
Inducing macroporosity in hydrogels using hydrogen peroxide as a blowing agent†
N. Mac
Kenna
and
A.
Morrin
*
School of Chemical Sciences, National Centre for Sensor Research, Dublin City University, Dublin 9, Ireland. E-mail: aoife.morrin@dcu.ie
First published on 18th August 2016
Abstract
A new gas blowing method to induce a macroporous structure in pH-responsive hydrogel materials with basic functional groups is reported by a new technique that generates oxygen bubbles via hydrogen peroxide decomposition to template the polymer. This overcomes pH limitations associated with the more traditional approach of using a carbon dioxide gas blowing agent. This new approach is shown to effectively induce a macroporous structure which overcomes the diffusional limitation of bulk hydrogels and, as a result, dramatically increases swelling rates. The hydrogel comprises an aliphatic diamine, Jeffamine®, cross-linked with polyethylene glycol diglycidyl ether (PEGDGE) in a single simple polymerisation step, generating a polymeric network with pendant basic groups. This cross-linking reaction requires protonation of the amine groups, which precludes it from being compatible with the carbon dioxide gas blowing method as this requires a low pH for the decomposition of carbonate. To overcome this, the production of oxygen bubbles in situ via a catalytic decomposition of hydrogen peroxide on silver nanoparticles is used to induce a macroporous structure in the hydrogel. This method was shown to successfully induce a macroporous structure whereby interconnected pores from sub-micron up to 0.5 mm diameter are created within the hydrogel. This new method of inducing macroporosity is described here in terms of the hydrogel polymerisation conditions and the nature of the porosity is characterised in terms of its ability to overcome the diffusional limitations of bulk hydrogels. Finally, rapid, reversible pulsatile pH-sensing is demonstrated using these hydrogels.
Introduction
One of the principal limitations of responsive hydrogel systems in many instances is the diffusionally-dependant rate of swelling limitation that they present and hence their associated slow rate of response.1,2 Whilst a slow response is desirable for some applications, such as long-term drug delivery, many applications require rapid swelling responses. For example, rapid response times are necessary in sensing and drug delivery and this becomes even more critical when immediate intervention may be required. Use of hydrogels as self-actuating pumps and valves in MEMS microfluidic devices also necessitates fast swelling for efficient flow control.It is well known and understood that faster response times can be achieved by reducing the diffusion path length as the swelling rate is primarily diffusion controlled. This is easily achieved via preparation of hydrogels with reduced size dimensions. Baldi et al.3 showed that reduction of the depth of their phenylboronic acid-based hydrogel microvalve from 500 µm to 30 µm decreased the opening time from over 4 h to 7 min in their microelectromechanical (MEMS) device. Bates et al. reduced the response time of their piezo-resistive pressure sensor from 20 h to 0.34 h by reducing hydrogel thickness from 400 µm to 50 µm. Zhang et al.4 reduced the thickness of their optical glucose sensor by decreasing the amount of hydrogel bilayers incorporated. They observed a decrease in response time from approximately 1.5 to 0.3 min when the bilayer number was reduced from 90 to 30. Micro- and nano-sized gels exhibit fast response times due to their size reduction in at least one dimension. Several recent reviews are available which discuss their synthesis and areas of application.5–7
Macroporous hydrogels exhibit significantly faster response times than diffusion-dependant systems. They possess an interconnected, macro-sized pore system and imbibe water rapidly by capillary action through the open channels.8 Various synthesis methods exist to induce macroporosity including ice-templating,9 surfactant-templating10 and gas blowing techniques. Ice-templating involves polymerising a hydrogel at cold temperatures (∼−18 °C) to create large ice crystals which produce large, interconnected pores upon thawing. Cross-linking occurs in non-frozen liquid channels where the soluble monomers/polymers are concentrated. Sahiner et al.11 reported that their cryogel based on 2-acrylamido-2-methylpropane sulfonic acid (pAMPS) reached equilibrium swelling almost 3600 times faster than the corresponding conventional hydrogel and has since applied these gels to persistent uptake of chromate from aqueous environments.12 Surfactants have also been used as pore-forming agents. However, pores created using surfactants are typically small (<150 µm), resulting in a swelling time in the order of minutes rather than seconds. Shi et al. achieved an equilibrium swelling time of approximately 2000 s using sodium n-dodecyl sulfonate (SDS) micelle templating of a hydroxyethyl cellulose-based hydrogel and a sodium alginate-based hydrogel.13 Bao et al.14 used mechanically agitated surfactants such as sodium n-dodecyl benzene sulfate, cetyltrimethyl ammonium bromide and alkylphenol poly(oxyethylene) to form bubbles to prepare porous poly(sodium acrylic acid) superabsorbent resins which reached equilibrium swelling in approximately 1500 s.
Gas blowing techniques are frequently employed to synthesise hydrogels, typically through generating carbon dioxide bubbles by reacting sodium bicarbonate (NaHCO3) or sodium carbonate (Na2CO3) with acid to initiate foaming in the hydrogel precursors. Gas foaming with NaHCO3 was used by Gumusderelioglu et al.15 to generate macroporous poly(acrylamide) and poly(acrylamide-co-acrylic acid) gels for protein delivery. These hydrogels reached equilibrium swelling in ∼30 s and completed release of bovine serum albumin (BSA) within 1 h. Interestingly, when a chitosan interpenetrating network was introduced for improving mechanical strength, swelling time increased to 30 min and total release achievable decreased from 80% to 60%. Kuang et al.16 used gas foaming with NaHCO3 and acetic acid to prepare biodegradable polysaccharide-based macroporous hydrogels which reached equilibrium swelling in 60–440 s depending on monomer composition. Battig et al.17 also used this approach in the development of an aptamer-functionalised macroporous gel for the controlled release of growth factors. The aptamers in the hydrogel could sense and hybridise with fluorescently-labelled complementary strands and produce fluorescence after an incubation time of 5 min.
This work describes a novel gas blowing method involving the decomposition of hydrogen peroxide on silver nanoparticles to produce oxygen bubbles. This method overcomes the requirement of a low pH environment that is necessary for the CO2 gas blowing templating technique. As such, a pH-sensitive hydrogel comprising PEDGE–Jeffamine®18,19 was successfully templated with this method and the resulting macroporosity characterised with respect to the morphology, swelling kinetics and electrochemistry. Finally, rapid pulsatile pH-switching is demonstrated with this material.
Experimental
Materials
Poly(ethylene glycol)diglycidyl ether (PEGDGE, average Mn 526), silver nanopowder (<100 nm particle size), hydrogen peroxide solution (30% w/w in H2O), Pluronic® F-127, hexaamineruthenium(III) chloride (HARC) and sodium hydroxide were purchased from Sigma-Aldrich (Ireland). Jeffamine® EDR-148 polyetheramine was obtained from Huntsman Corporation (US). Untreated carbon cloth (44 × 48 yarns per inch, 99% carbon) was purchased from Fuel Cell Store (US) and phosphate buffer saline (PBS) tablets were purchased from Applichem (Germany). All chemicals were used as purchased and all aqueous solutions were prepared using deionised (DI) water (18 MΩ cm @ 298 K). The Ag/AgCl reference electrode and Pt gauze auxiliary electrode were purchased from CH Instruments, Inc. (UK) and Sigma-Aldrich (Ireland) respectively.Instrumentation
Optical images were recorded of hydrogels which had reached equilibrium swelling in DI water using a high resolution digital microscope (Keyence VHX-2000). Scanning electron micrographs were obtained using a Hitachi S3400V scanning electron microscope at an accelerating voltage of 20 kV. All samples were gold-sputtered for 90 s using a Quorum Technologies sputter coater (750T). A Deben Coolstage (UK) was attached to the SEM to keep the samples frozen at −10 °C throughout the analysis. All voltammetric analysis were performed on a CH potentiostat (CHI660C).Fabrication of PEDGE–Jeffamine® bulk hydrogel films
The fabrication of the hydrogel material is reported elsewhere.19,20 Briefly, the hydrogels were prepared by cross-linking Jeffamine® EDR-148 polyetheramine and PEGDGE in DI water. The molar ratio of epoxide to amine was maintained at 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0:1:0. Bulk hydrogel films were prepared by dropcasting the PEDGE–Jeffamine® precursor solution onto glass slides and placing cover slips on top. Various amounts of precursor solution (1 mL, 600 µL and 300 µL) were dropcast to prepare hydrogels with different heights (0.8 mm, 0.5 mm and 0.25 mm). All gels were placed in the refrigerator (4 °C) overnight to cure.
:0:1:0. Bulk hydrogel films were prepared by dropcasting the PEDGE–Jeffamine® precursor solution onto glass slides and placing cover slips on top. Various amounts of precursor solution (1 mL, 600 µL and 300 µL) were dropcast to prepare hydrogels with different heights (0.8 mm, 0.5 mm and 0.25 mm). All gels were placed in the refrigerator (4 °C) overnight to cure.
Fabrication of PEDGE–Jeffamine® macroporous hydrogels
Macroporous hydrogels were prepared by adding 2% (w/w) silver nanoparticles (0.02 g), PEGDGE (0.349 g), 1 M H2O2 (0.533 g) and Jeffamine® (0.098 g) sequentially into a glass vial, capping it quickly and vortexing for 10 s. Various Pluronic® loadings were added to the precursor solution (0, 5, 10, 20, 40 and 50% w/w) using a 10% (w/v) Pluronic® solution. The amount of H2O2 added was decreased as the Pluronic® loading increased so the cross-linking density of the network remained constant. The vials were then heated in an oven at a specified temperature for 20–45 min (depending on temperature investigated). The resulting polymer material was characterised by measuring the ratio of macroporous to bulk hydrogel after polymerisation using a Workzone® digital caliper. Bulk hydrogel was cut away from the macroporous material with a scalpel, and discs with different diameters (3.5, 12 and 20 mm) were then cut from the macroporous material.Investigation of swelling rate and response time of hydrogels
Swelling behaviour of hydrogel films and discs in water was determined as follows. Each disc was weighed (wdry) before immersion into the desired aqueous solution. At specific time intervals, the swollen discs were removed from solution, blotted dry with filter paper and weighed (wwet). The swelling ratio at various time intervals was calculated using the following relationship: | (1) |
Modification of carbon cloth electrodes with hydrogel
Carbon cloth strips (4 × 1 cm) were cut from a large sheet of carbon cloth. Off-the-shelf nail varnish was applied to the cloth to define the working electrode area (0.5 × 0.5 cm) and prevent solution from travelling up through the cloth via capillary action. Parafilm was wrapped around the top of each electrode to prevent gel from polymerising where the potentiostat connections attach.Macroporous and bulk hydrogel precursor solutions were prepared as described above, with an electrode pinned above the bottom of the glass vial, using the lid, to prevent hydrogel from polymerising on the electrode. Macroporous and bulk hydrogel modified carbon cloth electrodes were then cut from the polymerised gel with a scalpel. All gels were cut to an area of 9 × 9 × 9 mm when fully swollen.
Cyclic voltammograms were obtained by cycling bare and hydrogel-modified carbon cloth electrodes between −0.5 and 0.2 V vs. Ag/AgCl in 1 mM HARC in 1 M KCl at 0.1 V s−1.
pH-Driven pulsatile swelling of macroporous gels
Macroporous hydrogels were prepared and discs were cut (3.5 × 4 mm) from the macroporous gel using the Harris Uni-Core™ core sampler. The discs were weighed before being washed thoroughly by placing the gels in fresh DI water (approx. 100 mL) every day for a week. The hydrogels were blotted with filter paper and re-weighed to obtain their maximum swelling ratio due to water uptake. This was recorded as the swelling ratiowater uptake. The swelling ratio due to a change in pH (swelling ratiopH) of these equilibrated hydrogels was then calculated after 1 min immersion in fresh solutions (pHs 3 and 10). Finally, the specific swelling response due to the pH change was calculated according to eqn (2). The solutions for the pH study were prepared by mixing 10 mM stock solutions of HCl (pH 2) and NaOH (pH 12) to keep ionic strength constant.| Swelling response = Swelling ratiopH – Swelling ratiowater uptake | (2) |
Results and discussion
Fabrication of macroporous hydrogels
Macroporous hydrogels containing interconnecting macro-sized pores can rapidly absorb water by capillary action through the open channels. They exhibit significantly faster response times as the process of swelling is no longer diffusion controlled. Gas blowing techniques are frequently employed to synthesis these hydrogels, typically through generating carbon dioxide bubbles by reacting NaHCO3 or Na2CO3 with acid to initiate foaming. This strategy was attempted with the PEDGE–Jeffamine® system. However, no significant foam was produced. NaHCO3 reacts with acid to produce a salt and a carbonic acid which decomposes to water and carbon dioxide. When low concentrations of acetic acid were added, the overall pH of the gel precursors solution remained highly alkaline due to the high basic pH of the Jeffamine® (pH 11.6) and thus the NaHCO3 could not react to produce carbonic acid and subsequent carbon dioxide gas, resulting in no foam. On the other hand, high concentrations of acid resulted in protonation of the amines so they were no longer nucleophilic in nature and able to react with the oxirane groups of the PEGDGE.21 Amine nucleophiles require moderate alkaline pH values, usually at least pH 9 to be reactive.22 Thus, the hydrogel was unable to cross-link and polymerise at low pH. Foaming and crosslinking reactions must occur simultaneously to achieve the well-established porous structures and this could not be achieved using this gas blowing approach.Due to this pH limitation, an alternative method for synthesising macroporous hydrogels was explored involving the decomposition of hydrogen peroxide to produce oxygen bubbles (Fig. 1). Many metals behave as catalysts for the decomposition of hydrogen peroxide including Ag, Fe, Pt, Cu and Pd.23–26 Their reaction is often exploited for hydrogen peroxide detection due to its widespread usage in the food industry, pharmaceutical, industrial and environmental analysis. Metal nanoparticles such as Ag, Au, Pt and Pd exhibit excellent catalytic activity for hydrogen peroxide due to their larger specific surface area compared to bulk metal.27 In this instance, Ag nanoparticles (<100 nm) were added to the PEGDGE–Jeffamine® hydrogel precursors whereby, in the presence of the silver, decomposition of hydrogen peroxide occurs, producing oxygen bubbles. It is likely in this instance that this is by way of a catalytic reaction.
 | ||
| Fig. 1 Schematic showing the mechanism for this oxygen gas blowing technique within a hydrogel precursor solution whereby a macroporous PEGDGE–Jeffamine® hydrogel is formed. The oxygen gas bubbles are generated via the decomposition of hydrogen peroxide by silver nanoparticles within the solution. | ||
In the presence of the Ag nanoparticles, dilute hydrogen peroxide was incorporated into the precursor solution. This resulting in a rapid foaming as the hydrogen peroxide decomposed upon interaction with the Ag nanoparticles. The rate of hydrogen peroxide decomposition is known to accelerate with increasing pH and temperature.28 These conditions are provided naturally by the PEGDGE–Jeffamine® system due to the alkaline pH of the Jeffamine® and the heat generated by the oxirane ring-opening of the PEGDGE. Additionally, polymerisation was not affected by the presence of Ag and hydrogen peroxide, thus indicating this to be a viable option for fabricating macroporous hydrogels. For a more detailed discussion on the polymerisation reaction itself, the reader is referred to Mac Kenna et al.19 and Yoshioka & Calvert.29
In order for this gas blowing technique to be viable, the oxygen bubbles produced within the precursor solution during the decomposition of H2O2 needed to remain as discrete bubbles for the duration of the polymerisation which takes 8 h approx. when carried out at room temperature. As such, a foam stabiliser, Pluronic® F-127 was added to the precursor solution. During the decomposition, once oxygen bubbles formed, they migrated towards the top of the hydrogel precursor solution but were prevented from escaping into the vial atmosphere due to the presence of Pluronic®. Thus, the oxygen bubbles assembled in the form of discrete bubbles from the top of the solution down, which served to template the resulting hydrogel. After polymerisation, two distinct materials were produced – macroporous hydrogel formed in the upper portion through the oxygen bubble template and bulk, non-porous hydrogel formed at the bottom of the material which was free from gas bubbles. The depth of the macroporous portion of the resulting material was dependent on several parameters, including the ability of the Pluronic® to stabilise the oxygen bubbles, aswell as temperature (Fig. 2). Fig. 2a is a bar chart showing that the proportion of macroporous to bulk hydrogel formed increases with increasing Pluronic® loading when hydrogels are cured at room temperature. It can be seen that as the Pluronic® loading increased from 0 to 40%, the depth of bulk region decreased from 4.3 (±0.1) mm to 2.7 (±0.2) mm while the depth of macroporous region = increased from 0.0 (±0.0) mm to 4.5 (±0.1) mm. This indicates that the Pluronic® was successfully stabilising the oxygen bubbles and was more efficient at the higher loadings.
 | ||
| Fig. 2 (a) Bar chart showing the proportion of bulk to macroporous hydrogel formed with increasing Pluronic® loading. Polymerisation carried out at room temperature. (b) Bar chart showing the proportion of bulk to macroporous hydrogel formed at various polymerisation temperatures using a 40% loading of Pluronic®. Inset: Images showing the resulting hydrogel material whereby the proportion of bulk (dark grey) to macroporous (light grey) material can be seen. n = 3 for all data presented. Hydrogel heights measured using a ruler with a precision of ±0.05 mm. | ||
PEGDGE–Jeffamine® hydrogels are typically polymerised at room temperature, however higher temperatures can be used to accelerate the epoxy–amine reaction.29,30 Consequently, polymerisation temperature was investigated to determine if greater volumes of macroporous gel could be induced when the polymerisation rate was increased. Thus, hydrogels were prepared with a 40% w/w Pluronic® loading and placed immediately into a heated oven to polymerise. Fig. 2b again consists of a bar chart displaying the proportion of macroporous versus bulk hydrogel formed upon polymerisation at different temperatures. As the temperature was increased from 20–140 °C, the polymerisation time was reduced from ∼8 h to 15 min which had a dramatic effect on the resulting hydrogel morphology. The depth of the macroporous gel increased almost 4 times, from 4.5 (±0.1) mm to 17.0 (±0.6) mm indicating that more bubbles were present during the shorter polymerisation times. This was likely due to the fact that the polymerisation occurred before coalescing of bubbles and loss of oxygen to the atmosphere could occur. For example, at 20 °C, it can be seen in the images in Fig. 2b that the pores at the top of the hydrogels are extremely large (up to several mm in diameter), resulting from bubble coalescence (see also ESI 1†). As the temperature is increased, the porosity becomes more uniform and the diameter of the pores decreases dramatically. It is also likely that the elevated temperatures increased the rate of oxygen decomposition, resulting in a greater amount of oxygen being producing at early times. As well as increasing the portion of macroporous the material, the depth of the undesired bulk simultaneously decreased because of this, from 2.7 (±0.3) mm to 1.7 (±0.2) mm when comparing room temperature to 120 °C polymerisation temperature. Thus, the optimum conditions for polymerisation was a 40% loading of Pluronic® to ensure good stabilisation of the gas bubbles and a polymerisation temperature of 120 °C to ensure polymerisation time was short and the rate of oxygen decomposition was fast so that the bubbles were stable and did not have time to coalesce. Using these conditions, the smallest, most uniform pore size resulted in the hydrogel. It should be noted that at temperatures greater than 120 °C, a change in hydrogel colour from grey to yellow indicated possible thermal degradation. Therefore, a polymerisation temperature of 120 °C was used for all subsequent experiments. Upon curing at this temperature, the bulk and macroporous components of the material were easily separated, to give free-standing macroporous hydrogels.
Morphology of hydrogels
The morphologies of the bulk and macroporous hydrogels were compared using optical microscopy and SEM. Fig. 3 shows optical images of fully swollen bulk and macroporous hydrogels. The bulk hydrogel appears smooth, with no obvious level of porosity. In comparison, the macroporous hydrogel is highly porous, with pores visible at the surface as large as 2500 µm in the swollen state.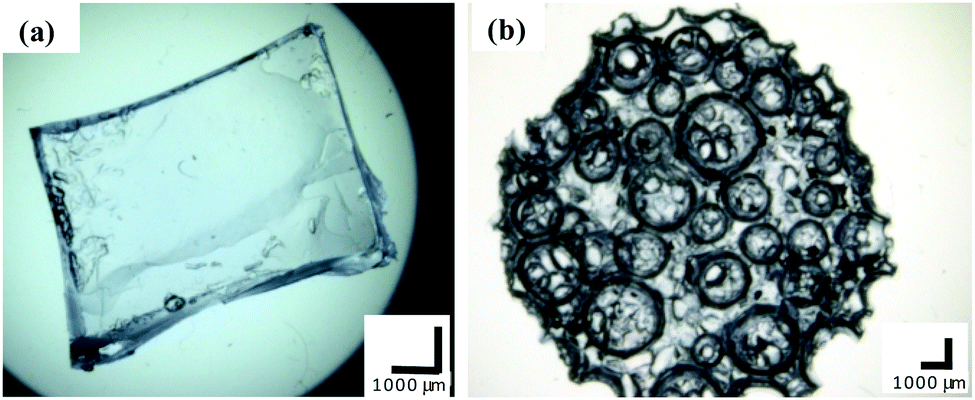 | ||
| Fig. 3 Optical microscope images of (a) bulk and (b) macroporous hydrogel (40% Pluronic® loading, polymerisation temperature 120 °C). All scale bars = 1000 µm. | ||
To gain a greater insight into the pore structure of the hydrogels, SEM analysis was carried out under vacuum. Fig. 4a–d display images of a dehydrated and fully hydrated bulk and macroporous hydrogel. In the bulk material, no pores are visible in the dry state and small pores ranging from 5–60 µm are induced when fully swollen. In comparison, the SEM images of the dehydrated and fully hydrated macroporous hydrogel show the macroporous structure to be comprised of an interconnecting pore system with typical pore size greater than 100 µm. Pore diameter size ranged from 10–500 µm in the dehydrated gel and 200–1000 µm in the swollen hydrogel. The thickness of the polymer walls ranged from approximately 200–250 µm in the dehydrated hydrogel and 25–40 µm in the swollen hydrogel. Upon swelling, the size of the pores significantly increased, pore walls became thinner and the channels became wider. The shape of the pores is mainly spherical but some distortion is evident, particularly when swollen. It is likely the distortion occurs due to the rapid pressure changes as the oxygen is generated and travels through the foam. The pores of the swollen macroporous gels are almost 28 times larger than the corresponding bulk gel demonstrating the effectiveness of this new gas blowing technique.
 | ||
| Fig. 4 SEM images of (a) a dehydrated bulk hydrogel and (b) a fully swollen bulk hydrogel. (c) A dehydrated macroporous and (d) a fully swollen macroporous hydrogel. Images (a) and (b) have Mag: ×160 and scale bar: 300 µm. Images (c) and (d) have Mag: ×50 and scale bar: 1 mm. | ||
Effect of hydrogel morphology on swelling kinetics
The swelling behaviours of these bulk and macroporous hydrogels were monitored to investigate the differences in swelling kinetics of these materials. Fig. 5a shows the swelling behaviour of hydrogel films of varying thicknesses (0.25–0.8 mm) immersed in water over 45 min. The rate of hydrogel swelling was observed to be slow and dependent on the depth dimension of the hydrogel, which would be expected for bulk hydrogel thick films. It can be seen that for films of all depths, the main increase in swelling ratio occurs within approx. the first 15 min of its immersion in water at room temperature. Within this region, the swelling ratio linearly increases with t1/2, indicating Fickian-type diffusion at this stage whereby the rate of diffusion is slower that the rate of relaxation of the polymer chains. After this initial increase, the rate of swelling decreases and becomes more characteristic of a relaxation-controlled swelling whereby the swelling ratio linearly increases in proportion to t. Equilibrium swelling is reached after 250 min approx. for the thinnest film of depth 0.25 mm (ESI 2†). The time to reach swelling equilibrium increases with film depth, and equilibrium swelling is not reached for the film with the greatest depth (0.8 mm) within 250 min. The swelling behavior of the macroporous hydrogels is shown in Fig. 5b. In contrast to the bulk materials, the macroporous hydrogels were observed to swell rapidly. The rate of swelling was observed to be independent of hydrogel size, which is indicative of a macroporous structure. This size-independent rapid swelling has been observed in other macroporous hydrogel systems and is attributed to the inter-connected macroporous structure of the hydrogel network31,32 whereby the rate of uptake of water is now dependent on the diameter of the capillary channels. The overall time required to reach equilibrium swelling increased with hydrogel size but all 3 hydrogels were fully swollen within 20 s. Equilibrium swelling ratio increased with gel size as would be expected as larger hydrogels possess a greater swelling capacity. Moving from the bulk films to the macroporous materials, the time to reach equilibrium swelling decreased from several hundred min to less than 20 s and the rate of swelling becomes independent of size. This dramatic improvement validates the formation of a truly macroporous structure generated by this new gas blowing method. | ||
| Fig. 5 (a) Effect of decreasing depth of bulk hydrogels on swelling response in DI water. (b) Effect of gel diameter on the gravimetric swelling response of macroporous hydrogels in water. All hydrogels had a height of 4 mm in the initial dehydrated state. | ||
Voltammetry on hydrogel-modified carbon cloth electrodes
Voltammetric analysis of carbon cloth-modified bulk and macroporous hydrogels was performed to demonstrate how the different diffusional properties influence voltammetry of a bulk solution redox species. The choice of carbon cloth as the electrode arises from earlier work19,20 as it conforms with the hydrogel and thus maintains electrical contact with the hydrogel as it swells and contracts. Electrochemical properties of the bulk and macroporous hydrogel coatings were compared. Hydrogels were prepared in solution as before, where the carbon cloth was dipped into the solution during the polymerisation. The adhered gel was then cut to the desired size. Upon swelling, both bulk and macroporous hydrogels were 9 × 9 × 9 mm. ESI 3† shows a photograph of both carbon cloth modifications.Fig. 6 shows the voltammetric responses of the bare carbon cloth electrodes, and bulk and macroporous hydrogel-modified carbon cloth electrodes in [Ru(NH3)6]3+/2+. The bare electrode showed reversible, diffusion-limited voltammetric behaviour as expected. The ΔEp was below 59 mV at the bare carbon cloth indicating an influence of thin layer voltammetry due to a surface confined species, possibly due to the porous nature of the electrode. However, the influence of this is reduced when the electrode is modified. While the bulk hydrogel prevented the diffusion of HARC to the electrode over the time-scale of the experiment, the voltammetry at the macroporous hydrogel-modified electrode had similar behaviour to the bare electrode. This indicates that over long timescales, the macroporous hydrogel does not present a diffusional barrier to the bulk solution [Ru(NH3)6]3+/2+ redox probe. Over shorter timescales, i.e., at faster scan rates, the effect of the capillary walls of the macroporous polymer were seen whereby the rate of diffusion of the redox probe to the electrode surface is slower at this electrode compared to the bare electrode. This is evident from the scan rate study performed (0.001–0.050 V s−1) on the bare carbon cloth, and macroporous hydrogel-modified carbon cloth (Fig. 6b). Both electrodes displayed a linear correlation between peak current (ip) and square root of scan rate, indicative of diffusion-controlled processes dominating for both electrodes. The non-zero intercepts however indicate that the voltammetry is again not completely driven by mass transfer and that adsorption onto, or confinement of the redox species in the carbon cloth may be playing a small role. Using the Randles–Sevcik equation, the diffusion coefficient, D for [Ru(NH3)6]3+/2+ at the bare carbon electrode is given by 10.30 × 10−5/A cm s−1 and the D for the modified electrode is 5.96 × 10−5/A cm s−1 where A is the area of the electrode (cm2). (Note: absolute values for D are not calculated, as the total surface area of the carbon cloth electrodes have not been determined). This means that the value of D for [Ru(NH3)6]3+/2+ is less than 50% lower in the presence of the macroporous material. This again confirms the presence of a macroporous films whereby the influence of the thin walls of the capillary-sized pores is observed by way of a decrease in rate of diffusion, when compared to a bare electrode. Given that a bulk film of the hydrogel of equivalent thickness would completely impede diffusion of the [Ru(NH3)6]3+/2+ species to the electrode surface, a reduction in rate of this order is indicative of the presence a macroporous open structure within the thick hydrogel film.
 | ||
| Fig. 6 (a) Cyclic voltammograms of the bare, bulk and macroporous hydrogel modified electrodes in HARC at a scan rate of 0.01 V s−1. (b) Dependence of the anodic peak current on the square root of the scan rate for the bare electrode (y = 0.002726x − 8.367 × 10−5) and the macroporous hydrogel modified electrode (y = 0.002074x − 4.897 × 10−5). All experiments carried out in 1 mM HRC in 1 M KCl. | ||
Effect of pH on swelling response of macroporous gels
Pulsatile pH-dependant swelling was evaluated between pH 3 and 10 solutions with 1 min intervals (Fig. 7). Reversible, consistent swelling was observed for the 10 cycles investigated. The macroporous hydrogel was capable of rapidly absorbing and releasing the swelling medium upon alteration of the pH due to its macroporous, open structure. The hydrogels achieved the maximum swelling response of 18.3 (±0.5) in pH 3 but only partially deionised and de-swelled in pH 10 as the swelling response decreased to 12.8 (±0.3) instead of 0. Neutralisation effects could be responsible as the hydrogels absorb the pH 3 solution and then release it into the pH 10 solution as they equilibrate in the new environment and vice versa. Alternatively, the rate of swelling and de-swelling in hydrogel systems may not be equal. In this instance, the de-swelling rate to attain its equilibrium collapsed state was possibly slower than the swelling rate to reach its equilibrium swollen state. This has been seen previously in other macroporous gel systems.33 However, these fast and reproducible swelling transitions are highly desirable in hydrogel-based sensing systems as well as in other applications including the intelligent self-actuating valves and pumps in MEMS devices. | ||
| Fig. 7 Pulsatile pH-dependant swelling (pH 3) and de-swelling (pH 10) behaviour of macroporous hydrogels (1 min intervals) (n = 3). | ||
Conclusion
Macroporous hydrogels were successfully prepared using in situ generated oxygen bubbles produced via the decomposition of hydrogen peroxide by silver nanoparticles. To the author's knowledge, this approach has not been investigated previously and thus has been shown for the first time as a viable synthesis method for hydrogel systems which may be limited by pH or other templating restrictions. An interconnected macropore system with pore sizes ranging from 200–1000 µm was established. These hydrogels were shown to exhibit rapid swelling in water where equilibrium swelling in water was reached in less than 20 s. This is a dramatic improvement of more than 1500-fold in terms of time required to reach swelling equilibrium when compared to their bulk counterparts. The rate of swelling was found to be independent on size, confirming its macroporous structure. These hydrogels retained their pH-sensitivity after templating and displayed rapid reproducible, well-behaved swelling transitions upon pulsatile pH switching. In addition to increasing the practicality of these materials in for sensing applications, the rapid pH-sensitive response could easily be exploited in drug delivery, as intelligent valves in MEMS devices or in numerous other applications.Acknowledgements
The authors wish to thank the Irish Research Council for financial assistance provided under the ‘EMBARK initiative’.References
- S. Chaterji, I. K. Kwon and K. Park, Prog. Polym. Sci., 2007, 32, 1083–1122 CrossRef CAS PubMed.
- Y. Pan, K. Shi, Z. Liu, W. Wang, C. Peng and X. Ji, RSC Adv., 2015, 5, 78780–78789 RSC.
- A. Baldi, Y. D. Gu, P. E. Loftness, R. A. Siegel and B. Ziaie, J. Microelectromech. Syst., 2003, 12, 613–621 CrossRef.
- X. Zhang, Y. Guan and Y. Zhang, Biomacromolecules, 2012, 13, 92–97 CrossRef CAS PubMed.
- A. Doering, W. Birnbaum and D. Kuckling, Chem. Soc. Rev., 2013, 42, 7391–7420 RSC.
- J. M. G. Swann and A. J. Ryan, Polym. Int., 2009, 58, 285–289 CrossRef CAS.
- A. L. Liu and A. J. Garcia, Ann. Biomed. Eng., 2016, 44, 1946–1958 CrossRef PubMed.
- H. V. Chavda, R. D. Patel, I. P. Modhia and C. N. Patel, Int. J. Pharm. Invest., 2012, 2, 134–139 CrossRef CAS PubMed.
- H. Bai, A. Polini, B. Delattre and A. P. Tomsia, Chem. Mater., 2013, 25, 4551–4556 CrossRef CAS PubMed.
- H. Tokuyama and A. Kanehara, Langmuir, 2007, 23, 11246–11251 CrossRef CAS PubMed.
- N. Sahiner and F. Seven, RSC Adv., 2014, 4, 23886–23897 RSC.
- N. Sahiner, S. Demirci, M. Sahiner and S. Yilmaz, J. Appl. Polym. Sci., 2016, 133, 43438 Search PubMed.
- X. Shi, Jietang and A. Wang, J. Appl. Polym. Sci., 2015, 132, 42027 Search PubMed.
- J. Bao, S. Chen, B. Wu, M. Ma, Y. Shi and X. Wang, J. Appl. Polym. Sci., 2015, 132, 41298 Search PubMed.
- M. Gumusderelioglu, D. Erce and T. T. Demirtas, J. Mater. Sci.: Mater. Med., 2011, 22, 2467–2475 CrossRef CAS PubMed.
- J. Kuang, K. Y. Yuk and K. M. Huh, Carbohydr. Polym., 2011, 83, 284–290 CrossRef CAS.
- M. R. Battig, Y. Huang, N. Chen and Y. Wang, Biomaterials, 2014, 35, 8040–8048 CrossRef CAS PubMed.
- P. Calvert, P. Patra and D. Duggal, in Electroactive Polymer Actuators and Devices, ed. Y. BarCohen, 2007, vol. 6524, p. M5240 Search PubMed.
- N. Mac Kenna, P. Calvert and A. Morrin, Analyst, 2015, 140, 3003–3011 RSC.
- N. Mac Kenna, P. Calvert, A. Morrin, G. G. Wallace and S. E. Moulton, J. Mater. Chem. B, 2015, 3, 2530–2537 RSC.
- P. Y. Bruice, Organic Chemistry, Prentice Hall, NJ, US, 2013 Search PubMed.
- G. T. Hermanson, Bioconjugate Techniques, Academic Press, UK, 2013 Search PubMed.
- A. Goodison, L. Gonzalez-Macia, A. J. Killard and A. Morrin, MRS Online Proc. Libr., 2014, vol. 1626, mrsf13-1626-k1608-1632 (1627 pages) Search PubMed.
- H. Lee, H.-J. Lee, D. L. Sedlak and C. Lee, Chemosphere, 2013, 92, 652–658 CrossRef CAS PubMed.
- W.-J. Zhang, L. Bai, L.-M. Lu and Z. Chen, Colloids Surf., B, 2012, 97, 145–149 CrossRef CAS PubMed.
- S. A. G. Evans, J. M. Elliott, L. M. Andrews, P. N. Bartlett, P. J. Doyle and G. Denuault, Anal. Chem., 2002, 74, 1322–1326 CrossRef CAS PubMed.
- J. Yin, X. Qi, L. Yang, G. Hao, J. Li and J. Zhong, Electrochim. Acta, 2011, 56, 3884–3889 CrossRef CAS.
- G. Blanco-Brieva, M. P. de Frutos-Escrig, H. Martin, J. M. Campos-Martin and J. L. G. Fierro, Catal. Today, 2012, 187, 168–172 CrossRef CAS.
- Y. Yoshioka and P. Calvert, Exp. Mech., 2002, 42, 404–408 CrossRef CAS.
- C. Zhao, G. Zhang and L. Zhao, Molecules, 2012, 17, 8587–8594 CrossRef CAS PubMed.
- H. Omidian and K. Park, J. Bioact. Compat. Polym., 2002, 17, 433–450 CrossRef CAS.
- H. Omidian, K. Park and J. G. Rocca, J. Pharm. Pharmacol., 2007, 59, 317–327 CrossRef CAS PubMed.
- M. V. Dinu, M. Přádný, E. S. Drǎgan and J. Michalek, J. Polym. Res., 2013, 20, 285 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6qm00052e |
| This journal is © the Partner Organisations 2017 |