
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A new class of platinum(II) complexes with the phosphine ligand pta which show potent anticancer activity†
M. D.
Živković
a,
J.
Kljun
b,
T.
Ilic-Tomic
c,
A.
Pavic
c,
A.
Veselinović
d,
D. D.
Manojlović
e,
J.
Nikodinovic-Runic
*c and
I.
Turel
 *b
*b
aUniversity of Kragujevac, Faculty of Medical Sciences, Department of Pharmacy Svetozara Markovića 69, 34000 Kragujevac, Serbia
bUniversity of Ljubljana, Department of Chemistry and Biochemistry, Faculty of Chemistry and Chemical Technology, Večna pot 113, SI-1000 Ljubljana, Slovenia. E-mail: iztok.turel@fkkt.uni-lj.si
cUniversity of Belgrade, Institute of Molecular Genetics and Genetic Engineering, Vojvode Stepe 444a, 11000 Belgrade, Serbia. E-mail: jasmina.nikodinovic@gmail.com; jasmina.nikodinovic@imgge.bg.ac.rs
dUniversity of Niš, Department of Chemistry Faculty of Medicine, 18000 Niš, Serbia
eUniversity of Belgrade, Department of Analytical Chemistry, Faculty of Chemistry, Studentski trg 12-16, 11000 Belgrade, Serbia
First published on 17th October 2017
Abstract
The anticancer potential of sixteen platinum(II) complexes with general formulae [PtCl(hq)(S-dmso)] (1a–8a) and [PtCl(hq)(pta)] (1b–8b) (where hq is 5-chloro-7-iodo-8-quinolinol (clioquinol; cqH) (1a, 1b), 8-hydroxy-5-nitroquinoline (nitroxoline; nxH) (2a, 2b), 5,7-dichloro-8-quinolinol (3a, 3b), 5,7-diiodo-8-quinolinol (4a, 4b), 5,7-dibromo-8-quinolinol (5a, 5b), 5,7-dichloro-8-hydroxy-2-methyl-quinoline (6a, 6b), 8-hydroxyquinoline (7a, 7b) and 8-quinolinethiol (8a, 8b); dmso is dimethyl sulfoxide and pta is 1,3,5-triaza-7-phosphaadamantane) was determined through in vitro cytotoxicity assay in human fibroblasts (MRC5) and two carcinoma cell lines (A375 and A549) and embryotoxicity assay in a zebrafish model. Interactions with double stranded DNA through in vitro assay and a molecular docking study were examined. All complexes, except 6a, exhibited a high cytotoxic effect on MRC5 cells at a concentration of 10 μg mL−1 while 1b, 5a, 6a and 3b showed selective toxicity towards carcinoma cell lines. In general, pta-based complexes (series b) were more toxic according to the results of a MTT screen and the LC50 values obtained in zebrafish (Danio rerio) assay; they also induced higher oxidative stress in this model. Successful cellular uptake of complexes was shown by the ICP-MS methodology. The binding propensity of the complex with DNA obtained in in silico studies can be correlated with those from the experimental investigation. Compounds with the highest binding potential, according to the interaction energy value, were 1b, 3b, 6b and 5b. From observations of the DNA interaction ability and of the in silico assessment, no apparent DNA fragmentation was observed either on DNA extracted from the treated cancer cell line or from the zebrafish embryos.
Introduction
The development of metallodrugs and the study of their mode-of-action has become one of the most important fields of inorganic chemistry after the discovery of cisplatin and its antitumor activity in the 1960s.1 Cisplatin and its successors are the most often used metal-based drugs in cancer chemotherapy.2,3 The high general toxicity of cisplatin which induces severe side-effects, such as nephrotoxicity, neurotoxicity, ototoxicity, and emetogenesis,4 necessitates the development of novel antitumor metal-based therapeutic agents. Besides problems with their toxicity, another limitation of the clinical efficacy of platinum drugs is their ability to induce cellular resistance.5–7 Resistance to cisplatin is generally multifactorial and can be attributed to reduced drug accumulation, inactivation of drugs by interaction with thiol containing species, and changes in the proteins involved in apoptosis.8–10 Carboplatin and oxaliplatin, the second generation of platinum-based drugs, were developed to obtain better antitumor activity, increased solubility and lower toxic side effects compared to cisplatin.8,9 The interactions which are responsible for antitumor activity of Pt-based drugs are covalent binding to guanine N7 atoms or intercalation of aromatic ligands into cellular DNA. The resulting changes in the tertiary structure of DNA lead to cancer cell death through apoptosis.10–12 Despite all these drawbacks, the first-line chemotherapy for 12 different neoplasms with more than 50% of clinically applied anticancer regimens contains a platinum(II)-based compound.13–15Considering all the mentioned facts, our idea was to design new Pt-based anticancer compounds containing ligands which already exert biological properties expecting synergistic effects.16 Several 8-hydroxyquinolines (hqH) are already known for their antimicrobial and antiprotozoal activity and many of them are or were used in clinical practice. Their biological activity is often ascribed to their ability to bind di- and trivalent transition metal ions such as copper(II), zinc(II), and iron(III). The hydroxyquinolines are known to be proteasome and NFκB inhibitors, inducers of apoptosis of human cancer cell lines, and stimulators of macrophages to release tumour necrosis factor alpha as well as potential therapeutic agents in the treatment of Alzheimer's and Huntington's diseases.16 Clioquinol (5-chloro-7-iodo-8-hydroxyquinoline; cqH; Fig. 1) is a member of the family of 5,7-(di)halo substituted hqH, formerly antibacterial agents but now used mainly as antiseptics or disinfectants. It has been shown that clioquinol activity is copper dependent and that it induces cell death in leukaemia and myeloma cell lines through the inhibition of the proteasome.17–20
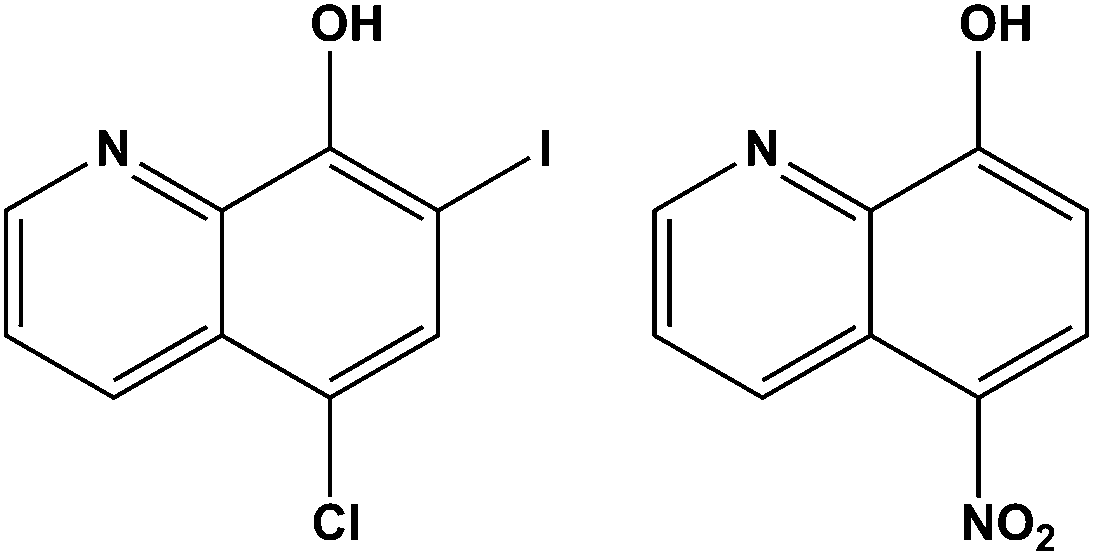 | ||
| Fig. 1 The clinically used 8-hydroxyquinoline agents clioquinol and nitroxoline. | ||
Interestingly, at non-cytotoxic concentrations the inhibitor impairs the degradation of the extracellular matrix and tumour cell invasion, revealing a specific anti-cancer mechanism not related to a general compound-induced cytotoxicity.21
Another member of the 8-hydroxyquinoline family already used in medicinal practice is nitroxoline (5-nitro-8-hydroxyquinoline, nxH; Fig. 1). This drug is an antibacterial agent used in the treatment of acute or recurrent urinary tract infections. Nitroxoline also showed appreciable bactericidal activity against growing and/or dormant tubercle bacilli.19,22 Also, it was discovered that nitroxoline is a potential selective and reversible inhibitor of cathepsin B which results in in vitro and in vivo tumor progression.23–25 The activity of nitroxoline was explained by the inhibition of extracellular and intracellular degradation of the extracellular matrix (ECM), reducing tumor cell spreading. These facts suggest that nitroxoline has great potential as a drug candidate in the treatment of cancer and other diseases connected with increased cathepsin B activity.23–25
On the other hand, the chemistry of second-row transition-metal complexes of 1,3,5-triaza-7-phosphaadamantane (pta) and related ligands has attracted much attention due to their potential as water-soluble catalysts.26,27 These complexes show high stability due to the strong metal–phosphorus bond. The ligand pta has become prominent in the bioinorganic scientific community as a ligand in the ruthenium–arene-pta (RAPTA) class of compounds developed by Dyson's group. Among the hundreds of reported compounds of this class, several compounds have achieved great success at the preclinical development stage as potential antimetastatic agents, thermoresponsive anticancer drugs and as agents for targeted cancer therapy targeting thiol and selenol containing proteins.27 The coordination chemistry of other precious metal–pta complexes is very well developed as numerous other rhodium, osmium and iridium organometallic compounds as well as rhodium and gold coordination compounds have been investigated for their catalytic and medicinal properties. Interestingly, reports of palladium and platinum are relatively scarce and in the case of platinum, complexes almost exclusively contain structures of bis-pta complexes (or complexes bearing one pta and another phosphine ligand).28,29 Some of these compounds were evaluated as potential medicinal agents; however, these studies had limited scope because of the very low aqueous solubility of these compounds.
In the current study we have continued this line of research, focusing on the synthesis, characterization and biological evaluation of the series of [PtCl(hq)(S-dmso)] type complexes, as well as their pta analogs [PtCl(hq)(pta)], all of them differing in the bidentate ligand with the quinoline moiety. Cytotoxicity against human cell lines and embryotoxicity in a zebrafish model were evaluated. The cellular Pt content was monitored by means of ICP-MS. The DNA-interaction ability of the selected complexes in silico and DNA fragmentation patterns of the treated cells were examined. The in silico DNA binding affinities of the studied complexes were obtained on the basis of a molecular docking study.
Results and discussion
Synthesis and characterization of 8-hydroxyquinoline Pt(II) complexes
Sixteen Pt(II) complexes with general formulae [PtCl(hq)(S-dmso)] (1a–8a) and [PtCl(hq)(pta)] (1b–8b) (where hq is 5-chloro-7-iodo-8-quinolinol (clioquinol; cqH) (1a, 1b), 8-hydroxy-5-nitroquinoline (nitroxoline; nxH) (2a, 2b), 5,7-dichloro-8-quinolinol (3a, 3b), 5,7-diiodo-8-quinolinol (4a, 4b), 5,7-dibromo-8-quinolinol (5a, 5b), 5,7-dichloro-8-hydroxy-2-methyl-quinoline (6a, 6b), 8-hydroxyquinoline (7a, 7b) and 8-quinolinethiol (8a, 8b); dmso is dimethyl sulfoxide and pta is 1,3,5-triaza-7-phosphaadamantane) have been synthesized (Fig. 2). The structures of all Pt(II) complexes were confirmed by elemental microanalyses, ESI-HRMS, NMR (1H and 31P), IR and UV-vis spectroscopy. Spectroscopic data of these complexes were correlated with those previously reported for 1a, 3a, 4a, 5a, 7a and 8a.16,30–32 Due to the low solubility of these complexes in deuterated solvents 13C NMR spectra were not recorded. The crystal structures of complexes α-2a, β-2a, β-6a, β-1b, and β-4b (see the definition of α and β isomers in Table 1) were confirmed by X-ray single crystal diffraction analysis.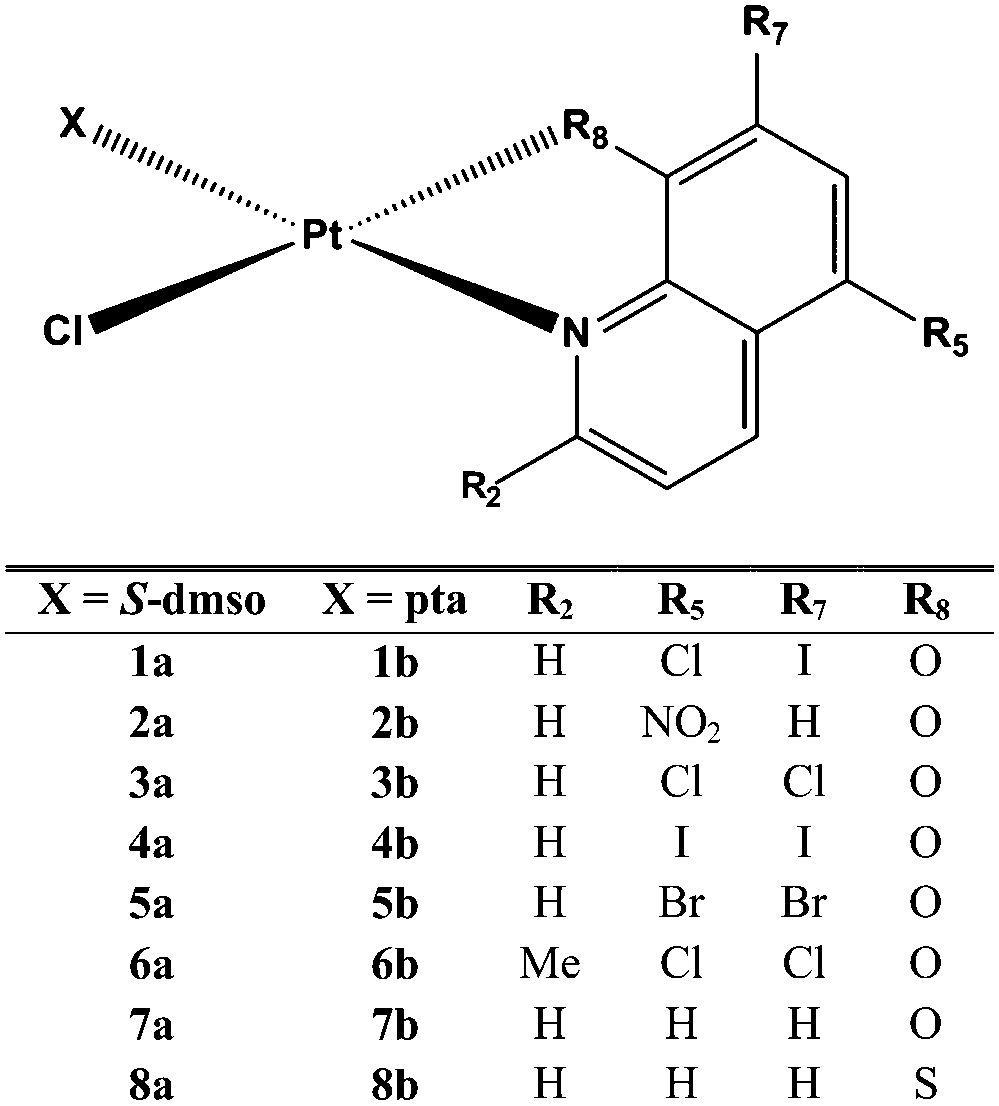 | ||
| Fig. 2 Structural representation of the investigated Pt(II) complexes (X is S-dmso for 1a–8a and pta for 1b–8b; dmso is dimethyl sulfoxide and pta is 1,3,5-triaza-7-phosphaadamantane). | ||
|
|
|||||
|---|---|---|---|---|---|
| [PtCl(hq)(S-dmso)] complex | Isomer | Relative energy (kcal mol−1) | [PtCl(hq)(pta)] complex | Isomer | Relative energy (kcal mol−1) |
| N.B.: The substitution of the S-coordinated dmso for the P-coordinated pta and the exchange of the N,O-ligands 1–7 for the N,S-ligand 8 mean that compounds which are structural analogs have different (even opposite) configuration indices. In order to facilitate the visualization we have defined the isomers as isomers α and β. The exact nomenclature according to IUPAC recommendations was added to the ESI (Table S1). Absolute energy baselines (in hartree): a −0.03878; b 0.027597; c −0.07739; d 0.00054; e −0.04089; f −0.09277; g −0.05503; h −0.00169; i 0.031101; j 0.005383; k −0.00719; l 0.070383; m 0.029125; n −0.02058; o 0.015464; p 0.069494. | |||||
| 1a | α | 15.9387 | 1b | α | 42.7315 |
| β | 0.0a | β | 0.0i | ||
| 2a | α | 0.0b | 2b | α | 42.9091 |
| β | 23.2072 | β | 0.0j | ||
| 3a | α | 58.0942 | 3b | α | 42.5878 |
| β | 0.0c | β | 0.0k | ||
| 4a | α | 56.8756 | 4b | α | 41.5913 |
| β | 0.0d | β | 0.0l | ||
| 5a | α | 56.5951 | 5b | α | 41.2826 |
| β | 0.0e | β | 0.0m | ||
| 6a | α | 128.8302 | 6b | α | 142.4817 |
| β | 0.0f | β | 0.0n | ||
| 7a | α | 53.9608 | 7b | α | 38.5925 |
| β | 0.0g | β | 0.0o | ||
| 8a | α | 119.4878 | 8b | α | 106.2298 |
| β | 0.0h | β | 0.0p | ||
The reaction pathway for the preparation of complexes 1a–8a and 1b–8b is shown in Scheme 1. The platinum(II) complexes 1a–8a were obtained by reacting the platinum precursor [PtCl2(dmso)2] with 1.05 molar equivalents of the corresponding ligand hqH by addition of 1.2 equivalents of sodium methoxide as a base. All these reactions were performed in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 methanol/acetone mixture. The solutions were filtered through Celite and the complexes were isolated as yellow solids after the evaporation of the solvents. The purity of the obtained complexes was above 98% and as such was suitable for further biological evaluation. Crystals of 6a were obtained by slow evaporation of the 1:2 methanol/acetone solvent mixture.
:2 methanol/acetone mixture. The solutions were filtered through Celite and the complexes were isolated as yellow solids after the evaporation of the solvents. The purity of the obtained complexes was above 98% and as such was suitable for further biological evaluation. Crystals of 6a were obtained by slow evaporation of the 1:2 methanol/acetone solvent mixture.
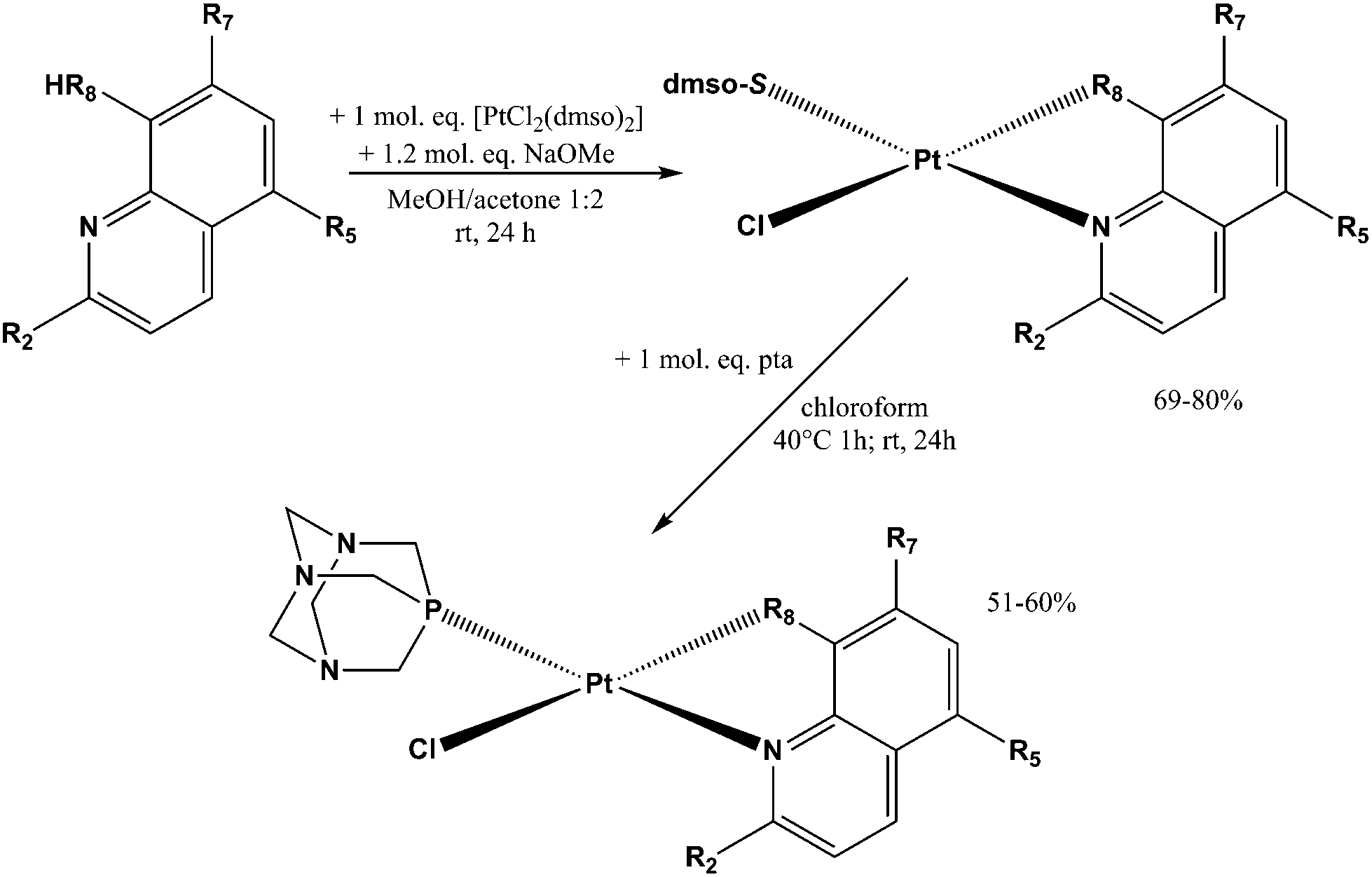 | ||
| Scheme 1 Schematic representation of the reactions for the synthesis of 1a–8a and 1b–8b. | ||
An exception was complex 2a, where the synthesis resulted in a mixture of regioisomers in a α:β ratio of 5:1 as evidenced by the 1H NMR spectrum (Fig. S1†). Attempts to synthesize or isolate each isomer by column chromatography on silica or alumina were not successful. Nevertheless, both isomers crystalized from a methanol/dichloromethane (DCM) solution where the major product was α-2a isomer crystallized as big yellow prismatic crystals while the minor β-2a product crystallized as much smaller and thin orange needle-like crystals.
Since we observed the presence of two geometric isomers of compound 2a, we searched the Cambridge Structural Database (CSD) for structural data of chemically related compounds with the fragment [PtCl(hq)]. At present (data retrieved on 11.4.2017), the database contains only six crystal structures of five different compounds containing this structural fragment. Complex 7a reported by Meng et al.32 adopts the α conformation, complex 4a also reported by Meng et al. is a co-crystal of both regioisomers (though the authors might not have noticed it, as this fact is not addressed in the paper at all), Kato et al. reported the structure of the complex (8-hqHH)[Pt(hq)Cl2], which is not relevant for regioisomer discussion,33 the papers of Ferraz et al.30 and Santos et al.16 both report the crystal structure of α-1a, and finally Yue et al. report the crystal structure of [PtCl(2-methyl-8-hydroxyquinolinato)(S-tetrahydrothiophene) which adopts the β conformation. Intrigued by the inconsistencies between the reported crystal structures as well as our own results and since we were unable to obtain crystals suitable for X-ray structural analysis of all novel compounds, we complemented spectroscopic and structural data with calculations of relative energies for α and β forms of complexes (Table 1) in order to unequivocally determine the structures of compounds. Complexes in which the oxygen/sulfur atom from the hq− ligand is at trans position relative to the chloride atom coordinated to the Pt(II) ion (β isomer; Table 1) were calculated to be lower in energy than the corresponding α isomer with the exception of 2a complex. These results suggest that the β isomer is energetically preferred, which well mirrors the experimentally observed structures in the reactions of complexation. On the other hand, the energy of the β form of 2a is higher than that of α, indicating that in this case α mode of coordination is energetically favorable (Table 1), though the energetic difference between the two isomers is the smallest in this case and can thus account for the concomitant presence of both isomers in solution.
Drawing definite conclusions is in this case very difficult and ours are supported by the following facts. Firstly, the crystals in the previous reports were obtained directly from the reaction mixtures,32 by attempting to crystallize a different complex from a dmso solution,30 or the crystallization conditions are not reported,16 while in our case pure isolated compounds (except 2a) were used for crystallization experiments. Secondly, based on our computational study, the experimental observation of both isomers in varying ratios in crude reaction mixtures and our structural studies, we could conclude that the reported crystal structures of α isomers are the result of crystallizations of minor regioisomers.
Complexes 1b–8b were obtained from the corresponding complexes 1a–8a in reactions with one molar equivalent of the pta ligand in chloroform. The reaction mixtures were heated for 1 h and then stirred at room temperature for the next 24 h, in order to obtain better yield and purity of the products, as prolonged heating resulted in increased amounts of side-products. Purifications of these complexes were performed on chromatographic columns with alumina as the stationary phase and acetone/DCM in different ratios as eluents. The column was first eluted with 3% acetone/DCM to remove the excess of the hq ligand, which was followed by elution with 20% acetone/DCM in which Rf factors of all complexes are in the range 0.65–0.80 while the starting [PtCl2(hq)(dmso)], as a common impurity present in all syntheses, has an Rf factor of 0.30–0.40. The solvents were rotary evaporated and we have tried to obtain crystals from solid residues. A mixture of DCM, in which the compounds are well soluble, and the nonpolar solvent n-hexane (the solvent mixture ratio was approximately 3:1) was used for crystallization (Fig. 3).
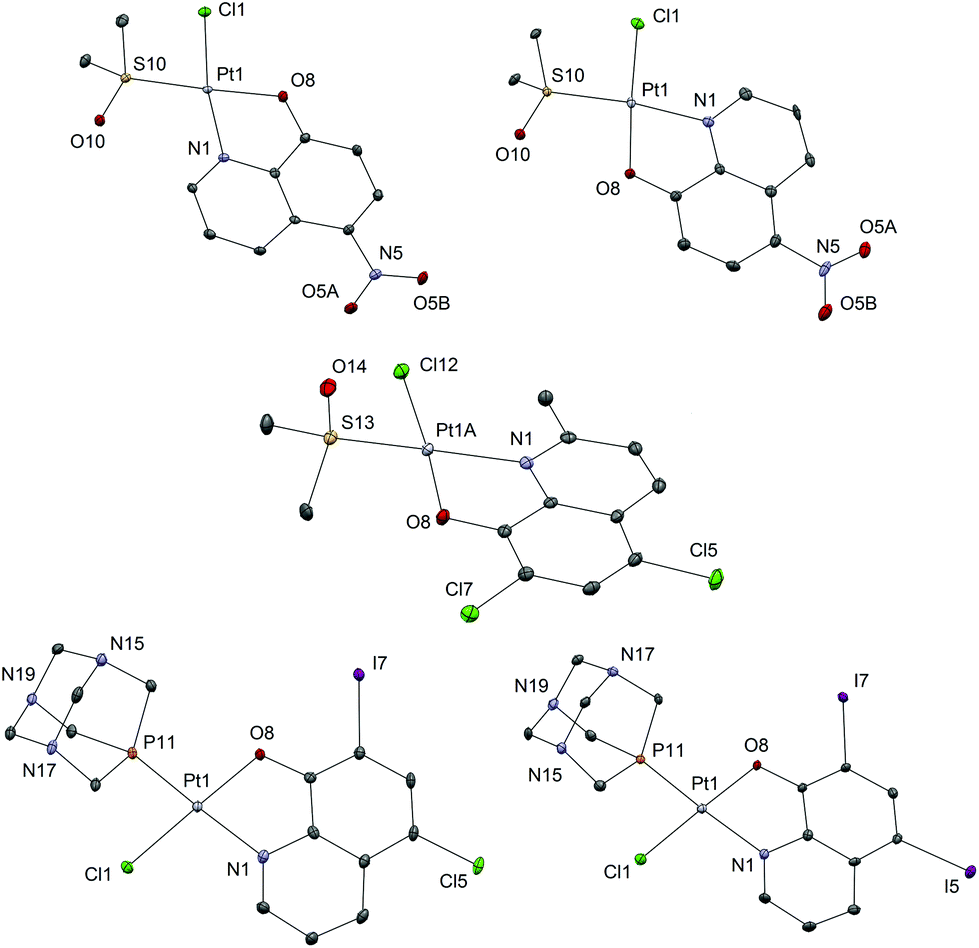 | ||
| Fig. 3 Crystal structures of complexes: top – α-2a, β-2a; middle – β-6a; bottom – β-1b and β-4b with heteroatom labelling. Thermal ellipsoids are shown at the 30% probability level. Hydrogen atoms are omitted for better clarity of presentation. For complex β-6a only one of the three crystallographically independent complex molecules is shown (for all three, see Fig. S2). | ||
All the investigated Pt(II) complexes have a square-planar geometry with the bidentately coordinated hq ligand. The complexes adopt a slightly distorted square-planar geometry as evidenced by the τ4 and τ′4 square-planar geometry indices (Table 2). There are no strong supramolecular interactions to explain the distortion which is most likely due to the steric bulk of the S-dmso and pta ligands in which the coordinated S and P atoms have tetrahedral geometry. To further support this explanation, the distortion is most notable in the case of compound 6a, bearing the only hydroxyquinoline ligand with a substituent at position 2 (methyl). The observed bond lengths and angles fall within the common range of values. Interestingly, complex 6a crystallizes with three crystallographically independent complex molecules with one co-crystallized solvent molecule (acetone), a phenomenon quite rarely observed (Fig. S2†). The chemical shifts of the aromatic protons of the ligands (7.19–9.65 ppm) are mostly shifted upfield upon bidentate coordination of hq oxygen and nitrogen atoms to Pt(II) (7.11–9.11 ppm). The singlet at 2.62 ppm for the methyl protons of free dmso was shifted downfield (3.55–3.67 ppm) after its coordination to the Pt(II) species in 1a–8a. Monodentate coordination of the pta ligand through the phosphorus atom in 1b–8b was detected by the appearance of the resonance in the region 4.40–4.48 ppm which belongs to the N–CH2–P methylene protons. This resonance is at 4.06 ppm for the free pta ligand. Correspondingly, in the 31P NMR spectrum of 1b–8b complexes, a single resonance peak at δ −97.4 ppm for the free pta ligand is shifted downfield to the region −64.10 to −61.41 ppm upon coordination of the pta ligand through the phosphorus atom.
| Cpd | Pt-N | Pt-O | Pt-Cl | Pt-P | τ 4 | τ′4 |
|---|---|---|---|---|---|---|
| τ 4 = [360° − (α + β)]/(360° − 2·θ); τ4′ = [(β − α)/(360° − θ)] + [(180° − β)/(360° − θ)]; α and β are the two greatest valence angles of the coordination center, θ is a tetrahedral angle of 109.471°. For the ideal square-planar geometry τ4 and τ′4 values are 0, and for the ideal tetrahedral geometry τ4 and τ′4 values are 1. | ||||||
| 1b | 2.063(4) | 2.010(3) | 2.308(2) | 2.198(2) | 0.070 | 0.089 |
| 4b | 2.079(5) | 2.007(4) | 2.298(2) | 2.198(2) | 0.066 | 0.083 |
Since dmso, a commonly used co-solvent in biological assays, is known to have deleterious effects on clinically used platinum agents such as cisplatin,36 we monitored the stability of complexes 1b and 4a in dmso solution over a period of 7 days by 1H NMR. The samples were kept at room temperature and in the dark between measurements. No changes in the NMR spectra of the studied complexes were observed (Fig. S3†). Considering this, we concluded that all the investigated Pt(II) complexes are stable in this solvent in the timeframe of the biological experiments and are suitable for further investigations.
Cell viability and uptake assays
The antiproliferative (cytotoxic) potentials of all sixteen Pt(II) complexes as well as cisplatin (Cis) were screened on human lung fibroblasts (MRC5) and melanoma (A375) by MTT assay after 48 h treatment (Fig. 4). All complexes, except 6a, exhibited a high cytotoxic effect on MRC5 cells at a concentration of 10 μg mL−1 (Fig. 4A). 3a, 2b, 6b, 8a, 8b and Cis caused 10–40% cell death at a concentration of 5 μg mL−1 on this model of normal cell lines (Fig. 4A). Generally, the tested compounds exhibited similar profiles of antiproliferative activity against the melanoma cell line (Fig. 4B), with 1b, 5a, 6a and 3b showing a certain selectivity towards the carcinoma cell line. In general, pta-derivatives appeared to be more cytotoxic. This is in line with previous observations that transition-metal complexes containing one or more pta ligands had improved aqueous solubility and therefore greater biological activity.28,29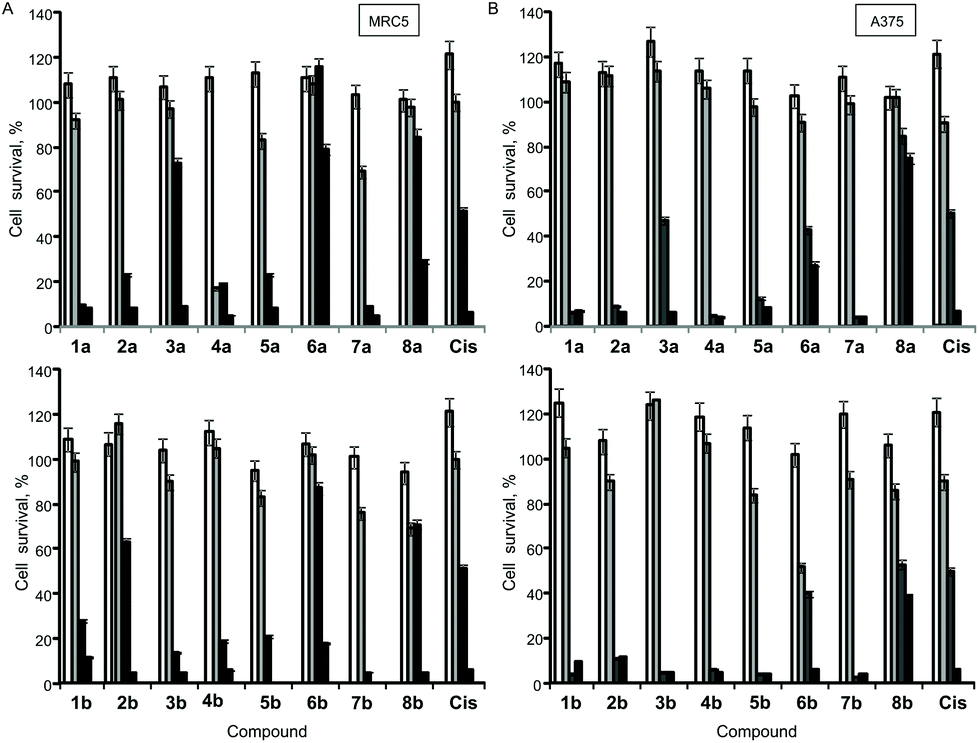 | ||
Fig. 4
In vitro cytotoxicity (antiproliferative effect) profile of Pt(II) complexes (1–8a/b) and cisplatin (Cis) against: (A) human lung fibroblast (MRC5) and (B) melanoma (A375) cell lines (□, 0.1 μg mL−1;  , 1 μg mL−1; , 1 μg mL−1;  , 5 μg mL−1 and ■, 10 μg mL−1 concentration of the tested compound). , 5 μg mL−1 and ■, 10 μg mL−1 concentration of the tested compound). | ||
Based on this initial screen considering selectivity towards cancer cells, four pairs of complexes were selected for further evaluation of the anticancer potential, making sure that both dmso- and pta-counterparts were included. IC50 values were determined against MRC5, A375 (melanoma) and human lung cancer (A549) by MTT assay (Table 3). Generally, the selected compounds showed a selectivity index comparable to cisplatin, but 1b, 5b, and 6a had IC50 values 2–2.5-fold higher against cancer cell lines (Table 3). 5b (5,7-dibromo-8-quinolinol-pta), 3b (5,7-dichloro-8-quinolinol-pta) and 1a (5-chloro-7-iodo-8-quinolinol-dmso) were the most cytotoxic, with IC50 values comparable to that of cisplatin. These three complexes and their corresponding counterparts were used in uptake experiments to ensure their ability to enter the cells and to compare it to that of cisplatin (Fig. 5).
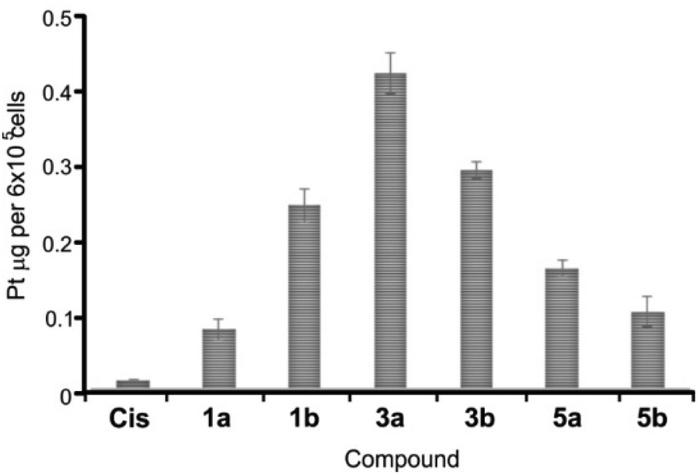 | ||
| Fig. 5 Platinum content in A549 cells. Cells were treated with 2 μM cisplatin (Cis) and selected complexes for 2 h at 37 °C. Data are shown as mean values ± standard deviations. | ||
| Compound | Cell lines | ||
|---|---|---|---|
| MRC5 | A375 | A549 | |
| 1a | 3.2 ± 0.1 | 4.1 ± 0.1 | 2.5 ± 0.1 |
| 1b | 7.2 ± 0.2 | 2.9 ± 0.1 | 4.3 ± 0.2 |
| 3a | 13.4 ± 0.4 | 11.5 ± 0.3 | 11.5 ± 0.3 |
| 3b | 4.2 ± 0.2 | 3.3 ± 0.1 | 2.5 ± 0.1 |
| 5a | 6.5 ± 0.3 | 4.1 ± 0.1 | 7.4 ± 0.4 |
| 5b | 3.6 ± 0.1 | 2.9 ± 0.1 | 1.5 ± 0.1 |
| 6a | 18.7 ± 0.5 | 9.3 ± 0.3 | 8.4 ± 0.5 |
| 6b | 12.2 ± 0.4 | 12.2 ± 0.5 | 9.8 ± 0.3 |
| Cis | 3.3 ± 0.1 | 4.1 ± 0.2 | 2.5 ± 0.1 |
In these experiments, 2 μM cisplatin and selected Pt(II) complexes were used since this concentration is close to IC50 values for Cis, 1a, 3b and 5b (Table 3). Upon exposure to 2 μM solutions for 2 h, the cellular content of Pt metal substantially increased in comparison with the cells that were treated with the vehicle solvent (dmso) that had Pt content close to zero. Therefore, facile uptake of both Cis and Pt(II) complexes occurred within 2 h of exposure (Fig. 5). Notably, all the tested complexes showed an increased ability to internalize in comparison with Cis, especially 3a, that had the highest IC50 value of 11.5 μM and showed the best uptake, suggesting that the activity of the compounds only partially depends on the ability to enter the cells. Complexes containing a bulkier pta-ligand (b series) were less readily taken up by cells in comparison with dmso-based complexes, with the exception of 1b (Fig. 5). A recent study also showed successful cellular uptake and equal distribution between the nucleus and mitochondria of 4a (5,7-diiodo-8-hydroxyquinoline complex) in HepG2 cells.32
Previously, the investigation of a series of four platinum(II) complexes with 8-hydroxyquinoline derivatives, including 1a, 3a and 7a, showed that cytotoxic activity for several tumor cell lines – HBL-100 (breast), HeLa (cervix), SW1573 (non-small cell lung carcinoma) and WiDr (colon carcinoma) – strongly depends on the substituents on the quinoline ring.16 Thus, in the case of the HLB-100, SW1573 and WiDr cell lines, a high biological activity was obtained for 8-hydroxyquinoline complexes with hydrogen or iodine at position 7, while the chlorine atom at the same position reduces the cytotoxicity. However, the HeLa cell line showed a different response where the substitution at position 7 of 8-hydroxyquinoline by a chlorine atom increases the cytotoxic activity of the platinum complex. The complex with the non-substituted 8-hydroxyquinoline was the most active in all the tested cell lines.16 Moreover, the effect of iodine substitution was studied in the case of complexes 7a and 4a (hq ligand and diiodo substituted analog, respectively). The inclusion of iodine caused an increase in cytotoxicity with a decrease of IC50 values from 9–30 μM to 4–12 μM (by a factor of 1.8–4.3) in five selected human cancer cell lines (BEL-7404, hepatoma; HepG2, liver; NCI-H460, lung; T-24, bladder carcinoma, A549, lung carcinoma) while retaining the same toxicity level on normal liver cells HL-7702 (IC50 = 25 μM). The most sensitive of the tested cell lines, HepG2 was subjected to cell cycle analysis which showed cell cycle arrest in the S phase with higher S phase arrest for the iodo-substituted compound. This was supported by the down-regulation of cdc25A, cyclin B, cyclin A, and CDK2 and the up-regulation of p53, p27, and p21, which was determined by the western blot assay. Complex 4a also acted as a telomerase inhibitor by interacting with G-quadruplexes and triggering cell senescence and apoptosis. Mitochondrial dysfunction was also observed in the treated cells, which might suggest an induction of mitochondrion-mediated apoptotic pathways, a mechanism not unknown for platinum-based agents.37 Moreover, the inclusion of iodine improved cellular uptake of Pt species and the authors suggest that halogenation of selected scaffolds in metal-based drug design is an interesting strategy.
In line with a previous study,16 we have confirmed that 1a (8-hydroxyquinoline complexes with chlorine at position 5 and iodine at position 7) was the most potent in terms of IC50 values. In addition, the pta-based complex (1b) showed similar activity; however, the selectivity of both complexes was quite poor (Table 3). Furthermore, the lung cancer cell line again appeared to be more sensitive in comparison with melanoma, like SW1573 in the previous study in comparison with breast and cervical cancer cells. The obtained results showed that the substitution pattern had an important effect on the antiproliferative activity and selectivity of the complexes depending on the cell line.
Interactions of Pt(II) complexes with DNA
It was established that Pt(II) complexes bind to DNA via coordination bonds distorting the normal three-dimensional structure of the double helix.38,39 This subsequently induces cell death through apoptosis.40 Given that DNA is often the primary target of Pt(II)-based antitumour complexes,41 we have investigated the reactivity of complexes 1a and 1b with the model compound 5′-GMP by 1H NMR: the ability of Pt(II) complexes 1–8 to interact with double stranded high molecular weight DNA was investigated in vitro (Fig. 6A) and assessed in silico (Fig. 6B).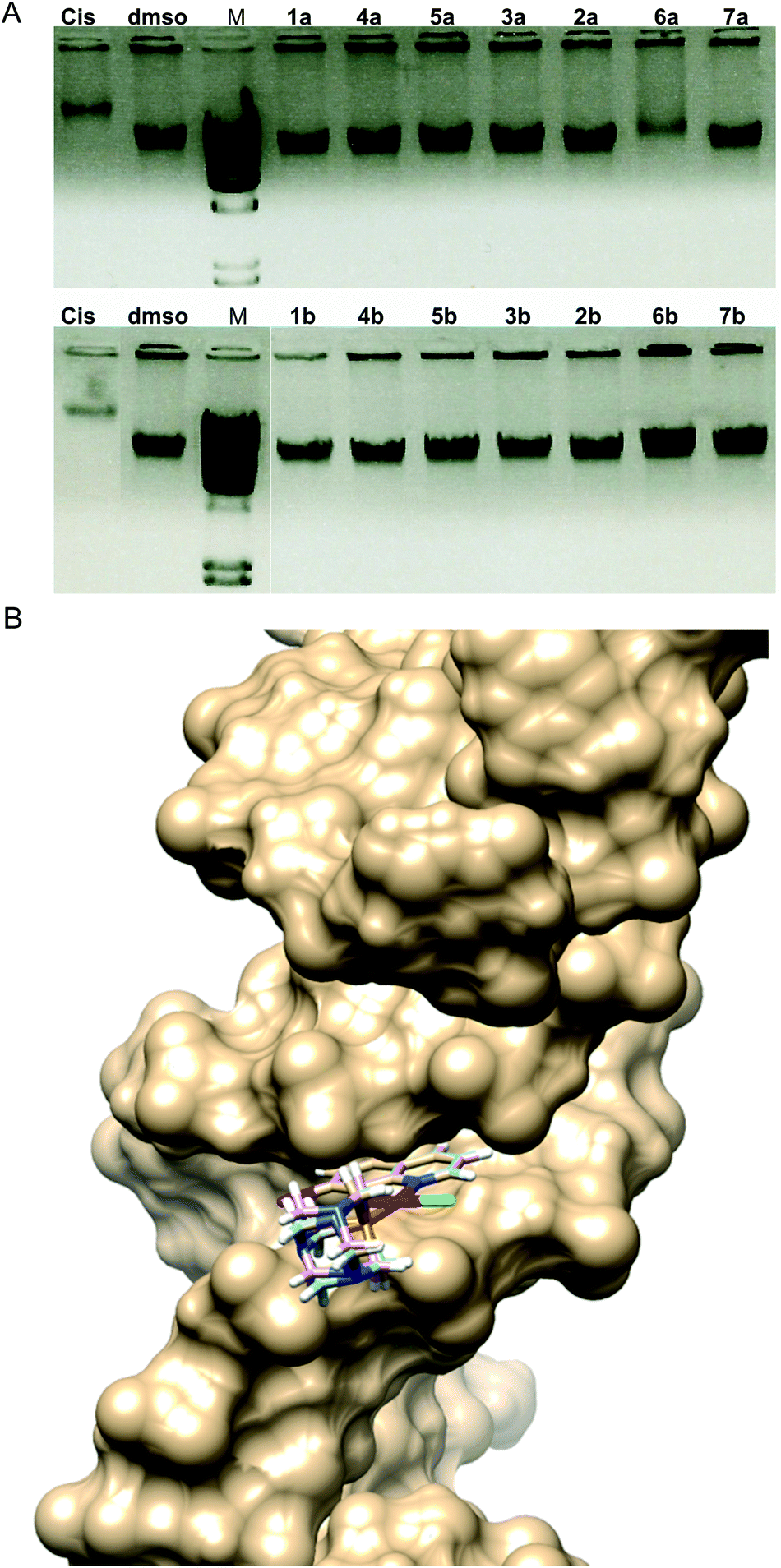 | ||
| Fig. 6 DNA interaction ability of Pt(II) complexes (A) 1–7a/b in comparison with cisplatin (Cis) and dmso control (M-λ-HinDIII high molecular weight marker, Promega); and (B) computational docking model illustrating the interactions between compounds 1b, 3b and 5b and the minor groove of DNA. | ||
As evidenced by 1H NMR experiments both 1a and 1b form adducts with 5′-GMP within a few hours as we observe a second set of signals assigned to 5′-GMP. However, the relative ratio of this set of signals compared to the free 5′-GMP decreases significantly within 24 hours with free 5′-GMP signals again becoming predominant after 72 hours in the case of 1a (Fig. S4†). In the case of 1b (Fig. S5†) the second set of 5′-GMP signals remains minor throughout the experiment; however, a third set of hydroxyquinoline signals is observed which becomes predominant after 24 h. Due to the known aqueous chemistry of platinum(II) coordination compounds and the recent discoveries of the combined solvolytic effect of dmso and water42 it is safe to assume that the chlorido and the 5′-GMP ligands are released and a stable aqua or S-dmso adduct is formed. As such, this weak interaction might not prove significant in more complex biological systems in the presence of other ligands with potentially higher affinity for metal ions (inorganic anions, amino acid residues, etc.).
Electrophoresis patterns in the presence of 25 μg mL−1 concentration of Pt(II) complexes showed that most of the complexes did not have the ability to interact with DNA in vitro in aqueous solution at physiological pH 7.4 (Fig. 6A). This was somewhat surprising due to the fact that an 8-hydroxyquinoline conjugated system is expected to bind DNA via intercalation. Only complex 6a showed similar interaction to that of cisplatin, while 1b exhibited a slight ability to interact with DNA (approx. 10% in comparison with the dmso control). It is worth mentioning that complexes 8a and 8b could not be included in this experiment due to their differing spectral characteristics that may have caused the interference with the signal (Fig. S6†).
In vitro DNA interaction pattern results may imply that complexes 1b and especially 6a could exhibit irreversible DNA degradation which may lead to cell death by necrosis, therefore indicating a potentially higher general toxicity of these compounds. However, both compounds exhibited lower antiproliferative activities in cytotoxicity assays in comparison with cisplatin (Table 3). On the other side, complexes 5b and 3b showed comparable and better cytotoxicity profiles in comparison with cisplatin, and no apparent DNA interaction ability. From the interaction with high molecular weight double stranded DNA in vitro, cytotoxicity against human cell lines could not be predicted, nor could DNA as a cellular target be completely excluded. Therefore, we examined the DNA-interaction ability of the selected complexes in silico and DNA fragmentation pattern of the treated A549 cells, as well as zebrafish embryos (Fig. S7†).
All information related to in silico DNA binding affinity for the studied compounds was obtained using a molecular docking study. The predicted top-ranking pose with the complex's lowest energy was used to suggest the best possible geometry of compounds inside the DNA double helix and the highest binding affinity of DNA. MolDock Score, Docking Score, Rerank Score, van der Waals (VdW) and van der Waals (Hbond) Score, Steric interactions, and Ligand efficiency (LE1 and LE3) were used as scoring functions. Top ranked poses according to the used scoring functions are presented in Table 4.
| Cpd | Hbond | VdW | E-Inter | LE1 | LE3 | Pose energy | Rerank | MolDock |
|---|---|---|---|---|---|---|---|---|
| E-Inter – Inter-energy of pose. LE1 – Ligand efficiency calculated as MolDock Score divided by heavy atom count. LE3 – Ligand efficiency calculated as Rerank Score divided by heavy atom count. | ||||||||
| 1a | −5 | −129.067 | −98.269 | −509.929 | −333.842 | −95.280 | −630.430 | −96.887 |
| 1b | −478.944 | −652.659 | −123.771 | −424.062 | −277.349 | −103.852 | −693.372 | −106.015 |
| 3a | −5 | −132.847 | −100.705 | −517.327 | −342.509 | −97.098 | −650.766 | −98.292 |
| 3b | −473.265 | −622.397 | −123.413 | −417.075 | −275.087 | −102.485 | −687.718 | −104.269 |
| 5a | −205.606 | −497.911 | −94.029 | −478.949 | −287.765 | −89.249 | −546.753 | −91.002 |
| 5b | −142.906 | −448.058 | −116.990 | −448.534 | −262.665 | −110.866 | −656.663 | −112.134 |
| 6a | −5 | −213.766 | −103.317 | −491.222 | −281.537 | −96.211 | −563.073 | −98.244 |
| 6b | −49.584 | −487.517 | −130.261 | −432.598 | −264.487 | −111.168 | −687.666 | −112.475 |
The more the negative value obtained from the applied score function, the greater the binding propensity of the complex with DNA, which can be correlated with the obtained experimental values for anticancer activity. According to Rerank Score, compounds with the highest binding potential were 1b (−693.372 kcal mol−1), 3b (−687.718 kcal mol−1), 6b (−687.666 kcal mol−1) and 5b (−656.663 kcal mol−1). The obtained results for van der Waals interactions of complexes with DNA revealed that among the studied complexes compound 1b has the highest interaction energy (−652.659 kcal mol−1) followed by compounds 3b (−622.397 kcal mol−1) and the lowest interaction was determined for compound 1a (−129.067 kcal mol−1).
Hydrogen bonds formed between a complex and DNA are important for metal complex–DNA binding and for other possible activities, such as antiviral or antibacterial.43 According to DNA (Hbond) Score values, compounds with the highest interaction energy were 6b and 3b (−478.944 kcal mol−1 and −473.265 kcal mol−1). Also docking studies determined that compounds 1a, 3a and 6a didn't form hydrogen bonds with DNA. Therefore, it can be concluded that the obtained results are in correlation with the presented experimental results for IC50 values (3b having IC50 values between 2.5 and 4.2 μM; Table 3), but to a much lower extent with in vitro DNA interaction ability experiments, with 6a showing some interaction (Fig. 6A).
The best poses for the selected complexes according to in vitro and in silico studies in relation to their overall values for interaction with DNA are presented in Fig. 6B. The minimum energy docked pose revealed that all complexes are very well fitted into the DNA minor groove. Docking poses suggest that complexes and DNA base pairs are arranged in such a way that they have effective π–π stacking interactions. These interactions can lead to higher van der Waals interactions with the DNA functional groups which define the stability of the groove, making the AT regions more preferable regions of the dodecamer.44 Complexes exhibited additional stabilization through the strong intermolecular hydrogen bonding interaction between the C-2 carbonyl oxygen of T and the N-3 nitrogen of A. The best binding pose in the AT stretches of the minor groove was indicative of an extensive H-bonding network. Structural analysis of the docking positions gave insight into the binding pattern of the complexes. According to the obtained results for the preferable conformation of the complex inside DNA, only the hydroxyquinoline (hq) part of the complex interacted with DNA. This fact can be correlated with the fact that the studied complexes had a different mechanism of action in comparison with cisplatin. Calculations revealed that in most cases, preferable atoms from the studied complex molecule's ligands that interacted with DNA were halogen atoms.
In accordance with observations on the DNA interaction ability and from the in silico assessment, no apparent DNA fragmentation was observed either on DNA extracted from the treated cancer cell line or from the zebrafish embryos, while cisplatin was shown to induce DNA damage under the same conditions (Fig. S7†). Indeed, it is generally accepted that binding of cisplatin to genomic DNA (gDNA) in the cell nucleus is largely responsible for its antitumor properties. The damage induced upon binding of cisplatin to gDNA interferes with normal transcription and/or DNA replication mechanisms, and eventually, these disruptions in DNA processing trigger cytotoxic processes that lead to cell death.45 However, it is known that cisplatin forms a large amount of adducts in mitochondrial DNA lacking histones.46 In any event, previous to cisplatin binding to genomic or mitochondrial DNA a loss of chloride groups is required. The N7 atoms of guanine and adenine located in the major groove of the double helix are the most accessible and reactive nucleophilic sites for platinum binding to DNA.47,48 In accordance with our observations, Kemp and co-workers have reported that DNA binding experiments have shown no significant differences in the DNA adducts formed between many of the DNA intercalators.49,50 This suggests that all DNA adducts formed by the bidentately coordinated platinum(II) family of metal complexes are similarly cytotoxic and/or that DNA binding may not be the sole mechanism that determines their cytotoxicity. Therefore, it can be hypothesised that differing cellular uptake levels or differing intracellular drug transport may determine how much of each DNA intercalator reaches its target intact, thereby affecting cytotoxicity.
Toxicity of Pt(II) complexes (1a, 1b, 3a, 3b, 5a and 5b) in zebrafish embryos
The effects of complexes on development of Danio rerio embryos were examined at concentrations close to IC50 values determined in the cytotoxicity assessment (Table 3). The selection of the complexes to be tested in zebrafish embryo model was based on the highest selectivity between MRC5 and the cancer cells. We exposed 4 h old embryos to various concentrations of Pt(II)-complexes and the obtained results revealed different toxicity profiles between pta-based and dmso-based platinum complexes (Table 5). According to the LC50 values obtained in zebrafish assay (Table 5) the tested complexes are ranked by their embryotoxicity as follows: 3b > 5b > 1b > 1a > 5a > 3a, which clearly demonstrates higher toxicity of pta-based complexes (series b) in comparison with dmso-based complexes (series a), mirroring the results of the MTT screen (Fig. 4). While dmso-based complexes had adverse effects on embryo development at concentrations of 10 μg mL−1 (5a) and ≥5 μg mL−1 (1a) and had LC50 values higher than the IC50 values obtained in the cell lines assay, pta-based complexes induced embryo mortality already at concentrations ≥1.5 μg mL−1 (1b and 3b) and ≥2 μg mL−1 (5b) (Fig. 7A) and showed LC50 values comparable to those on cell lines. Interestingly, complexes 1b and 3b induced similar defects in embryos such as skeletal deformities and pericardial edema (Fig. 7B), whereas 3b appeared to be more cardiotoxic than 1b, inducing slower embryo heart beat rate, absence of caudal circulation and, consequently, higher pericardial edema. On the other hand, complex 5b had a different milder teratogenic effect, inducing only notochord deformity without effects on the development of other organs (Fig. 7B). | ||
Fig. 7 Effects of platinum(II) complexes (dmso-based: 1a, 3a and 5a in comparison with pta-based: 1b, 3b and 5b) on the development of zebrafish embryos: (A) at 0.5 to 10 μg mL−1 concentration at 96 hours post fertilization (■ – normal embryos,  – teratogenic embryos, and □ – lethal embryos) and (B) images of zebrafish embryos at 96 hpf exposed to selected Pt(II) complexes. Depending on the type and concentration of platinum complexes, treated zebrafish embryos developed pericardial edema (3b), skoliosis (1b and 3b), malformed jaw (5a) or malformed notochord (5b). – teratogenic embryos, and □ – lethal embryos) and (B) images of zebrafish embryos at 96 hpf exposed to selected Pt(II) complexes. Depending on the type and concentration of platinum complexes, treated zebrafish embryos developed pericardial edema (3b), skoliosis (1b and 3b), malformed jaw (5a) or malformed notochord (5b). | ||
| Compound | LC50 (μg mL−1) | EC50 (μg mL−1) |
|---|---|---|
| a 3a and cisplatin were nontoxic at the tested concentration range (1–10 μg mL−1); LC50 – the concentration causing the mortality of 50% of the zebrafish embryos; EC50 – the concentration with a negative effect on the development of 50% of the zebrafish embryos (mortality and teratogenicity). | ||
| 1a | 3.53 | 3.73 |
| 1b | 2.82 | 2.21 |
| 3a | >10a | >10a |
| 3b | 1.48 | 1.46 |
| 5a | 8.92 | 6.89 |
| 5b | 2.47 | 2.23 |
| Cis | >10a | >10a |
Although cisplatin appeared to be nontoxic on developing zebrafish embryos, treatments under 10 μg mL−1 cisplatin prevented embryo hatching up to 120 hours post fertilization (hpf) that is regarded as a toxicity parameter. Hatching success is supposed to be a sensitive endpoint of the zebrafish embryo in toxicity assay, since no hatched embryos had a lethal outcome.51 While it has been demonstrated that platinum and platinum nanoparticles had an adverse impact on the hatching success in developing zebrafish embryos,51,52 we have recently shown that other Pt(II) complexes, such as {[Pt(1,3-pd)Cl]2(μ-pz)}Cl2·2LiCl·2H2O, may adversely influence zebrafish hatching as well.39 On the other hand, none of the platinum(II)-complexes tested in this study interfered with hatching success.
The most cytotoxic complexes (1a, 1b, 3a, 3b, 5a and 5b) were also tested for the ability to induce the oxidative stress response in zebrafish and to compare it to that of Cis (Fig. 8). Reactive oxygen species (ROS) production at sublethal concentration and concentration close to LC50 concentration values (Table 5) was significantly higher in embryos treated with pta-based complexes in comparison with that of dmso-based ones (from 2.5-fold for 1avs.1b to 10-fold for 3avs.3b) and in comparison with Cis (Fig. 8B).
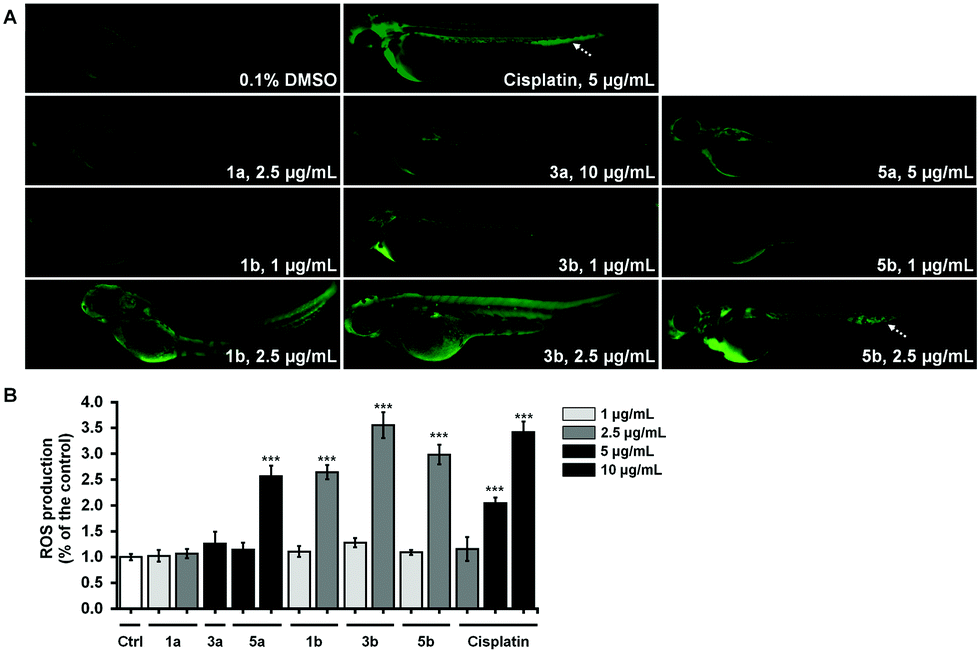 | ||
| Fig. 8 Effects of platinum(II) complexes (dmso-based: 1a, 3a and 5a in comparison with pta-based: 1b, 3b and 5b) on ROS production in zebrafish embryos (A) with the increased ROS production in the caudal hematopoietic tissue marked by a dashed arrow. ROS levels were determined by measuring the fluorescence of individual zebrafish embryos (B). Statistically significant differences between the control (0.1% DMSO) and treated groups are denoted with asterisks (*P < 0.5, **P < 0.01; ***P < 0.001; Student's t-test). | ||
All three pta-based complexes markedly increased embryos’ ROS level at the IC50 values, thus indicating that this may be one of the mechanisms of their activity. Notably, embryos upon cisplatin treatment displayed an intensive fluorescence in the region of caudal hematopoietic tissue at doses of 5 μg mL−1 and 10 μg mL−1 (Fig. 8A), indicating its possible myelosuppressive effect. Dose-dependent myelosuppression, particularly neutropenia, is one of the major side effects of cisplatin therapy in cancer patients, restricting its therapeutic use.53 On the other hand, none of the tested complexes (except 5b at 2.5 μg mL−1) increased ROS production in the caudal hematopoietic tissue.
Cisplatin was approved by the Food and Drug Administration in 1978 in spite of some dose limiting side effects; hence data on cisplatin toxicology in wild type zebrafish that emerged as a model later is not readily available in the literature. It was shown that 50 μM and 100 μM cisplatin significantly reduced hair-cell survival up to 81% and 55%, respectively, while at a concentration of 400 μM, cisplatin induced a lethal outcome in embryos.54 It is noteworthy that 5b, which demonstrated preferential cytotoxicity against cancer cell lines, did not provoke significant in vivo toxic response on the zebrafish embryo model at concentrations close to IC50 values for certain cell lines (i.e. lung cancer), making it promising for further development as a potential therapeutic agent.
Experimental section
Materials and methods
Dimethyl sulfoxide (dmso), 1,3,5-triaza-7-phosphaadamantane (pta), 5-chloro-7-iodo-8-quinolinol (clioquinol), 8-hydroxy-5-nitroquinoline (nitroxoline), 5,7-dichloro-8-quinolinol, 5,7-diiodo-8-quinolinol,5,7-dibromo-8-quinolinol, 5,7-dichloro-8-hydroxy-2-methyl-quinoline, 8-hydroxyquinoline, 8-quinolinethiol, and K2[PtCl4] were obtained from Sigma-Aldrich. The complex cis-[PtCl2(dmso)2] was synthesized according to the published procedure.55The NMR spectra were recorded on a Bruker Advance II 500 spectrometer (at room temperature and 500.1 MHz) by using TMS as an internal standard. Infrared spectra were recorded with a PerkinElmer Spectrum 100 FTIR spectrometer equipped with a Specac Golden Gate Diamond ATR as a solid sample support. UV–vis spectra were collected on a PerkinElmer LAMBDA 750 UV/vis/near-IR spectrophotometer, while the samples were prepared in chloroform with a concentration of 1 × 10−4 mol L−1. Elemental analyses for carbon, hydrogen and nitrogen parameters were performed using a PerkinElmer 2400 II instrument. HRMS were recorded using an Agilent 6224 Accurate Mass TOF LC/MS instrument. X-ray diffraction data were collected using an Oxford Diffraction SuperNova diffractometer with a Mo or Cu microfocus X-ray source with mirror optics and an Atlas detector. The structures were solved by direct methods implemented in SIR9256 and refined by a full-matrix least-squares procedure based on F2 using SHELXL-97.57 All non-hydrogen atoms were refined anisotropically. The hydrogen atoms were placed at calculated positions and treated using appropriate riding models. The programs Mercury and Platon were used for data analysis and figure preparation.58,59 Complete crystallographic data are given in the ESI file in Tables S2 and S3.† The crystal structures of compounds α-2a, β-2a, 6a, 1b, and 4b have been submitted to the CCDC and have been allocated deposition numbers CCDC 1483253–1483257.†
Synthesis and characterization of Pt(II) complexes
0.25 mmol (105.56 mg) of cis-[PtCl2(dmso)2] were dissolved in 15 mL of a 1:2 mixture of methanol and acetone. To this solution were added 1.05 molar equivalents of the corresponding hqH ligand and 1.2 molar equivalents of sodium methoxide (acting as a base). The obtained mixture was stirred at room temperature for 24 h. The solvent was then rotary evaporated and the obtained solid product represented the corresponding Pt(II) complex of sufficient purity with yields between 70 and 80%. The purity of each Pt(II) complex (>98%) was checked by elemental microanalysis, ESI-HRMS and NMR spectroscopy.
[PtCl(5-chloro-7-iodo-8-hydroxyquinolinato)(S-dmso)] (1a). Yield 78% (0.1116 g), yellow solid. 1H NMR (CDCl3, 500.10 MHz): δ 9.41 (dd, J = 5.3, 1.1 Hz, 1H, C2H), 8.59 (d, J = 1.2 Hz, 1H, C4H), 7.95 (s, 1H, C6H), 7.46 (dd, J = 8.5, 5.4 Hz, 1H, C3H), 3.68 (s, 6H, CH3-dmso). IR selected bands (cm−1, ATR): 3007, 1551, 1488, 1454, 1397, 1377, 1127, 1117, 1043, 1027, 805. UV–vis (λ (nm) (ε (L mol−1 cm−1)) c = 1 × 10−4 mol L−1, CHCl3): 287 (33
000), 431 (4900). ESI-HRMS (CH3CN) m/z (found for [M + H]+ (calc.)): 611.8550 (611.8453). Anal. calc. for C11H10Cl2INO2PtS: C, 21.55; H, 1.64; N, 2.28. Found: C, 21.11; H, 1.92; N, 1.96.
[PtCl(8-hydroxy-5-nitroquinolinato)(S-dmso)] (2a). Yield: 78% (0.0923 g), yellow solid. The presence of two isomers is evident in the NMR spectrum of 2a in CDCl3. 1H NMR (CDCl3, 500.10 MHz): δ 9.72 and 9.56 (2 × dd, J = 8.9, 1.1 Hz, J = 5.3, 1.2 Hz, 2 × 1H), 8.71 and 8.67 (2 × d, J = 9.1 Hz, J = 9.2 Hz, 2 × 1H), 7.82 and 7.75 (2 × dd, J = 8.9, 5.3 Hz, J = 9.0, 5.1 Hz, 2 × 1H), 7.18 and 7.08 (2 × d, J = 9.1 Hz, J = 9.1 Hz, 2 × 1H), 3.67 and 3.56 (s, 2 × 6H, CH3-dmso) ppm. IR selected bands (cm−1, ATR): 3060, 3017, 1569, 1504, 1485, 1282, 1129, 817. UV–vis (λ (nm) (ε (L mol−1 cm−1)) c = 1 × 10−4 mol L−1, CHCl3): 286 (23
700), 328 (9800), 430 (12000). ESI-HRMS (CH3CN) m/z (found for [M + H]+ (calc.)): 496.9822 (496.9724). Anal. calc. for C11H11ClN2O4PtS: C, 26.54; H, 2.23; N, 5.63. Found: C, 26.96; H, 2.23; N, 5.58.
[PtCl(5,7-dichloro-8-hydroxyquinolinato)(S-dmso)] (3a). Yield: 69% (0.0841 g), yellow solid. 1H NMR (CDCl3, 500.10 MHz): δ 9.50 (d, J = 4.5 Hz, 1H, C2H), 8.68 (d, J = 8.5 Hz, 1H, C4H), 7.69 (s, 1H, C6H), 7.53 (dd, J = 5.5, 5.5 Hz, 1H, C3H), 3.66 (s, 6H, CH3-dmso) ppm. IR selected bands (cm−1, ATR): 3029, 3011, 1458, 1381, 1372, 1128, 1119, 1034, 890. UV–vis (λ (nm) (ε (L mol−1 cm−1)) c = 1 × 10−4 mol L−1, CHCl3): 276 (39
000), 428 (5100). ESI-HRMS (CH3CN) m/z (found for [M + H]+ (calc.)): 519.9194 (519.9097). Anal. calc. for C11H10Cl3NO2PtS: C, 25.32; H, 1.93; N, 2.68. Found: C, 25.69; H, 1.93; N, 2.64.
[PtCl(5,7-diiodo-8-hydroxyquinolinato)(S-dmso)](4a). Yield: 75% (0.1231 g), yellow solid. 1H NMR (CDCl3, 500.10 MHz): δ 9.33 (dd, J = 5.4, 1.1 Hz, 1H, C2H), 8.41 (dd, J = 8.5, 1.1 Hz, 1H, C4H), 8.32 (s, 1H, C6H), 7.40 (dd, J = 8.5, 5.4 Hz, 1H, C3H), 3.69 (s, 6H, CH3-dmso) ppm. IR selected bands (cm−1, ATR): 3011, 1485, 1388, 1375, 1361, 1125, 1035. UV–vis (λ (nm) (ε (L mol−1 cm−1)) c = 1 × 10−4 mol L−1, CHCl3): 290 (42
000), 353 (2200), 434 (6100). ESI-HRMS (CH3CN) m/z (found for [M + H]+ (calc.)): 705.7933 (705.7932). Anal. calc. for C11H10ClI2NO2PtS: C, 18.75; H, 1.43; N, 1.99. Found: C, 19.02; H, 1.26; N, 1.98.
[PtCl(5,7-dibromo-8-hydroxyquinolinato)(S-dmso)] (5a). Yield: 78% (0.1304 g), yellow solid. 1H NMR (CDCl3, 500.10 MHz): δ 9.49 (dd, J = 5.4, 1.1 Hz, 1H, C2H), 8.64 (dd, J = 8.5, 1.1 Hz, 1H, C4H), 8.00 (s, 1H, C6H), 7.54 (dd, J = 8.6, 5.4 Hz, 1H, C3H), 3.69 (s, 6H, CH3-dmso) ppm. IR selected bands (cm−1, ATR): 3087, 3022, 3002, 1495, 1454, 1370, 1126, 1117, 1030, 914, 871. UV–vis (λ (nm) (ε (L mol−1 cm−1)) c = 1 × 10−4 mol L−1, CHCl3): 278 (16
000), 347 (2100), 428 (4400). ESI-HRMS (CH3CN) m/z (found for [M + H]+ (calc.)): 607.8181 (607.8082). Anal. calc. for C11H10Br2ClNO2PtS: C, 21.64; H, 1.65; N, 2.29. Found: C, 21.97; H, 1.42; N, 2.18.
[PtCl(5,7-dichloro-8-hydroxy-2-methyl-quinolinato)(S-dmso)] (6a). Yield: 80% (0.1179 g), yellow solid. 1H NMR (CDCl3, 500.10 MHz): δ 8.46 (d, J = 8.7 Hz, 1H), 7.53 (s, 1H), 7.32 (d, J = 8.7 Hz, 1H), 3.55 (s, 6H, CH3-dmso), 3.21 (s, 3H, CH3) ppm. IR selected bands (cm−1, ATR): 3011, 1554, 1499, 1448, 1366, 1136, 1022, 969. UV–vis (λ (nm) (ε (L mol−1 cm−1)) c = 1 × 10−4 mol L−1, CHCl3): 284 (35
000), 332 (2900), 433 (4900). ESI-HRMS (CH3CN) m/z (found for [M + H]+ (calc.)): 533.9353 (533.9250). Anal. calc. for C12H12Cl3NO2PtS: C, 26.90; H, 2.26; N, 2.61. Found: C, 27.11; H, 2.21; N, 2.59.
[PtCl(8-hydroxyquinolinato)(S-dmso)] (7a). Yield: 79% (0.0546 g), yellow solid. 1H NMR (CDCl3, 500.10 MHz): δ 9.40 (dd, J = 5.3, 1.2 Hz, 1H), 8.35 (dd, J = 8.3, 1.0 Hz, 1H), 7.52–7.41 (m, 2H), 7.07 (d, J = 8.0 Hz, 2H), 3.64 (s, 6H, CH3-dmso) ppm. IR selected bands (cm−1, ATR): 3014, 2916, 1504, 1470, 1382, 1320, 1122, 1112, 1033, 818. UV–vis (λ (nm) (ε (L mol−1 cm−1)) c = 1 × 10−4 mol L−1, CHCl3): 268 (38
000), 413 (4700). ESI-HRMS (CH3CN) m/z (found for [M + H]+ (calc.)): 451.9955 (451.9857). Anal. calc. for C11H12ClNO2PtS: C, 29.18; H, 2.67; N, 3.09. Found: C, 28.95; H, 2.29; N, 2.83.
[PtCl(8-quinolinethiolato)(S-dmso)] (8a). Yield: 71% (0.0435 g), orange solid. 1H NMR (CDCl3, 500.10 MHz): δ 9.89 (dd, J = 5.4, 1.5 Hz, 1H), 8.39 (dd, J = 8.2, 1.4 Hz, 1H), 7.81 (dd, J = 7.4, 1.0 Hz, 1H), 7.58–7.54 (m, 2H), 7.49 (t, J = 7.7 Hz, 1H), 7.42–7.35 (m, 1H), 3.63 (s, 6H, CH3-dmso) ppm. IR selected bands (cm−1, ATR): 3067, 3016, 2915, 1501, 1306, 1218, 1134, 1032, 984. UV–vis (λ (nm) (ε (L mol−1cm−1)) c = 1 × 10−4 mol L−1, CHCl3): 265 (19
500), 479 (4100). ESI-HRMS (CH3CN) m/z (found for [M + H]+ (calc.)): 469.9101 (469.9771). Anal. calc. for C11H11ClNOPtS2: C, 28.12; H, 2.79; N, 2.98. Found: C, 28.03; H, 2.41; N, 3.03.
[PtCl(5-chloro-7-iodo-8-hydroxyquinolinato)(pta)] (1b). Yield: 60% (0.0421 g), yellow solid. 1H NMR (CDCl3, 500.10 MHz): δ 8.94–8.92 (m, 1H, C2H), 8.61 (dd, J = 8.6, 1.2 Hz, 1H, C4H), 7.85(s, 1H, C6H), 7.65 (ddd, J = 8.6, 4.9, 1.3 Hz, 1H, C3H), 4.59 (s, 6H, N–CH2–N), 4.46 (d, J = 2.1 Hz, 6H, N–CH2–P) ppm. 31P NMR (202 MHz, CDCl3): δ −61.56 ppm. IR selected bands (cm−1, ATR): 2934, 2866, 1484, 1440, 1364, 1238, 1012, 966, 942. UV–vis (λ (nm) (ε (L mol−1 cm−1)) c = 1 × 10−4 mol L−1, CHCl3): 288 (27
000), 444 (4800). ESI-HRMS (CH3CN) m/z (found for [M + H]+ (calc.)): 690.9179 (690.9082). Anal. calc. for C15H16Cl2IN4OPPt: C, 26.03; H, 2.33; N, 8.09. Found: C, 25.74; H, 2.58; N, 8.31.
[PtCl(8-hydroxy-5-nitroquinolinato)(pta)] (2b). Yield: 52% (0.0349 g), yellow solid. 1H NMR (CDCl3, 500.10 MHz): δ 8.91–8.89 (m, 2 × 1H), 8.46 (dd, J = 8.6, 1.2 Hz, 1H), 8.23 (s, 1H), 7.64–7.61 (m, 1H), 4.59 (s, 6H, N–CH2–N), 4.46 (d, J = 2.1 Hz, 6H, N–CH2–P). 31P NMR (202 MHz, CDCl3): δ −61.41 ppm. IR selected bands (cm−1, ATR): 2922, 2853, 1568, 1504, 1482, 1454, 1295, 1277, 1236, 1008, 967, 943. UV–vis (λ (nm) (ε (L mol−1 cm−1)) c = 1 × 10−4 mol L−1, CHCl3): 270 (17
000), 333 (8100), 449 (12400). ESI-HRMS (CH3CN) m/z (found for [M + H]+ (calc.)): 579.0477 (579.0558). Anal. calc. for C15H17ClN5O3PPt: C, 31.23; H, 2.97; N, 12.14. Found: C, 31.62; H, 3.08; N, 12.45.
[PtCl(5,7-dichloro-8-hydroxyquinolinato)(pta)] (3b). Yield: 54% (0.0485 g), yellow solid. 1H NMR (CDCl3, 500.10 MHz): δ 8.96 (dd, J = 6.5, 2.6 Hz, 1H, C2H), 8.62 (dd, J = 8.6, 1.2 Hz, 1H, C4H), 7.63 (ddd, J = 8.6, 5.0, 1.3 Hz, 1H, C3H), 7.56 (s, 1H, C6H), 4.59 (s, 6H, N–CH2–N), 4.45 (d, J = 1.9 Hz,6H, N–CH2–P) ppm. 31P NMR (202 MHz, CDCl3): δ −62.27 ppm. IR selected bands (cm−1, ATR): 2927, 2912, 1489, 1445, 1371, 1365, 1012, 972, 964, 942. UV–vis (λ (nm) (ε (L mol−1 cm−1)) c = 1 × 10−4 mol L−1, CHCl3): 279 (40
500), 348 (2000), 441 (3700). ESI-HRMS (CH3CN) m/z (found for [M + H]+ (calc.)): 598.9827 (598.9726). Anal. calc. for C15H16Cl3N4OPPt: C, 29.99; H, 2.68; N, 9.33. Found: C, 30.30; H, 2.87; N, 8.99.
[PtCl(5,7-diiodo-8-hydroxyquinolinato)(pta)] (4b). Yield: 52% (0.0545 g), yellow solid. 1H NMR (CDCl3, 500.10 MHz): δ 8.90–8.88 (m, 1H, C2H), 8.44 (dd, J = 8.6, 1.2 Hz, 1H, C4H), 8.22 (s, 1H, C6H), 7.62 (ddd, J = 8.6, 4.9, 1.4 Hz, 1H, C3H), δ 4.60 (s, 6H, N–CH2–N), 4.47 (d, J = 2.0 Hz, 6H, N–CH2–P) ppm. 31P NMR (202 MHz, CDCl3): δ −61.41 ppm. IR selected bands (cm−1, ATR): 2930, 2908, 2861, 1478, 1440, 1358, 1014, 964, 941. UV–vis (λ (nm) (ε (L mol−1 cm−1)) c = 1 × 10−4 mol L−1, CHCl3): 290 (35
000), 445 (5600). ESI-HRMS (CH3CN) m/z (found for [M + H]+ (calc.)): 781.8462 (781.8427). Anal. calc. for C15H16ClI2N4OPPt: C, 22.99; H, 2.06; N, 7.15. Found: C, 23.21; H, 1.87; N, 7.44.
[PtCl(5,7-dibromo-8-hydroxyquinolinato)(pta)] (5b). Yield: 55% (0.0496 g), yellow solid. 1H NMR (CDCl3, 500.10 MHz): δ 8.92–8.90 (m, 1H, C2H), 8.55 (d, J = 1.3 Hz, 1H, C4H), 7.83 (s, 1H, C6H), 7.64–7.61 (m, 1H, C3H), 4.59 (s, 6H, N–CH2–N), 4.46 (d, J = 2.1 Hz, 6H, N–CH2–P) ppm.31P NMR (202 MHz, CDCl3): δ −62.38 ppm. IR selected bands (cm−1, ATR): 2936, 2866, 1484, 1444, 1364, 1236, 1113, 1012, 965, 942. UV–vis (λ (nm) (ε (L mol−1 cm−1)) c = 1 × 10−4 mol L−1, CHCl3): 283 (26
000), 349 (3300), 441 (6500). ESI-HRMS (CH3CN) m/z (found for [M + H]+ (calc.)): 686.8805 (686.8706). Anal. calc. for C15H16Br2ClN4OPPt: C, 26.12; H, 2.34; N, 8.13. Found: C, 26.34; H, 2.22; N, 7.99.
[PtCl(5,7-dichloro-8-hydroxy-2-methyl-quinolinato)(pta)] (6b). Yield: 58% (0.0421 g), yellow solid. 1H NMR (CDCl3, 500.10 MHz): δ 8.42 (d, J = 8.7 Hz, 1H), 7.47 (d, J = 3.4 Hz, 1H), 7.31 (dd, J = 8.7, 0.9 Hz, 1H), 4.56 (s, 6H, N–CH2–N), 4.49 (d, J = 2.5 Hz, 6H, N–CH2–P) ppm. 31P NMR (202 MHz, CDCl3): δ −64.10 ppm. IR selected bands (cm−1, ATR): 2914, 2870, 1551, 1444, 1425, 1365, 1277, 1239, 1011, 969, 943. UV–vis (λ (nm) (ε (L mol−1 cm−1)) c = 1 × 10−4 mol L−1, CHCl3): 284 (29
000), 350 (2000), 431 (5000). ESI-HRMS (CH3CN) m/z (found for [M + H]+ (calc.)): 612.9967 (612.9866). Anal. calc. for C16H18Cl3N4OPPt: C, 31.26; H, 2.95; N, 9.11. Found: C, 31.52; H, 2.78; N, 8.88.
[PtCl(8-hydroxyquinolinato)(pta)] (7b). Yield: 54% (0.0406 g), yellow solid. 1H NMR (CDCl3, 500.10 MHz): δ 9.41–9.40 (m, 1H), 8.35 (dd, J = 8.4, 1.2 Hz, 1H), 7.52–7.44 (m, J = 8.3, 4.9, 1.5 Hz, 2H), 7.07 (dd, J = 7.9, 3.3 Hz, 2H), 4.58 (s, 6H, N–CH2–N), 4.45 (d, J = 1.9 Hz, 6H, N–CH2–P) ppm. 31P NMR (202 MHz, CDCl3): δ −63.58 ppm.IR selected bands (cm−1, ATR): 2932, 2871, 1500, 1464, 1375, 1319, 1277, 1240, 1011, 970, 944. UV–vis (λ (nm) (ε (L mol−1 cm−1)) c = 1 × 10−4 mol L−1, CHCl3): 273 (22
000), 426 (4000). ESI-HRMS (CH3CN) m/z (found for [M + H]+ (calc.)): 532.0471 (532.0441). Anal. calc. for C15H18ClN4OPPt: C, 33.87; H, 3.41; N, 10.54. Found: C, 33.45; H, 3.28; N, 10.47.
[PtCl(8-quinolinethiolato)(pta)] (8b). Yield: 51% (0.0400 g), orange solid. 1H NMR (CDCl3, 500.10 MHz): δ 9.83–9.81 (m, 1H), 8.37 (dd, J = 8.2, 1.2 Hz, 1H), 7.81 (d, J = 7.3 Hz, 1H), 7.62–7.59 (m, 1H), 7.45 (t, J = 7.7 Hz, 1H), 7.38 (d, J = 7.8 Hz, 1H), 4.55 (s, 6H, N–CH2–N), 4.42 (d, J = 1.9 Hz, 6H, N–CH2–P) ppm. 31P NMR (202 MHz, CDCl3): δ −61.85 ppm. IR selected bands (cm−1, ATR): 3046, 2914, 2867, 1497, 1275, 1238, 1218, 1011, 969, 944. UV–vis (λ (nm) (ε (L mol−1 cm−1)) c = 1 × 10−4 mol L−1, CHCl3): 272 (21
000), 464 (4700). ESI-HRMS (CH3CN) m/z (found for [M + H]+ (calc.)): 545.0412 (545.0269). Anal. calc. for C15H18ClN4SPPt: C, 32.82; H, 3.49; N, 10.21. Found: C, 33.20; H, 3.29; N, 9.85.
Reactivity of complexes towards the DNA model compound 5′-guanosine monophosphate
Compounds 1a and 1b (2–3 mg) were dissolved in 630 μL of dmso-d6. An equimolar amount of 5′-GMP disodium salt was dissolved in approx. 70 μL of D2O and added to the NMR tube containing the solution of a complex. 1H NMR spectra were recorded prior to the addition of 5′-GMP and after 5/10 min (1a/1b), 20, 45, 75 min, 3, 5, 24, 72 h, and 1 week.Cell viability assays
Cell viability was tested by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) colorimetric assay as previously described.60 Assays were carried out after 48 h of cell incubation in media containing test compounds at concentrations ranging from 0.1 μg mL−1 to 10 μg mL−1. Compounds were used from the stock solutions freshly prepared in dmso. The results are presented as percentage of the control (cells treated with dmso) that was arbitrarily set to 100%. The percentage viability values were plotted against the log of concentration and a sigmoidal dose response curve was calculated by non-linear regression analysis using GraphPad Prism software version 5.0 for Windows (GraphPad Software, CA, USA). From these curves, IC50 values were obtained.Platinum cellular uptake assay
The Pt content was measured using inductively coupled plasma mass spectrometry (ICP-MS) according to a previously published procedure61 but with minor modifications. A549 cells (6 × 105 per plate) were seeded into 60 mm plates in a total volume of 5 ml of RPMI medium per plate, cultured for 24 h, and then exposed to 2 μM of the investigated compounds for 2 h at 37 °C. The degree of confluence in the case of A549 cells at the end of exposure was nearly 80%. After exposure, the compound-containing medium was removed, and the cells were washed with 5 ml PBS and then lysed in 1.12 mL of sub-boiled nitric acids for 1.5 h at room temperature. After this period, an aliquot of 0.28 ml was transferred into a 10 mL tube and after adding the internal standards, diluted with Milli-Q water.The measurements of Pt in samples were carried out on an ICP-MS (iCAP Q, Thermo Scientific X series 2, UK). A platinum stock solution containing 1.000 g L−1 of this element was used to prepare standard solutions for the ICP-MS measurements. As internal standards were used the following: 6Li, 45Sc, 115In and 159Tb (VHG standards, Manchester, UK). The system was controlled using Qtegra Instrument Control software. Instrument conditions for the determinations are given in Table S4.† Each sample was analyzed in triplicate and each analysis consisted of three replicates. The coefficients of variation for single measurements were below 10%.
In vitro DNA binding by gel electrophoresis assay
Genomic DNA (gDNA) from Pseudomonas aeruginosa PAO1 was purified with a DNeasy tissue kit (Qiagen, Hilden, Germany) and it was used as model for high molecular weight double stranded DNA. The quality and the concentration of DNA were estimated by measuring UV absorbance with a NanoVue Plus spectrophotometer (GE Healthcare, Freiburg, Germany). The ability of the used Pt(II) complexes and cisplatin to bind gDNA from P. aeruginosa was examined by agarose gel electrophoresis as previously described.62 Briefly, for the gel electrophoresis experiments, gDNA (500 ng) was treated with the tested compounds (25 μg mL−1) in phosphate buffer (pH 7.4), and the contents were incubated for 12 h at 37 °C, then subjected to gel electrophoresis on 0.8% (w/v) agarose gel containing 0.1 μg mL−1 of ethidium bromide in TAE buffer (40 mM Tris acetate/1 mM EDTA, pH 7.4) buffer at 60 V for 2 h. Gels were visualized and analyzed using a Gel Doc EZ system (Bio-Rad, Life Sciences, Hercules, USA) equipped with the Image Lab™ software.Molecular docking
For in silico analysis of DNA–metal complex interactions, the semi-empirical quantum chemistry method (PM6) was applied for the geometry optimization of the studied Pt(II) complexes.63 The PM6 method was chosen because of its excellent compromise between computational time and description of electronic correlation.64 This semi-empirical level of theory has already been successfully applied to metal complex geometry optimization in similar docking studies.65,66 All geometry optimization calculations were performed using the Gaussian 09 molecular package.67 As a model to study the interaction between the metal complex and DNA the structure of the B-DNA dodecamer d(CGCGAATTCGCG)2 (PDB:1BNA) was used.68 The flexible compound structures were docked into rigid DNA structures using the Molegro Virtual Docker (MVD v. 2013.6.0.1).69 MVD has been successfully applied in docking studies of metal complexes in DNA.65,70 Hydrogen bonds, and hydrophilic and hydrophobic interactions between the studied complexes and DNA were calculated. The binding site was computed with a grid resolution of 0.3 Å. The MolDock SE as a search algorithm was used with the number of runs set at 100. The parameters of the docking procedure were: population size 50, maximum number of iterations 1500, energy threshold 100.00 and maximum number of steps 300. The number of generated poses was 5. The estimation of Pt(II) complexes and DNA interactions was described by the MVD-related scoring functions: MolDock Score, Docking Score, Rerank Score, Hbond Score, van der Waals (VdW) and van der Waals (Hbond) Score in relation to DNA interaction, Steric interactions and Ligand efficiency (LE1 and LE3). A maximum population of 100 and a maximum number of iterations of 10000 were used for each run and the best five poses were retained. Visualization of the docked pose has been done by using the CHIMERA (http://www.cgl.ucsf.edu/chimera/) molecular graphics program.
Toxicity against zebrafish embryos
Zebrafish embryo-toxicity assessments were carried out according to general rules of the OECD Guidelines for the Testing of Chemicals.71 All experiments involving zebrafish were performed in compliance with the European directive 86/609/EEC and the ethical guidelines of the Guide for the Care and Use of Laboratory Animals of the Institute for Molecular Genetics and Genetic Engineering, University of Belgrade.Adult wild type zebrafish (Danio rerio) obtained from a commercial supplier (Pet Centar, Belgrade, Serbia) and maintained under laboratory conditions for several months were used in this study. A preliminary embryotoxicity screen was carried out using eight different Pt(II) complexes (1a, 1b, 3a, 3b, 5a, 5b, 6a, and 6b) at three different concentrations (1, 2.5 and 10 μg mL−1) using 20 embryos per concentration. Based on the results of this screen, LC50 and EC50 values were determined for 6 different Pt(II) complexes (1a, 1b, 3a, 3b, 5a, and 5b) testing 7 different concentrations (0.5, 1, 1.5, 2, 2.5, 5 and 10 μg mL−1) and 30 embryos per concentration. Embryos were treated at 4 hours post fertilization (hpf) with seven concentrations of Pt(II) complexes (0.5, 1, 1.5, 2, 2.5, 5 and 10 μg mL−1) using 30 embryos per concentration. dmso (0.1%, v/v) was used as a negative control. Embryos were placed to 24-well plates containing 1000 μL test solution, 10 embryos per well, and incubated at 28 °C.
All embryos were staged and evaluated using standard procedures as previously described.71,72 Apical endpoints (Table S5†) were recorded at 24, 48, 72, 96 and 114 h post fertilization (hpf) using an inverted microscope (CKX41; Olympus, Tokyo, Japan). Based on the results of this assay, LC50 and EC50 values were determined.
The evaluation of ROS production in the zebrafish model
Generation of intracellular ROS in zebrafish embryos upon Pt-complex treatments was determined by using an oxidation-sensitive fluorescent probe dye, 2′,7′-dichlorodihydrofluorescein diacetate (DCF-DA), as described previously.73 Briefly, embryos were exposed to different concentrations of the tested compounds in a period of 6–72 hpf; afterwards embryos were treated with a DCFH-DA solution (20 μg mL−1) and incubated for 1 h in the dark at 28.5 °C. After incubation, the embryos were rinsed in fresh embryo water and anesthetized before visualization. The images of stained embryos were observed using a fluorescent microscope (Olympus BX51, Applied Imaging Corp., San Jose, CA, USA) and the fluorescence intensity of individual larvae was quantified using the Image J program.DNA fragmentation assessment
For the analysis of cellular DNA degradation, A495 cells (2 × 105 cells) were exposed to concentrations equal to the IC50 of cisplatin and selected Pt(II) complexes (1a, 1b, 3a, 3b, 5a, and 5b) for 48 h. The DNA was subsequently extracted from the cells and analyzed by agarose electrophoresis (1%, w/v, agarose gel in a TAE buffer (40 mM Tris acetate/1 mM EDTA, pH 7.4) at 60 V for 2 h) as described previously.74To examine the effect of the platinum complex on zebrafish cellular DNA, a total of ten zebrafish embryos have been exposed to LC50 concentrations (the concentration causing the mortality of 50% of the embryos; Table 5) of six platinum complexes (1a, 1b, 3a, 3b, 5a, and 5b) for 48 h (from 6 to 54 hpf). Cisplatin was used as a positive control at a concentration of 10 μg mL−1. The DNA was subsequently extracted from the zebrafish embryos using a DNeasy Blood and Tissue kit (Qiagen, Hilden, Germany) according to the manufacturer's instructions and analyzed by agarose electrophoresis (0.8%, w/v, agarose gel in a TAE buffer (40 mM Tris acetate/1 mM EDTA, pH 7.4) at 70 V for 30 min).
Conclusions
Anticancer platinum complexes with their unique physico-chemical properties still prevail in the field of metals in cancer chemotherapy with their use escalated and being sustained over decades.75 Although a huge number of studies have been performed with various platinum compounds (including cisplatin) in the last five decades, there are still many questions that are unsolved. It is therefore extremely important to bring new insights into this field not only to better understand old drugs but also to design new potential drugs more successfully. The sixteen complexes assessed in this study exhibited different effects and activity in comparison with cisplatin. Evidence is shown that DNA is not the main target of the platinum-hq complexes but further work is needed to elucidate their exact mode of action. On the other side, pta containing Pt(II) complexes showed greater potential in terms of selectivity towards cancer cells, a mode-of-action which involves increased ROS generation and better embryotoxic profile in comparison with dmso containing counterparts, with [PtCl(5,7-dibromo-8-hydroxyquinolinato)(pta)] (5b) being the most promising complex from this study.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work has been financially supported by the Ministry of Education and Science, Republic of Serbia, under Grant No. 172036 and 173048 and the programme grant P1-0175 (I. T.) and the postdoctoral research grant Z1-6735 (to J. K.) from the Slovenian Research Agency. The EN→FIST Centre of Excellence, Trg OF 13, SI-1000 Ljubljana, Slovenia, is acknowledged for the use of a SuperNova diffractometer. M. Ž. is grateful to the Erasmus Mundus Action 2 Project Basileus V for a scholarship for post-docs with 6 months mobility period at the University of Ljubljana. Great gratitude also goes to the support from the bilateral Slovenian–Serbian project BI-RS/16-17-024.References
- B. Rosenberg, L. Vancamp, J. E. Trosko and V. H. Mansour, Nature, 1969, 222, 385 CrossRef CAS PubMed.
- N. Muhammad and Z. Guo, Curr. Opin. Chem. Biol., 2014, 19, 144 CrossRef CAS PubMed.
- B. Rosenberg, in Cisplatin: Chemistry and Biochemistry of a Leading Anticancer Drug, ed. B. Lippert, Wiley-VCH, New York, 1999, pp. 3–30 Search PubMed.
- Z. Siddik, Oncogene, 2003, 22, 7265 CrossRef CAS PubMed.
- M. A. Fuertes, C. Alonso and J. M. Pérez, Chem. Rev., 2003, 103, 645 CrossRef CAS PubMed.
- G. Chu, J. Biol. Chem., 1994, 269, 787 CAS.
- L. R. Kelland, Drugs, 2000, 59, 37 CrossRef.
- N. Farrell, Compr. Coord. Chem. II, 2003, 9, 809 Search PubMed.
- L. Kelland, Nat. Rev. Cancer, 2007, 7, 573 CrossRef CAS PubMed.
- Y. Jung and S. J. Lippard, Chem. Rev., 2007, 107, 1387 CrossRef CAS PubMed.
- D. Wang and S. J. Lippard, Nat. Rev. Drug Discovery, 2005, 4, 307 CrossRef CAS PubMed.
- X. Wang, Anti-Cancer Agents Med. Chem., 2010, 10, 396 CrossRef CAS PubMed.
- N. J. Wheate and J. G. Collins, Curr. Med. Chem.: Anti-Cancer Agents, 2005, 5, 267 CrossRef CAS PubMed.
- N. J. Wheate, S. Walker, G. E. Craig and R. Oun, Dalton Trans., 2010, 39, 8113 RSC.
- J. Zhang, L. Wang, Z. Xing, D. Liu, J. Sun, X. Li and Y. Zhang, Anti-Cancer Agents Med. Chem., 2010, 10, 272 CrossRef CAS PubMed.
- C. M. Santos, S. Cabrera, C. Ríos-Luci, J. M. Padrón, I. L. Solera, A. G. Quiroga, M. A. Medrano, C. Navarro-Ranninger and J. Alemán, Dalton Trans., 2013, 42, 13343 RSC and references cited therein.
- K. G. Daniel, D. Chen, S. Orlu, Q. C. Cui, F. R. Miller and Q. P. Dou, Breast Cancer Res., 2005, 7, R897 CrossRef CAS PubMed.
- X. Mao, X. Li, R. Sprangers, X. Wang, A. Venugopal, T. Wood, Y. Zhang, D. A. Kuntz, E. Coe, S. Trudel, D. Rose, R. A. Batey, L. E. Kay and A. D. Schimmer, Leukemia, 2009, 23, 585 CrossRef CAS PubMed.
- V. Prachayasittikul, S. Prachayasittikul, S. Ruchirawat and V. Prachayasittikul, Drug Des., Dev. Ther., 2013, 7, 1157 CrossRef PubMed.
- M. Gobec, J. Kljun, I. Sosič, I. Mlinarič-Raščan, M. Uršič, S. Gobec and I. Turel, Dalton Trans., 2014, 43, 9045 RSC.
- A. Mitrović, J. Kljun, I. Sosič, S. Gobec, I. Turel and J. Kos, Dalton Trans., 2016, 45, 16913 RSC.
- B. Murugasu-Oei and T. Dick, Int. J. Antimicrob. Agents, 2001, 18, 579 CrossRef CAS PubMed.
- B. Mirković, M. Renko, S. Turk, I. Sosič, Z. Jevnikar, N. Obermajer, D. Turk, S. Gobec and J. Kos, ChemMedChem, 2011, 6, 1351 CrossRef PubMed.
- B. Mirković, B. Markelc, M. Butinar, A. Mitrović, I. Sosič, S. Gobec, O. Vasiljeva, B. Turk, M. Čemažar, G. Serša and J. Kos, Oncotarget, 2015, 6(22), 19027 Search PubMed.
- I. Sosič, B. Mirković, K. Arenz, B. Štefane, J. Kos and S. Gobec, J. Med. Chem., 2013, 56(2), 521 CrossRef PubMed.
- A. D. Phillips, L. Gonsalvi, A. Romerosa, F. Vizza and M. Peruzzini, Coord. Chem. Rev., 2004, 248, 955 CrossRef CAS.
- B. S. Murray, M. V. Babak, C. G. Hartinger and P. J. Dyson, Coord. Chem. Rev., 2016, 306, 86 CrossRef CAS and references cited therein.
- A. D. Phillips, L. Gonsalvi, A. Romerosa, F. Vizza and M. Peruzzini, Coord. Chem. Rev., 2004, 248, 955 CrossRef CAS.
- J. Bravo, S. Bolaño, L. Gonsalvi and M. Peruzzini, Coord. Chem. Rev., 2010, 254, 555 CrossRef CAS.
- K. S. O. Ferraza, D. C. Reisa, J. G. Da Silvaa, E. M. Souza-Fagundesb, E. J. Baranc and H. Beraldo, Polyhedron, 2013, 63, 28 CrossRef.
- A. Casado-Sánchez, R. Gómez-Ballesteros, F. Tato, F. J. Soriano, G. Pascual-Coca, S. Cabrera and J. Alemán, Chem. Commun., 2016, 52, 9137 RSC.
- T. Meng, S.-F. Tang, Q.-P. Qin, Y.-L. Liang, C.-X. Wu, C.-Y. Wang, H.-T. Yan, J.-X. Donga and Y.-C. Liu, MedChemComm, 2016, 7, 1802 RSC.
- M. Kato, Y. Ogawa, M. Kozakai and Y. Sugimoto, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2002, 58, m147 Search PubMed.
- L. Yang, D. R. Powella and R. P. Houser, Dalton Trans., 2007, 955 RSC.
- A. Okuniewski, D. Rosiak, J. Chojnacki and B. Becker, Polyhedron, 2015, 90, 47 CrossRef CAS.
- S. J. Fischer, L. M. Benson, A. Fauq, S. Naylor and A. J. Windebank, Neurotoxicology, 2008, 29, 444 CrossRef CAS PubMed.
- S. P. Wisnovsky, J. J. Wilson, R. J. Radford, M. P. Pereira, M. R. Chan, R. R. Laposa, S. J. Lippard and S. O. Kelley, Chem. Biol., 2013, 20(11), 1323 CrossRef CAS PubMed.
- J. Reedijk, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 3611 CrossRef CAS PubMed.
- I. Turel and J. Kljun, Curr. Top. Med. Chem., 2011, 11(21), 2661 CrossRef CAS PubMed.
- R. C. Todd and S. J. Lippard, Metallomics, 2009, 1, 280 RSC.
- Y. Jung and S. J. Lippard, Chem. Rev., 2007, 107, 1387 CrossRef CAS PubMed; Y. Zhao, W. He, P. Shi, J. Zhu, L. Qiu, L. Lin and Z. Guo, Dalton Trans., 2006, 2617 RSC.
- H. P. Varbanov, D. Ortiz, D. Höfer, L. Menin, M. Galanski, B. K. Keppler and P. J. Dyson, Dalton Trans., 2017, 46, 8929 RSC.
- N. Raman, S. Sobha and L. Mitu, Monatsh. Chem., 2012, 143, 1019 CrossRef CAS; Q. Saquib, A. A. Al-Khedhairy, S. A. Alarifi, S. Dutta, S. Dasgupta and J. Musarrat, Int. J. Biol. Macromol., 2010, 47, 68 CrossRef PubMed.
- R. Filosa, A. Peduto, S. Di Micco, P. de Caprariis, M. Festa, A. Petrella, G. Capranico and G. Bifulco, Bioorg. Med. Chem., 2009, 17, 13 CrossRef CAS PubMed.
- M. A. Fuertes, C. Alonso and J. M. Pérez, Chem. Rev., 2003, 103(3), 645 CrossRef CAS PubMed.
- S. E. Miller and D. A. House, Inorg. Chim. Acta, 1991, 187, 125 CrossRef CAS.
- X. L. Yang and A. H. Wang, Pharmacol. Ther., 1999, 83, 181 CrossRef CAS PubMed.
- V. Cepeda, M. A. Fuertes, J. Castilla, C. Alonso, C. Quevedo and J. M. Pérez, Anticancer Agents Med. Chem., 2007, 7, 3 CrossRef CAS PubMed.
- S. Kemp, N. J. Wheate, D. P. Buck, M. Nikac, J. G. Collins and J. R. Aldrich-Wright, J. Inorg. Biochem., 2007, 101, 1049 CrossRef CAS PubMed.
- S. Kemp, N. J. Wheate, M. J. Pisani and J. R. Aldrich-Wright, J. Med. Chem., 2008, 51(9), 2787 CrossRef CAS PubMed.
- R. Osterauer, N. Haus, B. Sures and H. R. Köhler, Chemosphere, 2009, 975 CrossRef CAS PubMed.
- P. V. Asharani, Y. Lianwu, Z. Gong and S. Valiyaveettil, Nanotoxicol., 2011, 43 CrossRef CAS PubMed.
- R. S. Go and A. A. Adjei, J. Clin. Oncol., 1999, 17, 409 CrossRef CAS PubMed.
- Y. Hirose, J. A. Simon and H. C. Ou, J. Assoc. Res. Otolaryngol., 2011, 12, 719 CrossRef PubMed.
- J. H. Price, A. N. Williamson, R. F. Schramm and B. B. Wayland, Inorg. Chem., 1972, 11, 1280 CrossRef CAS.
- A. Altomare, G. Cascarano, C. Giacovazzo, A. C. Guagliardi, M. C. Burla, G. Polidori and M. Camalli, J. Appl. Crystallogr., 1994, 27, 435 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed.
- A. L. Spek, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2009, 65, 148 CrossRef CAS PubMed.
- C. F. Macrae, P. R. Edgington, P. McCabe, E. Pidcock, G. P. Shields, R. Taylor, M. Towler and J. van De Streek, J. Appl. Crystallogr., 2006, 39, 453 CrossRef CAS.
- M. B. Hansen, S. E. Nielsen and K. Berg, J. Immunol. Methods, 1989, 119, 203 CrossRef CAS PubMed.
- A. E. Egger, C. Rappel, M. A. Jakupec, C. G. Hartinger, P. Heffeter and B. K. Keppler, J. Anal. At. Spectrom., 2009, 24, 51 RSC.
- V. T. Yilmaz, E. Gocmen, C. Icsel, M. Cengiz, S. Y. Susluer and O. Buyukgungor, J. Photochem. Photobiol., B, 2014, 131, 31 CrossRef CAS PubMed.
- E. A. Amin and D. G. Truhlar, J. Chem. Theory Comput., 2008, 4, 75 CrossRef CAS PubMed; J. J. P. Stewart, J. Mol. Model, 2007, 13, 1173 CrossRef PubMed.
- G. Frison and G. Ohanessian, J. Comput. Chem., 2008, 29, 416 CrossRef CAS PubMed.
- L. Senerovic, M. D. Zivkovic, A. Veselinovic, A. Pavic, M. I. Djuran, S. Rajkovic and J. Nikodinovic-Runic, J. Med. Chem., 2015, 58, 1442 CrossRef CAS PubMed.
- A. M. A. Alaghaz, B. A. El-Sayed, A. A. El-Henawy and R. A. A. Ammar, J. Mol. Struct., 2013, 1035, 83 CrossRef CAS; R. Cincinelli, L. Musso, S. Dallavalle, R. Artali, S. Tinelli, D. Colangelo, F. Zunino, M. De Cesare, G. L. Beretta and N. Zaffaroni, Eur. J. Med. Chem., 2013, 63, 387 CrossRef PubMed.
- M. J. Frisch, Gaussian 09, Wallingford, CT, 2004 Search PubMed.
- H. R. Drew, R. M. Wing, T. Takano, C. Broka, S. Tanaka, K. Itakura and R. E. Dickerson, Proc. Natl. Acad. Sci. U. S. A., 1981, 78, 2179 CrossRef CAS; K. Suntharalingam, O. Mendoza, A. A. Duarte, D. J. Mann and R. Vilar, Metallomics, 2013, 5, 514 RSC.
- R. Thomsen and M. H. Christensen, J. Med. Chem., 2006, 49, 3315 CrossRef CAS PubMed.
- A. A. Adeniyi and P. A. Ajibade, Molecules, 2013, 18, 10829 CrossRef PubMed.
- OECD guidelines for the testing of chemicals, Fish embryo acute toxicity (FET) test. OECD 2013, No 236.
- C. B. Kimmel, W. W. Ballard, S. R. Kimmel, B. Ullmann and T. F. Schilling, Dev. Dyn., 1995, 203, 253 CrossRef CAS PubMed.
- E. A. Kim, S. H. Lee, C. I. Ko, S. H. Cha, M. C. Kang, S. M. Kang, S. C. Ko, W. W. Lee, J. Y. Ko, J. H. Lee, N. Kang, J. Y. Oh, G. Ahn, Y. H. Jee and Y. J. Jeon, Carbohydr. Polym., 2014, 102, 185 CrossRef CAS PubMed.
- G. V. Kalayda, S. Komeda, K. Ikeda, T. Sato, M. Chikuma and J. Reedijk, Eur. J. Inorg. Chem., 2003, 4347 CrossRef CAS.
- G. Sava, A. Bergamo and P. Dyson, Dalton Trans., 2011, 40, 9069 RSC; J. Zhang, L. Wang, Z. Xing, D. Liu, J. Sun, X. Li and Y. Zhang, Anticancer Agents Med. Chem., 2010, 10, 272 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1483253–1483257. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7qi00299h |
| This journal is © the Partner Organisations 2018 |