RSC;
(g) W. Wu, R. Tang, Q. Li and Z. Li, Chem. Soc. Rev., 2015, 44, 3997 RSC;
(h) J. Mei, N. L. C. Leung, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, Chem. Rev., 2015, 115, 11718 CrossRef CAS PubMed.
- M. Liu, L. Zhang and T. Wang, Chem. Rev., 2015, 115, 7304 CrossRef CAS PubMed.
-
(a) M. Oda, H.-G. Nothofer, G. Lieser, U. Scherf and S. C. J. Mesker, Adv. Mater., 2000, 12, 362 CrossRef CAS;
(b) S. H. Chen, D. Katsis, A. W. Schmid, J. C. Mastrangelo, T. Tsutsui and T. N. Blanton, Nature, 1999, 397, 506 CrossRef CAS;
(c) H. Shi, B. M. Conger, D. Katsis and S. H. Chen, Mol. Cryst. Liq. Cryst., 1998, 24, 163 CrossRef CAS.
-
(a) J. Kumar, T. Nakashima and T. Kawai, J. Phys. Chem. Lett., 2015, 6, 3445 CrossRef CAS PubMed;
(b) E. M. Sanchez-Carnerero, A. R. Agarrabeitia, F. Moreno, B. L. Maroto, G. Muller, M. J. Ortiz and S. De La Moya, Chem. – Eur. J., 2015, 21, 13488 CrossRef CAS PubMed;
(c) J. C. Y. Ng, J. Liu, H. Su, Y. Hong, H. Li, J. W. Y. Lam, K. S. Wong and B. Z. Tang, J. Mater. Chem. C, 2014, 2, 78 RSC;
(d) H. Maeda, Y. Bando, K. Shimomura, I. Yamada, M. Naito, K. Nobusawa, H. Tsumatori and T. Kawai, J. Am. Chem. Soc., 2011, 133, 9266 CrossRef CAS PubMed;
(e) S. C. J. Meskers, E. Peeters, B. M. W. Langeveld-Voss and R. A. J. Janssen, Adv. Mater., 2000, 50, 3684 Search PubMed.
-
(a) J. P. Riehl, Chem. Rev., 1986, 86, 1 CrossRef CAS;
(b) F. S. Richardson and J. P. Riehl, Chem. Rev., 1977, 77, 6 CrossRef.
-
(a) H. Li, J. Chen, Y. Zhao, J. W. Lam, K. S. Wong, H. Wu, B. S. Li and B. Z. Tang, Mater. Horiz., 2014, 1, 518 RSC;
(b) H. Li, J. Cheng, H. Deng, E. Zhao, B. Shen, J. W. Y. Lam, C. Wong, K. S. Wu, B. S. Li and B. Z. Tang, J. Mater. Chem. C, 2015, 3, 2399 RSC;
(c) H. Li, J. Cheng, H. Deng, E. Zhao, B. Shen, J. W. Y. Lam, C. Wong, K. S. Wu, B. S. Li and B. Z. Tang, Sci. Rep., 2016, 6, 19277 CrossRef PubMed;
(d) J. C. Y. Ng, H. Li, Q. Yuan, J. Liu, C. Liu, X. Fan, B. S. Li and B. Z. Tang, J. Mater. Chem. C, 2014, 2, 4615 RSC.
-
(a) C.-J. Kuo, T.-Y. Li, C.-C. Lien, C.-H. Liu, F.-I. Wu and M.-J. Huang, J. Mater. Chem., 2009, 19, 1865 RSC;
(b) Y. Yuan, D. Li, X. Zhang, X. Zhao, Y. Liu, J. Zhang and Y. Wang, New J. Chem., 2011, 35, 1534 RSC;
(c) Y. Zhang, S. Lai, Q.-X. Tong, M.-F. Lo, T.-W. Ng, M.-Y. Chan, Z.-C. Wen, J. He, K.-S. Jeff, X.-L. Tang, W. M. Liu, C. C. Ko, P. Wang and C. S. Lee, Chem. Mater., 2012, 24, 61 CrossRef;
(d) B. Zhao, Y. Fang, Y. Xu, Q. Deng, T. Liu, W. Kan and L. Wang, Tetrahedron Lett., 2016, 57, 1825 CrossRef CAS.
-
(a) B. S. Li, K. K. L. Cheuk, D. Yang, J. W. Y. Lam, L. J. Wan, C. Bai and B. Z. Tang, Macromolecules, 2003, 36, 5447 CrossRef CAS;
(b) B. S. Li, K. K. L. Cheuk, L. Ling, J. Chen, L. X. Xiao, C. Bai and B. Z. Tang, Macromolecules, 2003, 36, 77 CrossRef CAS.
- H. Huang, Y. Wang, S. Zhuang, X. Yang, L. Wang and C. Yang, J. Phys. Chem. C, 2012, 116, 19458 CAS.
- Z. Wang, P. Lu, S. Chen, Z. Gao, F. Shen, W. Zhang and Y. Xu, J. Mater. Chem., 2011, 21, 5451 RSC.
- E. Molteni, G. Onida and G. Tiana, J. Phys. Chem. B, 2015, 119, 4803 CrossRef CAS PubMed.
- K. Watanable, H. Lida and K. Akagi, Adv. Mater., 2012, 24, 6451 CrossRef PubMed.
- M. Haeussler, A. J. Qin and B. Z. Tang, Polymer, 2007, 48, 6181 CrossRef CAS.
- Y. Zhang, S. Lai, Q.-X. Tong, M.-Y. Chan, T.-W. Ng, Z.-C. Wen, G. Q. Zang, S.-T. Lee, H.-L. Kwong and C. S. Lee, J. Mater. Chem., 2011, 21, 8206 RSC.
-
(a) A. Satrijo, S. C. J. Meskers and T. M. Swager, J. Am. Chem. Soc., 2006, 128, 9030 CrossRef CAS PubMed;
(b) K. Watanabe, I. Osaka, S. Yorozuya and K. Akagi, Chem. Mater., 2012, 24, 1011 CrossRef CAS;
(c)
T. Taniguchi and T. Usuki, Circular Dichoism Spectroscopy. Supramolecular Chemistry: From Molecules to Nanomaterials, John Wiley & Sons Ltd, New York, 2012 Search PubMed;
(d) G. Gottarelli, S. Lena, S. Masiero, S. Pieraccini and G. P. Spada, Chirality, 2008, 20, 471 CrossRef CAS PubMed.
-
(a) B. S. Li and C. Goh, Micron, 2010, 41, 227 CrossRef CAS PubMed;
(b) B. S. Li, S. Kang, K. K. C. Cheuk, L. Wan, L. Ling, C. Bai and B. Z. Tang, Langmuir, 2001, 20, 7598 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental section, 1H, 13C NMR spectra (Fig. S1–S8). DSC spectrum (Fig. S9). See DOI: 10.1039/c6qm00120c |
|
| This journal is © the Partner Organisations 2017 |
Click here to see how this site uses Cookies. View our privacy policy here.