
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Self-healing alginate–gelatin biohydrogels based on dynamic covalent chemistry: elucidation of key parameters
Asja
Pettignano
abc,
Marleen
Häring
a,
Luca
Bernardi
 c,
Nathalie
Tanchoux
b,
Françoise
Quignard
b and
David
Díaz Díaz
*ad
c,
Nathalie
Tanchoux
b,
Françoise
Quignard
b and
David
Díaz Díaz
*ad
aInstitute of Organic Chemistry, University of Regensburg, Universitätstrasse. 31, D-93040 Regensburg, Germany. E-mail: David.Diaz@chemie.uni-regensburg.de; Fax: +49 941 943-4121; Tel: +49 941 943-4373
bInstitut Charles Gerhardt, UMR 5253 CNRS/UM/ENSCM, Matériaux Avancés pour la Catalyse et la Santé, ENSCM, 8 rue Ecole Normale, 34296 Montpellier Cedex 5, France
cDepartment of Industrial Chemistry “Toso Montanari”, School of Science, Alma Mater Studiorum – University of Bologna, V. Risorgimento 4, 40136 Bologna, Italy
dInstitute of Advanced Chemistry of Catalonia (IQAC-CSIC), Jordi Girona 18-26, Barcelona 08034, Spain
First published on 29th July 2016
Abstract
In this work, gelatin was crosslinked with oxidized alginate in the presence of borax yielding a hybrid biohydrogel system with the ability to self-repair upon mechanical damage. A judicious balance between concentration, stoichiometric ratio of the two biopolymers and gelatin source was found necessary to achieve optimal self-healing properties of these biohydrogels. The pH was found to have a major influence on the reconstruction of the damaged hydrogel interface, confirming that the dynamic Schiff base linkages between the amine groups of gelatin and the aldehyde groups of oxidized alginate play a fundamental role on the healing process of the hybrid gels.
Introduction
Natural tissues have always been a great source of inspiration for material scientists and a striking evidence of this winning combination can be found in the growing research field on biomimetic materials. One of the most impressive properties of biological materials is their ability to regenerate and heal after the infliction of a physical damage. The origin of some well-known natural self-healing processes (e.g., merging of broken bones, cell-wall recovery upon deformation of wood) can be found in the presence of sacrificial bonds between molecules that, by breaking and reforming, allow both a better adaptation of the biomaterial to external conditions and the reparation of damaged material.1–4 Similarly, an emerging research area on the fabrication of self-healable gel networks relies on the concept of dynamic covalent chemistry.5–7 In contrast to covalently crosslinked gels, whose bonds are too stable to allow exchange reactions, gels based on dynamic bonds are able to reform bonds around a damaged zone, allowing the restoration of the original properties.7 This property finds a great value in biomedicine where new synthetic biomaterials with ability to autonomously repair mechanical damages will be particularly important to improve the performance and extend the lifetime of implants.8 Moreover, self-healing hydrogels that can be injected (thixotropic) are excellent candidates for cell therapy and drug delivery applications.9–12Considering the importance of an in-depth understanding of the self-healing properties of functional materials in order to maximize their range of possible applications, the present study aims to accurately investigate the self-repair properties of crosslinked gelatin-based hydrogels. Gelatin is a biodegradable protein, produced by hydrolytic degradation of collagen, widely used in different biomedical applications because of its ability to promote cell adhesion and proliferation.13–15 On the other hand, the uncontrollable partial renaturation of collagen-like triple helices of gelatin in aqueous media imposes its crosslinking in order to maintain the gel structure in the biological systems.16 An inexpensive, biodegradable and biocompatible crosslinker is exemplified by alginate, a polysaccharide derived from seaweeds, in its partial oxidized form.17 The formation of covalent bonds between the free amino groups of lysine and hydroxylysine residues of gelatin with the accessible aldehyde functions of oxidized alginate results in the formation of highly stable yet physiologically degradable gel networks (i.e., through Schiff base linkages) with improved cell adhesion, biocompatibility and biodegradability properties.18 The biomedical utility of this hybrid crosslinked polymer is well-known and several reports have demonstrated its potential as both drugs19–21 and cell carrier22–26 as well as wound healing implants.27,28 To the best of our knowledge, despite the usability of this system in different biomedical applications, proper characterization of the self-healing character of the in situ generated biohydrogel has not yet been reported.9,29,30 Other self-healing hydrogels based on reversible Schiff base connections have also been recently reported by other groups, which demonstrates an increasing scientific interest in this type of materials.10,11,29,30–40
In this work, alginate was oxidized to the corresponding dialdehyde and reacted with gelatin in the presence of sodium tetraborate (borax), yielding hybrid hydrogels with self-healing properties due to reversible Schiff base formation. The aim of this study was to provide a focused investigation to elucidate the key parameters with major influence on the healing process in these hybrid systems. The results derived from this study will be useful for the development of a more targeted research towards the practical uses of alginate–gelatin hydrogel systems.
Experimental section
Materials
Alginic acid sodium salt (low viscosity, 15–20 cP for 1% in H2O), gelatin from porcine skin (type A, gel strength ∼300 g Bloom), gelatin from bovine skin (type B, gel strength ∼225 g Bloom) and sodium tetraborate decahydrate (borax) were purchased from Sigma-Aldrich. Dialysis tubing cellulose membranes (MWCO 14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000, LOT 3110) were purchased from Sigma-Aldrich. All other reagents used were of analytical or equivalent grade. Milli-Q-purified water was used for the preparation of hydrogels.
000, LOT 3110) were purchased from Sigma-Aldrich. All other reagents used were of analytical or equivalent grade. Milli-Q-purified water was used for the preparation of hydrogels.
Periodate oxidation of sodium alginate
Oxidized alginate was prepared by following a previously reported procedure with slight modifications.41 Sodium alginate (80 mmol on monomer unit) was dissolved in deionized water (320 mL) and a solution of NaIO4 (40 mmol in 80 mL of deionized water) was added. The mixture was stirred at room temperature in the dark for 18 h. The degree of oxidation was obtained by determining the concentration of unconsumed periodate. Iodometric titration was performed on three aliquots (2.5 mL each) of the reaction mixture and the average value was reported. At the end of the reaction, ethylene glycol (20 mL) was added and stirred for 1 h to quench any unreacted periodate. The solution was dialyzed against distilled water for 3 days with at least 4 changes of water and freeze-dried.Preparation of oxidized alginate–gelatin hydrogels
The desired amount of the two biopolymers was weighted and separately dissolved in 0.1 M borax aqueous solution to obtain a final concentration of 5, 10 or 15% (w/v). Gelatin was heated up to 37 °C to be successfully dissolved. The alginate solution was transferred to the cylindrical polypropylene mould (i.e., 2–5 mL syringe of 12.5 mm in diameter and with the tip being cut-off) and the gelatin solution was subsequently added. The solution mixture was sonicated for ca. 2 min in an ultrasonic bath at room temperature (RT) and stirred by means of a rotating stirrer for at least 16 h to ensure good homogenization during gelation.Characterization methods
Fourier transform infrared (FT-IR) spectra were recorded at room temperature using an Excalibur FTS 3000 FT-IR spectrometer (Biorad) equipped with a single reflection ATR (attenuated total reflection) accessory (Golden Gate, Diamond). Wild Makroskop M420 optical microscope equipped with a Canon shot A640 digital camera was used for collecting pictures of the self-healing process. Ultrasound bath (USC200TH, VWR™) was used for homogenization of solution mixtures. Oscillatory rheology was performed with an AR 2000 Advanced rheometer (TA Instruments). A 1000 µm gap setting and 25 °C were used for the measurements in a plain-plate geometry (40 mm, stainless steel). Rheological measurements involved dynamic strain sweep (DSS) (moduli vs. strain), dynamic frequency sweep (DFS) (moduli vs. frequency) and dynamic time sweep (moduli vs. time) measurements. The self-healing behavior of model gels was investigated by several cycles of a 3-steps loop experiment involving: (1) application of a low shear strain (5% strain, 1 Hz, 5 min; gel state, G′ (storage modulus) > G″ (loss modulus)); (2) increase of the shear strain until the gel fractures (600% strain, 1 Hz, 2 min; viscous material, G′ < G″), and (3) return at the same rate to the initial strain % value (5% strain, 1 Hz, 10 min; recovered gel phase, G′ > G″).Results and discussion
Following the general procedure described in the literature,41 sodium alginate was first partially oxidized to the corresponding dialdehyde by employing sodium metaperiodate as oxidizing agent. The reaction consists in the specific oxidation of the free hydroxyl groups at C2 and C3 of the alginate monomeric units, leading to the formation of the corresponding dialdehyde (OxA) in 25% yield.41 A degree of oxidation of ca. 47% was determined by iodometric titration and the presence of aldehyde groups was confirmed by FT-IR analysis, showing the appearance of the typical weak symmetric vibration of aldehyde at 1735 cm−1 (Fig. 1). OxA was subsequently crosslinked with type B gelatin (GB) in the presence of 0.1 M borax, leading to the in situ formation of the expected hydrogel. Under these conditions, the crosslinking process is ascribed to Schiff base formation between the free amino groups of lysine or hydroxylysine of gelatin and the alginate aldehyde groups, assisted by the borate–diol complexation between borax and the hydroxyl groups of alginate.26 The sol-to-gel phase transition was completed within 5 min (confirmed by the absence of flow upon turning the container upside down) and was accompanied by a progressive darkening of the solution, from pale yellow to dark-orange, due to the formation of aldimine linkages (–CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N–) (Scheme 1).
N–) (Scheme 1).
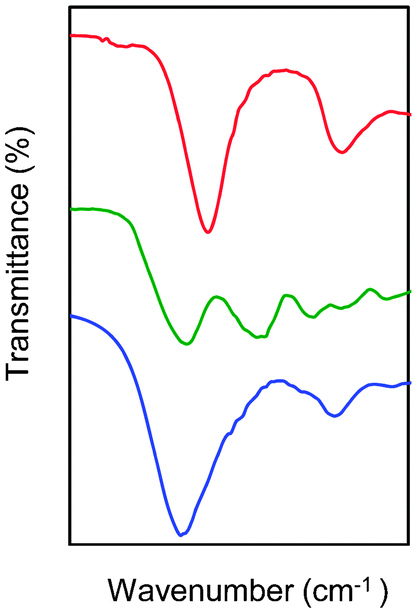 | ||
| Fig. 1 FT-IR spectra of oxidized alginate (top line, red), unmodified type B gelatin (middle line, green) and crosslinked OxA–GB15 hydrogel (bottom line, blue) as representative example. OxA–GB15 refers to crosslinked material prepared using 15% w/w solutions of the two biopolymers. | ||
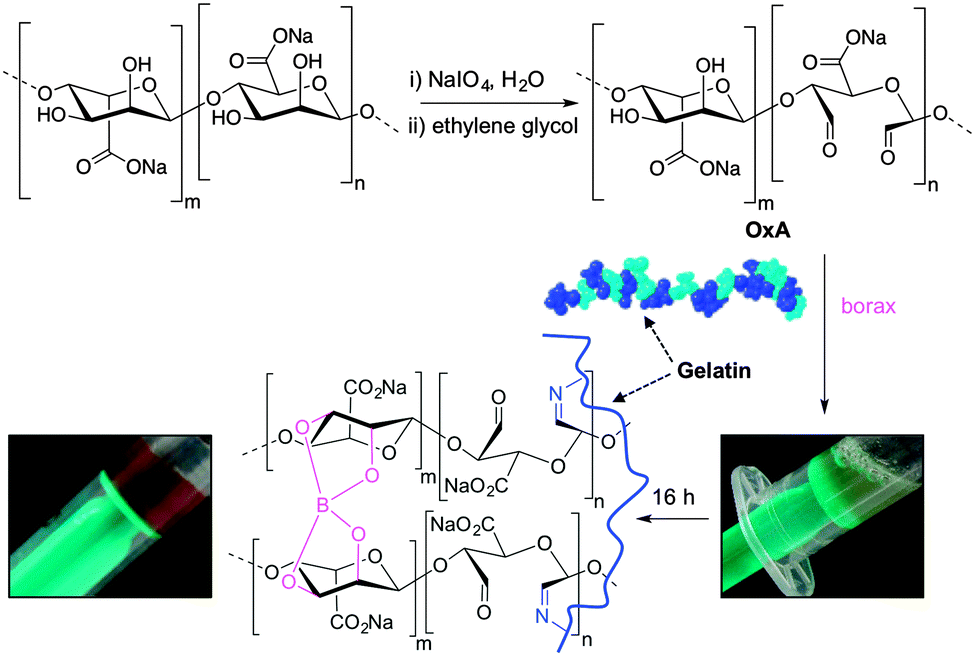 | ||
| Scheme 1 Schematic representation of the crosslinking of OxA and gelatin in the presence of borax. Inset pictures show the darkening and immobilization of the starting solution during the sol-to-gel transition. | ||
In agreement with previous studies,42 the crosslinking reaction was confirmed by comparative FT-IR analysis of OxA, GB and the obtained hydrogels (Fig. 1). The appearance of a new peak at 1636 cm−1, compared to the spectra of OxA and GB, is consistent with efficient Schiff base formation.
In order to study the effect of crosslinking on the self-healing capacity of gelatin gels, monoliths with different concentrations and compositions were obtained and their properties compared to those of unmodified gelatin as reference. Monolithic hydrogels were obtained by dissolving the desired amounts of OxA and GB in a borax solution and mixing them into a syringe with the tip being cut-off. These mixtures were homogenized during the gelation process by means of a rotating peg. A preliminary screening to determine the optimal stoichiometric ratio for the desired self-healing property was performed using 15% w/w solutions of OxA and GB. Three samples with different OxA:GB weight ratios (i.e., 1:1, 2:1, 1:2) were obtained and cut into several pieces (Fig. 2a). The pieces were subsequently reconnected by applying gentle pressure and left resting for 7 days (additional experiments indicated that this was an appropriate time to guarantee complete healing of the material, see below). Although all rebuilt monoliths were stable upon hanging, only the one prepared at 2:1 w/w OxA:GB showed both better healing at the cut interfaces and higher resistance to stretch (Fig. 2b), being eventually ruptured at a virgin area rather than at the reattached interfaces. Our observations were in good agreement with previous reports showing the increase of crosslinking degree with increased amounts of oxidized alginate.43 The self-healing process was also confirmed by rheological measurements (see below).
 | ||
| Fig. 2 (a) Monolith OxA–GB hydrogel cut in three pieces (OxA:GB = 2:1 w/w). The gel was prepared using 15% w/v solutions of OxA and GB and (b) severe stretching of the monolith after reconnecting the pieces (optimal healing time = 7 days). | ||
Thus, the 2:1 OxA:GB ratio was selected for further experiments. Subsequently, we studied the influence of concentration on the self-healing of OxA–GB hydrogels, by employing 5, 10 and 15% w/w solutions of the two biopolymers. The results indicated that concentration had a striking effect on the gel formation showing no gel transition at 5% w/w and remarkable differences on the gel properties at 10% and 15% w/w. The use of a 10% w/w concentration (OxA–GB10) led to soft, almost flat hydrogels (Fig. 3a), while the use of 15% w/w (OxA–GB15) allowed the preparation of highly regular and self-standing cylindrical monoliths (Fig. 3b). In order to demonstrate the possibility of fine-tuning the properties of OxA–GB hydrogels, both concentrations were used for additional comparative studies.
 | ||
| Fig. 3 Visual appearance of freshly prepared (a) OxA–GB10 and (b) OxA–GB15 monolithic hydrogels. | ||
To verify the optimal healing time, OxA–GB10 and OxA–GB15 gels were prepared in the absence and in the presence of Direct Blue 1 (the dye was added in the gelation step). Alternating dyed and non-dyed pieces were fused together by simply joining together the fresh-cut surfaces. Both gels were immediately able to form a stable self-supporting bridge (Fig. 4a and b). The progress of the dye diffusion through the gel networks was followed with time, showing a gradual darkening of the non-dyed pieces, which was almost complete after 6 days. The dynamic bonds between OxA and GB allow the continuous movement and re-crosslinking of the polymer chains, favoring the molecular reconstruction of the hydrogel network and the consequent diffusion of the dye through the cut areas. In general, this process was accompanied by a gradual smoothing of the external surface of the monoliths. It is worth mentioning that although the healed scars were still visible after healing especially in the OxA–GB15 hydrogel, due to imperfect contact between the pieces, the absence of clear interfaces was evident upon examination of the inner part of the rebuilt monolith (Fig. 4c).
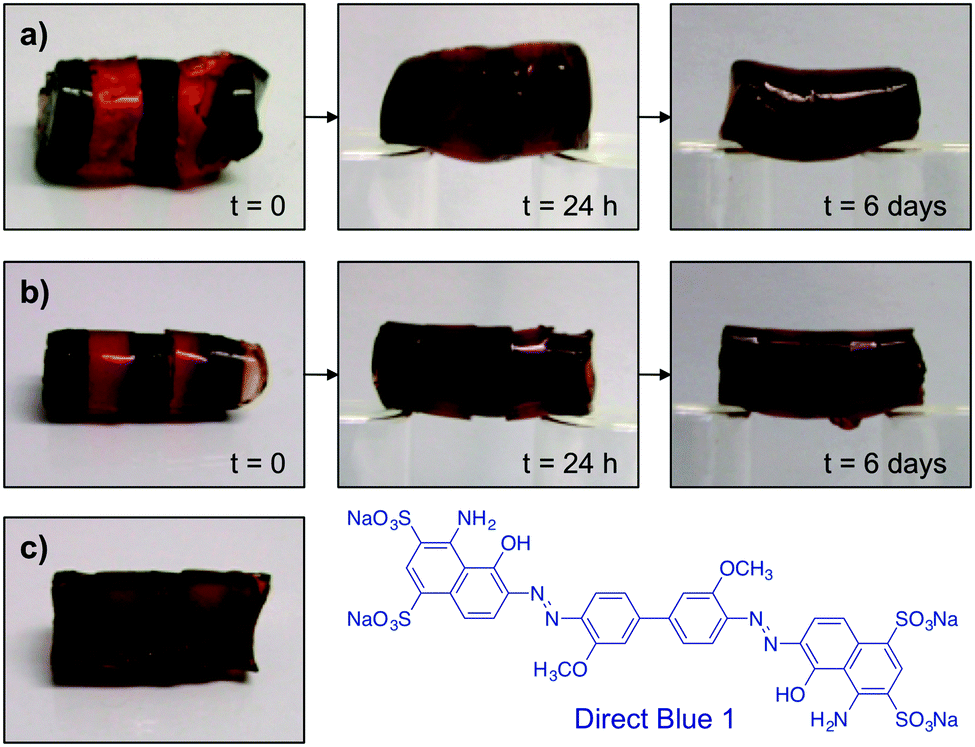 | ||
| Fig. 4 Visualization of the self-healing progress with time using monolithic hydrogels made of alternating dyed and non-dyed pieces: (a) OxA–GB10 hydrogel, (b) OxA–GB15 hydrogel, (c) internal cross-section of OxA–GB15 after self-healing. Direct Blue 1 was used as dye in this experiment. | ||
At this point, the resistance of OxA–GB10 and OxA–GB15 healed gels to different types of mechanical stresses (e.g., bridge formation, stretching, bending and hanging the cut pieces) was tested maintaining 6 days as the minimum healing time (Fig. 5). Although both samples performed satisfactorily, OxA–GB10 sample showed a much higher elastic elongation than OxA–GB15.
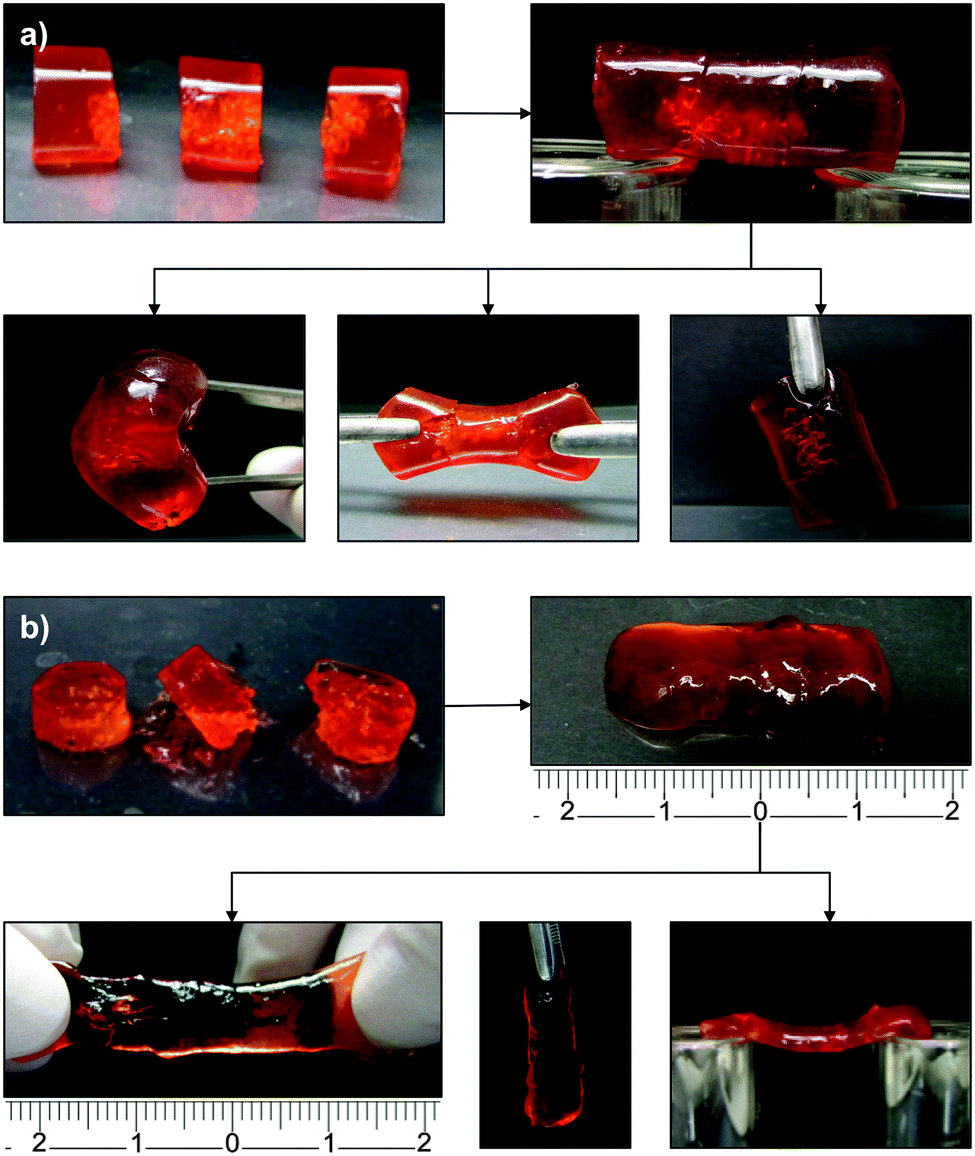 | ||
| Fig. 5 Resistance of (a) OxA–GB10 and (b) OxA–GB15 self-healed hydrogels to different types of mechanical stresses. | ||
To confirm the effect of the protein source on the self-healing ability of these hybrids, a new hydrogel (OxA–GA15) was prepared for comparison using 15% w/w solution of type A gelatin (GA) and OxA. Interestingly, after keeping in contact the pieces for 9 days only partial healing was observed (Fig. 6). This result indicates the superiority of GB for preparing OxA–gelatin hydrogels with self-healing ability. A possible explanation for this major difference might be related to the different processes used to isolated the protein in each case. While GA is derived from acid-cured tissue and has an isoelectric point (pI) of 7.0–9.0, GB is obtained from lime-cured tissue and has an pI of 4.7–5.2.44 When the pH of the medium is close to the pI, a quick aggregation of protein molecules takes place reducing the interaction with water molecules,45 which is important for the stabilization of the gel network. The pH of alginate solution in deionized water is about 7.2, which may favor the solvation of GB aggregates (compared to GA) inducing the formation of more stable hybrid networks. At the working pH, the positively charged NH3+ groups of GA can lead to partial unavailability of the amino groups of the proteins slowing down the dynamic bond formation and therefore the kinetics of the self-healing process.46
 | ||
| Fig. 6 Photographs showing (a) OxA–GA15 hydrogel freshly cut into several pieces and (b) hanging of the gel after 9 days of healing. | ||
In order to confirm that reversible Schiff base formation is indeed involved in the healing process, freshly cut OxA–GB15 hydrogels were subjected to different pH values before testing the self-healing ability. As shown in Fig. 7, monolithic hydrogels were cut into two pieces and immersed in a 0.1 M NaOH solution or in a 0.1 M HCl solution for 5 min. After this time, the two blocks were put back in contact for 48 h. The basic solution afforded the reconstruction of the hydrogel, which could be hung without visible cracks (Fig. 7a). This behavior was attributed to the favorable regeneration of the Schiff base linkage at basic pH. In sharp contrast, the hydrogel completely lose its self-healing ability after acidic treatment due to the limited Schiff base formation at low pH (Fig. 7b).
 | ||
| Fig. 7 Influence of pH on the self-healing ability of OxA–GB15: (a) pH 13.0 and (b) pH 1.26. Immersion time in the corresponding solutions = 5 min. | ||
The importance of aldehyde crosslinking for the self-healing phenomenon was demonstrated using unmodified GB as negative control. Similarly to the previous experiment, the monolithic GB hydrogel was cut into pieces and put back in contact applying gentle pressure. Although the material showed surface sticky properties, no self-healing was achieved after 8 days. The rebuilt pieces disintegrated into its original components under minor stretching showing a totally flat interface between the pieces that were in contact (Fig. 8). Interestingly, unmodified GB gel also showed the growth of mold after ca. 1 week, while the crosslinked hydrogels presented no signals of biodegradation after 1 month. This difference could also be due to the antiseptic properties provided by borax solutions.28
 | ||
| Fig. 8 Absence of self-healing ability in a GB monolithic hydrogel (10% w/v) after cutting and reconnecting the pieces. Visible white spots (right) correspond to the growth of mold within 1 week. | ||
A further comparison between the crosslinked and pristine gelatin was performed by punching a hole of ca. 3 mm × 3 mm in a gel film made of OxA–GB10 and in its gelatin control. While a hole made in an unmodified gelatin gel remained unchanged over time, the one in OxA–GB10 disappeared in 24 h (Fig. 9).
 | ||
| Fig. 9 Self-healing of OxA–GB10 hydrogel after making a small hole in the bulk material. | ||
The dynamic of this behavior could also be observed under an optical microscope by making a scratch on the gel surface and following its evolution with time. The damaged area in the gelatin control remained unchanged whereas the fracture in the OxA–GB10 sample practically vanished after 24 h. In good agreement with the pH-controlled equilibrium of the imine formation, the addition of one drop of 0.1 M NaOH on the damaged region accelerated the healing process leading to a reconstructed surface in ca. 5 min (Fig. 10a), while it had no effect on the control gel (Fig. 10b).
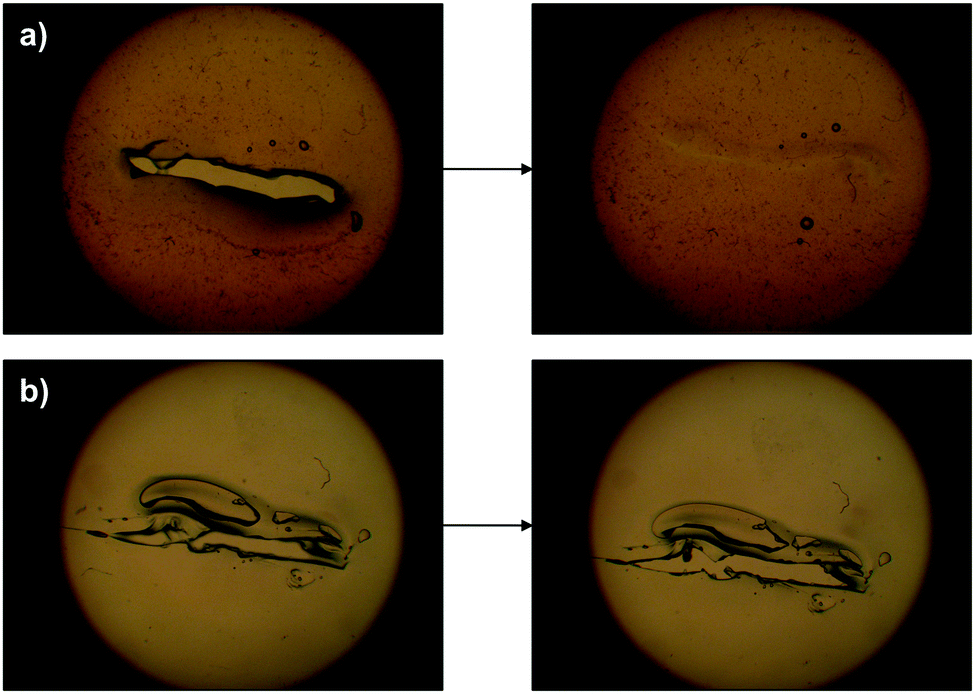 | ||
| Fig. 10 (a) Optical microscopy images showing the healing of a fractured OxA–GB10 gel sample. The process is accelerated by the addition of aqueous NaOH. (b) No healing was observed under the same conditions for the control GB sample. | ||
As the injectability of hydrogels is of major importance for numerous biomedical applications, this property was also recorded for the more rigid OxA–GB15 hydrogel. After complete gel formation, the gel sample was placed into a 5 mL syringe and continuously ejected through a 21-gauge needle (Fig. 11a). Due to shear stress during injection, the gel successfully passed through the needle without clogging, proving its thixotropic nature. The obtained filaments can be successfully reshaped into a disc by simple compression in a cylindrical mould for ca. 2 h (Fig. 11b).
 | ||
| Fig. 11 (a) Injectability property of OxA–GB15 and (b) reshape of the material as a monolithic disc using a cylindrical mould. | ||
Finally, the thixotropic behavior of crosslinked gels was also investigated by a rheological loop test. Based on preliminary dynamic strain sweep experiments, continuous time sweep measurements were performed to monitor the elastic response of the samples through alternating stress amplitudes of oscillatory forces and constant frequency. The loop test consisted in 3 steps: (1) application 5% strain at 1 Hz frequency for 5 min. During this step the gel nature of the sample was characterized by a G′ (storage modulus) value one order of magnitude higher than the G″ (loss modulus); (2) increase of the shear strain to 600% to ensure the rupture of the gel (G′ < G″) and maintained for 2 min; (3) reduction at the same rate to the initial 5% strain and maintained for 10 min to stabilize the recovered gel network (G′ > G″). The loop was repeated at least two times for all the samples. As expected, in contrast to the sharp and irreversible decrease of the moduli (ca. one order of magnitude) observed for the gelatin control (Fig. 12a), OxA–GB10 hybrid gel recovered more than 80% of the original moduli within few minutes, showing the same behavior through multiple cycles (Fig. 12b). These results correlate the observed macroscopic self-healing of the hybrid gel with the effective chain diffusion to the damages sites at the molecular level, providing the restoration of the overall consistence of the bulk sample.47
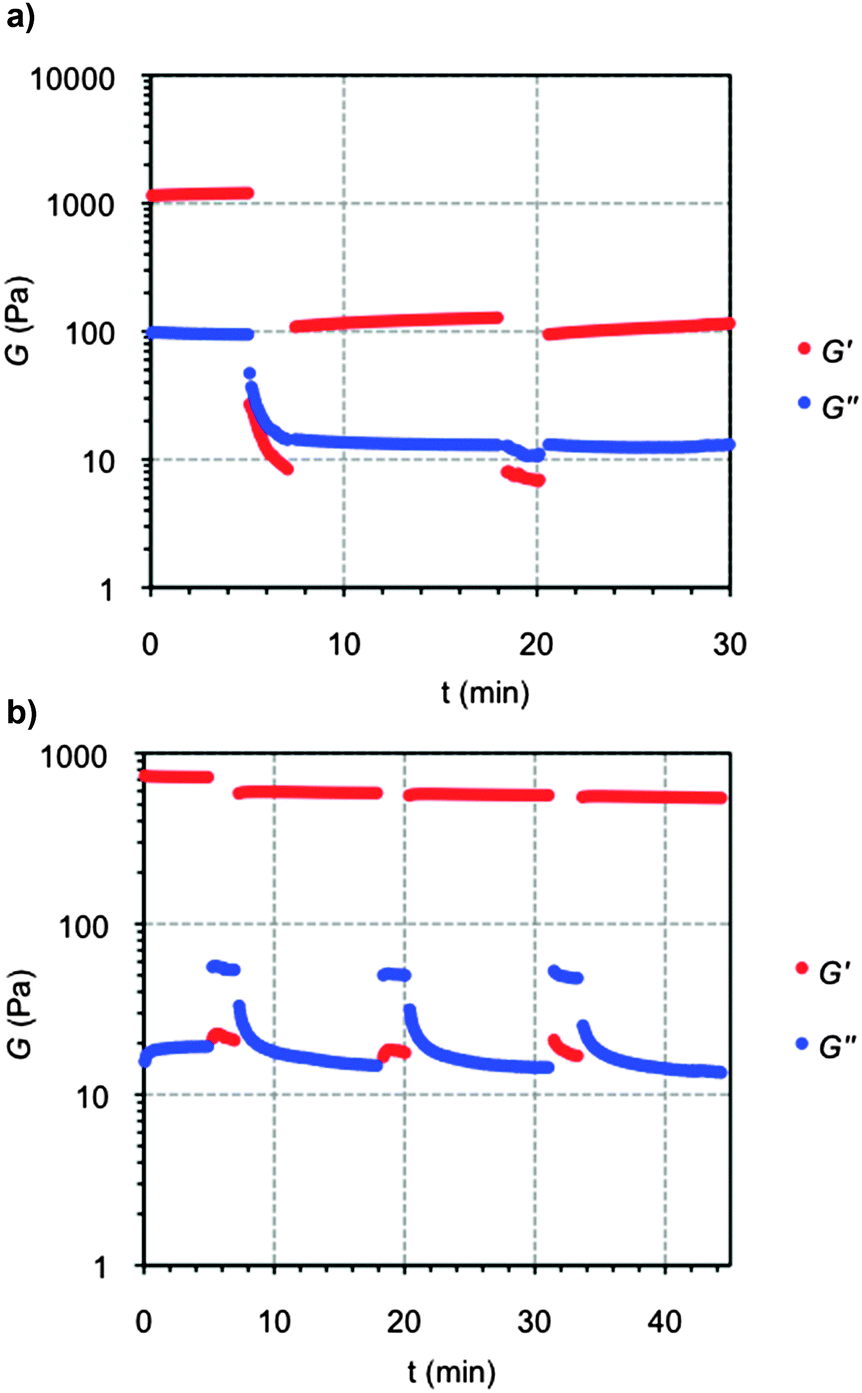 | ||
| Fig. 12 Representative 3-steps loop rheological test for (a) GB hydrogel and (b) OxA–GB10 hybrid hydrogel. | ||
Conclusions
Dynamic covalent chemistry is a powerful synthetic strategy to fabricate biohydrogels with self-healing properties. In this work, gelatin was crosslinked with oxidized alginate in the presence of borax yielding a hybrid biohydrogel system (OxA–GB) with the ability to self-repair upon mechanical damage. The foregoing results indicate a judicious balance between concentration, stoichiometric ratio of the two biopolymers and gelatin source (i.e., type A = GA; type B = GB) is necessary to achieve optimal self-healing properties of these biohydrogels. In this study, a 2:1 OxA:GB ratio and the use of 10 and 15% w/w solutions of OxA and GB provided the best results (OxA–GB10 and OxA–GB15 hydrogels, respectively). OxA–GB10 led to soft and almost flat hydrogels, while OxA–GB15 allowed the preparation of self-standing cylindrical monoliths. When the monoliths were cut into pieces, both OxA–GB10 and OxA–GB15 showed self-healing properties upon reconnection of the pieces. However, self-healed OxA–GB10 sample showed a much higher elastic elongation than OxA–GB15. The pH was found to have a major influence on the reconstruction of the damaged hydrogel interface, confirming that the dynamic Schiff base linkages between the amine groups of GB and the aldehyde groups of OxA play a fundamental role on the healing process. The self-healing properties of the OxA–GB system, with respect to the gelatin control, were also qualitatively examined by optical microscopy and quantified by rheological measurements. The results of this study can be relevant for a more targeted research on the potential applications of alginate–gelatin hydrogel systems.
Acknowledgements
This work was supported the University of Regensburg, the Deutsche Forschungsgemeinschaft (DFG, DI 1748/3-1) and the SINCHEM Joint Doctorate Programme selected under the Erasmus Mundus Action 1 Programme (FPA 2013-0037). We are thankful to the Laboratoire Charles Coulomb (Université de Montpellier & CNRS, Montpellier, France) for granting access to the rheometer and to M. Jean-Marc Fromental for assistance with the rheological measurements. D. D. D. thanks the DFG for the Heisenberg Professorship Award.References
- V. Amendola and M. Meneghetti, Nanoscale, 2009, 1, 74–88 RSC.
- P. Fratzl, J. R. Soc., Interface, 2007, 4, 637–642 CrossRef CAS PubMed.
- G. Fantner, E. Oroudjev, G. Schitter, L. Golde, P. Thurner, M. Finch, P. Turner, T. Gutsmann, D. Morse, H. Hansma and P. Hansma, Biophys. J., 2006, 90, 1411–1418 CrossRef CAS PubMed.
- J. Thompson, J. Kindt, B. Drake, H. Hansma, D. Morse and P. Hansma, Nature, 2001, 414, 773–776 CrossRef CAS PubMed.
- Y. Jin, C. Yu, R. Denman and W. Zhang, Chem. Soc. Rev., 2013, 42, 6634–6654 RSC.
- N. Roy, B. Bruchmann and J. Lehn, Chem. Soc. Rev., 2015, 44, 3786–3807 RSC.
- Z. Wei, J. Yang, J. Zhou, F. Xu, M. Zrínyi, P. Dussault, Y. Osada and Y. Chen, Chem. Soc. Rev., 2014, 43, 8114–8131 RSC.
- A. B. Brochu, S. L. Craig and W. M. Reichert, J. Biomed. Mater. Res., Part A, 2011, 96, 492–506 CrossRef PubMed.
- B. Yang, Y. Zhang, X. Zhang, L. Tao, S. Li and Y. Wei, Polym. Chem., 2012, 3, 3235–3238 RSC.
- Y. Zhang, L. Tao, S. Li and Y. Wei, Biomacromolecules, 2011, 12, 2894–2901 CrossRef CAS PubMed.
- Z. Wei, J. Yang, Z. Liu, F. Xu, J. Zhou, M. Zrínyi, Y. Osada and Y. Chen, Adv. Funct. Mater., 2015, 25, 1352–1359 CrossRef CAS.
- S. Hsu, F. Hsieh and T. Tseng, Neural Regener. Res., 2015, 10, 1922–1923 CrossRef PubMed.
- Y. Tabata and Y. Ikada, Adv. Drug Delivery Rev., 1998, 31, 287–301 CrossRef CAS.
- P. Neumann, B. Zur and Y. Ehrenreich, J. Biomed. Mater. Res., 1981, 15, 9–18 CrossRef CAS PubMed.
- M. Changez, V. Koul, B. Krishna, A. Dinda and V. Choudhary, Biomaterials, 2004, 25, 139–146 CrossRef CAS PubMed.
- G. Tronci, A. Neffe, B. Pierce and A. Lendlein, J. Mater. Chem., 2010, 20, 8875–8884 RSC.
- S. Khorshidi and A. Karkhaneh, Mater. Lett., 2016, 164, 468–471 CrossRef CAS.
- A. Sarker, J. Amirian, Y. Min and B. Lee, Int. J. Biol. Macromol., 2015, 81, 898–911 CrossRef CAS PubMed.
- Z. Dong, Q. Wang and Y. Du, J. Membr. Sci., 2006, 280, 37–44 CrossRef CAS.
- B. Balakrishnan and A. Jayakrishnan, Biomaterials, 2005, 26, 3941–3951 CrossRef CAS PubMed.
- E. Boanini and A. Bigi, J. Colloid Interface Sci., 2011, 362, 594–599 CrossRef CAS PubMed.
- U. Rottensteiner, B. Sarker, D. Heusinger, D. Dafinova, S. Rath, J. Beier, U. Kneser, R. Horch, R. Detsch, A. Boccaccini and A. Arkudas, Materials, 2014, 7, 1957–1974 CrossRef CAS.
- S. Khorshidi and A. Karkhaneh, Mater. Lett., 2016, 164, 468–471 CrossRef CAS.
- B. Sarker, J. Rompf, R. Silva, N. Lang, R. Detsch, J. Kaschta, B. Fabry and A. Boccaccini, Int. J. Biol. Macromol., 2015, 78, 72–78 CrossRef CAS PubMed.
- Y. Hu, L. Liu, Z. Gu, W. Dan, N. Dan and X. Yu, Carbohydr. Polym., 2014, 102, 324–332 CrossRef CAS PubMed.
- B. Balakrishnan, N. Joshi, A. Jayakrishnan and R. Banerjee, Acta Biomater., 2014, 10, 3650–3663 CrossRef CAS PubMed.
- B. Balakrishnan, M. Mohanty, P. Umashankar and A. Jayakrishnan, Biomaterials, 2005, 26, 6335–6342 CrossRef CAS PubMed.
- B. Balakrishnan, M. Mohanty, A. Fernandez, P. Mohanan and A. Jayakrishnan, Biomaterials, 2006, 27, 1355–1361 CrossRef CAS PubMed.
- Y. Zhang, B. Yang, X. Zhang, L. Xu, L. Tao, S. Li and Y. Wei, Chem. Commun., 2012, 48, 9305–9307 RSC.
- F. Ding, S. Wu, S. Wang, Y. Xiong, Y. Li, B. Li, H. Deng, Y. Du, L. Xiao and X. Shi, Soft Matter, 2015, 11, 3971–3976 RSC.
- S. Lü, C. Gao, X. Xu, X. Bai, H. Duan, N. Gao, C. Feng, Y. Xiong and M. Liu, ACS Appl. Mater. Interfaces, 2015, 7, 13029–13037 Search PubMed.
- Y. Gao, Q. Luo, S. Qiao, L. Wang, Z. Dong, J. Xu and J. Liu, Angew. Chem., Int. Ed., 2014, 53, 9343–9346 CrossRef CAS PubMed.
- N. Roy, E. Buhler and J.-M. Lehn, Polym. Int., 2014, 63, 1400–1405 CrossRef CAS.
- Z. Q. Lei, P. Xie, M. Z. Rong and M. Q. Zhang, J. Mater. Chem. A, 2015, 3, 19662–19668 CAS.
- F. Yu, X. Cao, J. Du, G. Wang and X. Chen, ACS Appl. Mater. Interfaces, 2015, 7, 24023–24031 CAS.
- Y.-H. Ma, J. Yang, B. Li, Y.-W. Jiang, X. Lu and Z. Chen, Polym. Chem., 2016, 7, 2037–2044 RSC.
- H. Li, J. Bai, Z. Shi and J. Yin, Polymer, 2016, 85, 106–113 CrossRef CAS.
- W. Yang, X. Wu, F. Liu, Y. Dou, Z. Hu and W. Hao, RSC Adv., 2016, 6, 34254–34260 RSC.
- T.-C. Tseng, L. Tao, F.-Y. Hsieh, Y. Wei, I.-M. Chiu and S.-H. Hsu, Adv. Mater., 2015, 27, 3518–3524 CrossRef CAS PubMed.
- U. Haldar, K. Bauri, R. Li, R. Faust and P. De, ACS Appl. Mater. Interfaces, 2015, 7, 8779–8788 CAS.
- B. Balakrishnan, S. Lesieur, D. Labarre and A. Jayakrishnan, Carbohydr. Res., 2005, 340, 1425–1429 CrossRef CAS PubMed.
- S. Khorshidi and A. Karkhaneh, Mater. Lett., 2016, 164, 468–471 CrossRef CAS.
- B. Sarker, D. Papageorgiou, R. Silva, T. Zehnder, F. Gul-E-Noor, M. Bertmer, J. Kaschta, K. Chrissafis, R. Detsch and A. Boccaccini, J. Mater. Chem. B, 2014, 2, 1470–1482 RSC.
- T. Aewsiri, S. Benjakul, W. Visessanguan and M. Tanaka, Int. J. Food Sci. Technol., 2008, 43, 685–693 CrossRef CAS.
- F. Vojdani, Methods of Testing Protein Functionality, Bury St. Edmunds Press, St Edmundsbury, 1st edn, 1996, pp. 11–60 Search PubMed.
- A. Saxena, K. Sachin, H. Bohidar and A. Verma, Colloids Surf., B, 2005, 45, 42–48 CrossRef CAS PubMed.
- S. Strandman and X. Zhu, Gels, 2016, 2, 16 CrossRef.
| This journal is © the Partner Organisations 2017 |