
In situ cross-linkable hyaluronic acid hydrogels using copper free click chemistry for cartilage tissue engineering†
Sang-Soo
Han‡
 a,
Hong Yeol
Yoon‡
b,
Ji Young
Yhee
b,
Myeong Ok
Cho
ac,
Hye-Eun
Shim
a,
Ji-Eun
Jeong
ad,
Dong-Eun
Lee
e,
Kwangmeyung
Kim
b,
Hwanuk
Guim
f,
John Hwan
Lee
g,
Kang Moo
Huh
*c and
Sun-Woong
Kang
*ad
a,
Hong Yeol
Yoon‡
b,
Ji Young
Yhee
b,
Myeong Ok
Cho
ac,
Hye-Eun
Shim
a,
Ji-Eun
Jeong
ad,
Dong-Eun
Lee
e,
Kwangmeyung
Kim
b,
Hwanuk
Guim
f,
John Hwan
Lee
g,
Kang Moo
Huh
*c and
Sun-Woong
Kang
*ad
aPredictive Model Research Center, Korea Institute of Toxicology, Daejeon, Korea
bCenter for Theragnosis, Biomedical Research Institute, Korea Institute of Science and Technology, Seoul, Korea
cDepartment of Polymer Science and Engineering, Chungnam National University, Daejeon, Korea. E-mail: khuh@cnu.ac.kr; Fax: +82-42-821-8910; Tel: +82-42-821-6663
dDepartment of Human and Environmental Toxicology Program, University of Science and Technology (UST), Daejeon, Korea. E-mail: swkang@kitox.re.kr; Fax: +82-42-610-8157; Tel: +82-42-610-8209
eAdvanced Radiation Technology Institute, Korea Atomic Energy Research Institute, Jeongeup, Korea
fCenter for Electron Microscopy Research, Korea Basic Science Institute, Daejeon, Korea
gDepartment of Chemical Engineering, Hanyang University, Seoul, Republic of Korea
First published on 25th October 2017
Abstract
We report a biocompatible and in situ cross-linkable hydrogel derived from hyaluronic acid via a bioorthogonal reaction and confirm the clinical potential of our hydrogel through in vivo cartilage regeneration. Gelation is attributed to copper-free click reactions between an azide and dibenzyl cyclooctyne. HA-PEG4-DBCO was synthesized and cross-linked via 4-arm PEG azide. The effects of the ratio of HA-PEG4-DBCO to 4-arm PEG azide on the gelation time, microstructure, surface morphology, equilibrium swelling, and compressive modulus were examined. The potential of a hydrogel as an injectable scaffold was demonstrated by the encapsulation of chondrocytes within the hydrogel matrix in vitro and in vivo. The results demonstrated that the hydrogel supported cell survival, and the cells regenerated cartilaginous tissue. In addition, these characteristics provide potential opportunities for the use of injectable hydrogels in tissue engineering applications.
Introduction
Hyaluronic acid (HA) is a biomaterial naturally found in the extracellular matrix and plays important roles in cellular functions as a regulator of cell adhesion and migration.1,2 Additionally, it has valuable properties such as biocompatibility and gel-forming ability and has functional groups that allow it to be easily modified.3 These properties make HA a great potential starting material for fabricating hydrogels for use in medical sciences and tissue engineering.4 Many recent studies of HA-based hydrogels have focused on the development of injectable hydrogels, which can be formed in situ after being injected into the body.5–8 These approaches offer many advantages for the adaptation of the implant after directly placing it at the defect site in the body and allow for the easy encapsulation of cells, drugs and other materials.9 In addition, no surgery is needed, resulting in a faster recovery.10Traditional HA-based hydrogels have been synthesized by using cross-linking agents such as glutaraldehyde,11 divinyl sulfone12 and carbodiimide,13 which are cytotoxic, and any unreacted agent must be removed before cell encapsulation.14 Over the past decade, many new cross-linking agents and methods have been developed.15 Leach, J. B., et al. synthesized an HA hydrogel using a photoinduced cross-linking method, which allowed for in situ cross-linking and uniform cell dispersion.16,17 However, the photoinduced cross-linking method triggers adverse effects on cells due to the toxicity of the photoinitiator and ultraviolet (UV) light. Michael addition reaction,18 Schiff base reaction,19 and enzymatic polymerization methods20 have also been reported, although these methods involve the multistep synthesis of functionalized HA, increasing the complexity of the system and resulting in a low reaction efficiency.
Click chemistry between the azido- and alkynyl-derivatives of HA with copper catalysts was recently revealed as an alternative approach for fabricating cross-linked hydrogels with a fast reaction rate, high efficiency and high chemoselectivity at physiologically relevant pH and temperature ranges.21,22 However, the use of copper, which is a cytotoxic element and may cause Alzheimer's disease and hepatitis,23,24 is required in click chemistry. As one click chemistry tool, the strain-promoted cyclooctyne–azide cycloaddition reaction has been widely applied in live cellular imaging, cell tracking, and non-living sample conjugation.25–27 Although the reaction rate of the strain-promoted cyclooctyne–azide cycloaddition reaction (10 mol−1 s−1) was lower than the copper-catalyzed azide–alkyne cycloaddition reaction (103 mol−1 s−1),28 the strain-promoted cyclooctyne–azide cycloaddition reaction does not require any cofactor and spontaneously conjugates molecules containing an azide group within physiological temperature and pH ranges.29 Thus, many injectable hydrogels using azide- and cyclooctyne-modified hyaluronic acid, chitosan, and dextran were designed and developed for application in tissue engineering.25,26,30,31 More importantly, these injectable hydrogels have shown good biocompatibility and helped the viability and proliferation of chondrocytes, fibroblasts and stem cells. However, recent studies have not fully demonstrated the potential of injectable hydrogels using copper-free click cross-linking, because these results have focused on in vitro conditions.
The objective of this study was to develop a HA-based injectable hydrogel via copper-free click chemistry and to confirm the clinical potential of our hydrogel through in vivo cartilage regeneration. To this end, we first synthesized dibenzocyclooctyl (DBCO)-modified HA via a simple 1-ethyl-3(3-dimethylaminopropyl)carbodiimide (EDC)/N-hydroxysuccinimide (NHS) coupling reaction. The HA-PEG4-DBCO hydrogels were fabricated by mixing the 4-arm PEG azide. The clinical potential, cytotoxicity, biocompatibility and biodegradation rate were confirmed in in vivo and in vitro experiments. Cartilage regeneration was evaluated using hydrogels with encapsulated chondrocytes.
Results and discussion
Synthesis of HA-PEG4-DBCO hydrogels
As shown in Scheme 1, HA-PEG4-DBCO hydrogels were prepared by cross-linking HA-PEG4-DBCO with 4-arm PEG azide. HA-PEG4-DBCO derivatives were prepared in a one-step reaction by coupling DBCO-PEG4-amine to HA using EDC and NHS reagents (Fig. 1, Step 1). | ||
| Scheme 1 Schematic illustrations of the in situ formation of a hydrogel. Hydrogel networks were generated via click chemistry during gel injection. | ||
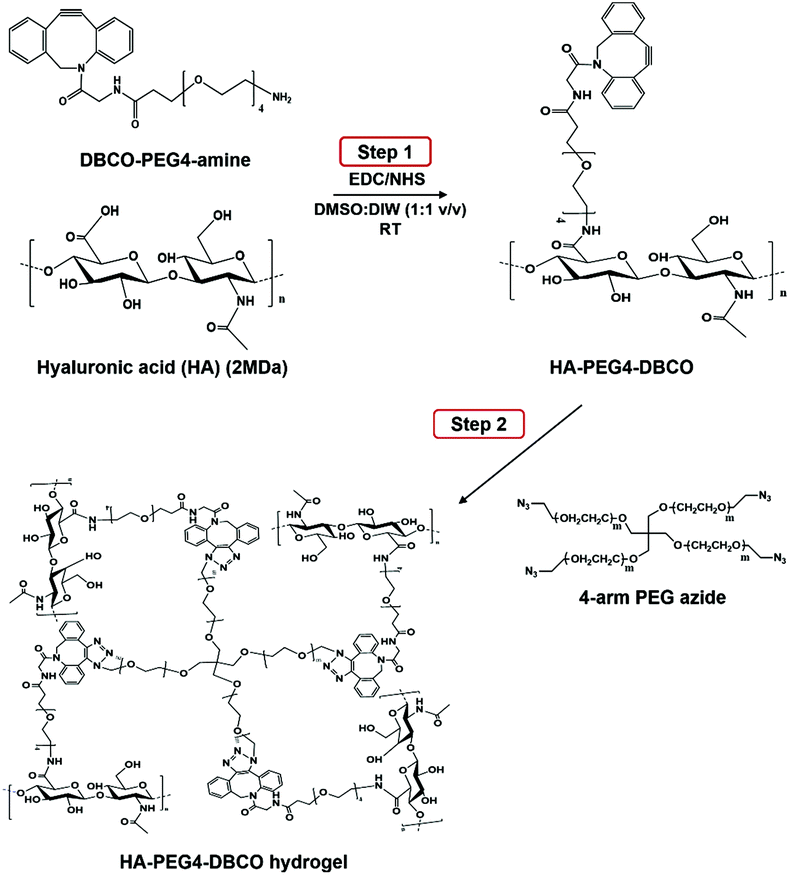 | ||
| Fig. 1 Synthesis of HA-PEG4-DBCO and in situ gel formation of a polymeric hydrogel composed of HA-PEG4-DBCO and 4-arm PEG azide conjugated by click chemistry. | ||
The chemical composition of HA-PEG4-DBCO was confirmed by proton nuclear magnetic resonance (1H-NMR) (Fig. 2B). The peak at 2 ppm indicated the N-acetyl glucosamine proton peak of native HA, and glucopyranosyl protons were assigned to 3–4 ppm.32 After the conjugation of HA and DBCO-PEG4-amine, resonances at 7.3–7.8 ppm verified the presence of DBCO protons.33 Based on these assignments, the degree of DBCO conjugation of HA-PEG4-DBCO was calculated to be 13% by comparing the integrated signal area of the protons between 2 ppm (–CH3 at HA) and 7.3–7.8 ppm (–CH at DBCO-PEG4–).
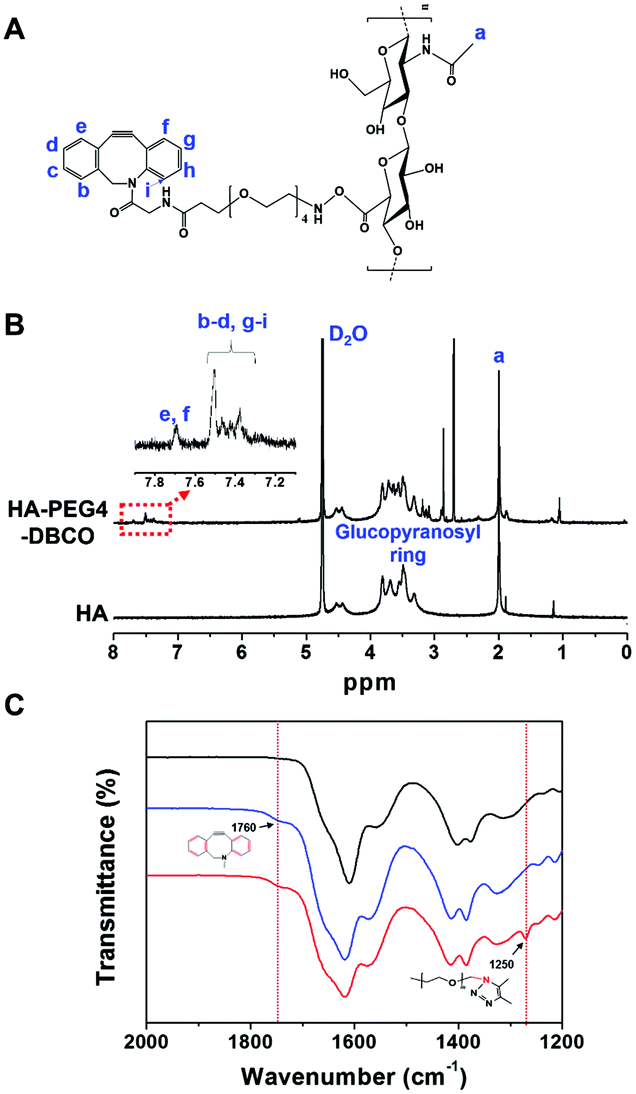 | ||
Fig. 2 Synthesis of HA-PEG4-DBCO and HA-DBCO hydrogels. (A) Chemical structure of HA-PEG4-DBCO. (B) 1H NMR spectra in D2O (300 MHz) of HA and HA-PEG4-DBCO. Proton points (a–i) shown in (A) appear in the NMR spectra in (B) as corresponding peaks. The N-acetyl glucosamine and glucopyranosyl proton peaks at 2 ppm and 3–4 ppm appear in native HA and HA-PEG4-DBCO. The expanded view of 7.2–7.8 ppm indicates the presence of DBCO. (C) ATR FT-IR spectral analysis of the HA (black line), HA-PEG4-DBCO (blue line), and HA-DBCO hydrogel (red line) in the range of 2000 to 1200 cm−1. The peak of aromatic C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bending appears at 1750 cm−1 in the spectrum of HA-PEG4-DBCO. In the spectrum of the HA-DBCO hydrogel, C–N stretching occurs at 1250 cm−1. C bending appears at 1750 cm−1 in the spectrum of HA-PEG4-DBCO. In the spectrum of the HA-DBCO hydrogel, C–N stretching occurs at 1250 cm−1. | ||
The HA-PEG4-DBCO hydrogels were synthesized by mixing HA-PEG4-DBCO and a 4-arm PEG azide cross-linker in deionized water (Fig. 1, Step 2). Fig. 2C shows the FT-IR spectra of HA, HA-PEG4-DBCO, and HA-PEG4-DBCO hydrogels. The band at 3290 cm−1 was assigned to the O–H stretching vibration, and the band at 2890 cm−1 appeared due to the C–H stretching vibrations. The peak at 1032 cm−1 corresponded to the C–O–C stretching. In the spectrum of HA-PEG4-DBCO, aromatic CC bending appeared at 1750 cm−1. After cross-linking HA-PEG4-DBCO using the 4-arm PEG azide, C–N was revealed at 1250 cm−1 due to C–N stretching in the spectrum of the HA-PEG4-DBCO hydrogel. Therefore, the synthesis of HA-PEG4-DBCO hydrogels was confirmed using a copper-free click chemistry reaction.
Characterization of HA-PEG4-DBCO hydrogels
The physical properties of the HA-PEG4-DBCO hydrogels were characterized by oscillatory rheology, mass and scanning electron microscopy (SEM) analyses. In the experiments, various ratios of HA-PEG4-DBCO/4-arm PEG azide concentrations were tested (Table 1).| Sample | Concentration of 4-arm PEG azide (mM) |
|---|---|
| HA-PEG4-DBCO/0 | 0 |
| HA-PEG4-DBCO/0.25 | 0.25 |
| HA-PEG4-DBCO/0.5 | 0.5 |
| HA-PEG4-DBCO/1 | 1 |
| HA-PEG4-DBCO/2.5 | 2.5 |
The gelation rate and elastic modulus of the hydrogels were monitored by rheological experiments via the storage modulus value (G′) and loss modulus value (G′′) (Fig. 3A). The gelation rate of the HA-PEG4-DBCO hydrogel was monitored at room temperature. When 10 mg ml−1 HA-PEG4-DBCO was mixed with the five different concentrations of 4-arm PEG azide, gelation occurred within 10–14 min. The gelation rate of HA-PEG4-DBCO/1 was fastest, which corresponded to a significantly faster gelation rate than that of HA-PEG4-DBCO/2.5.
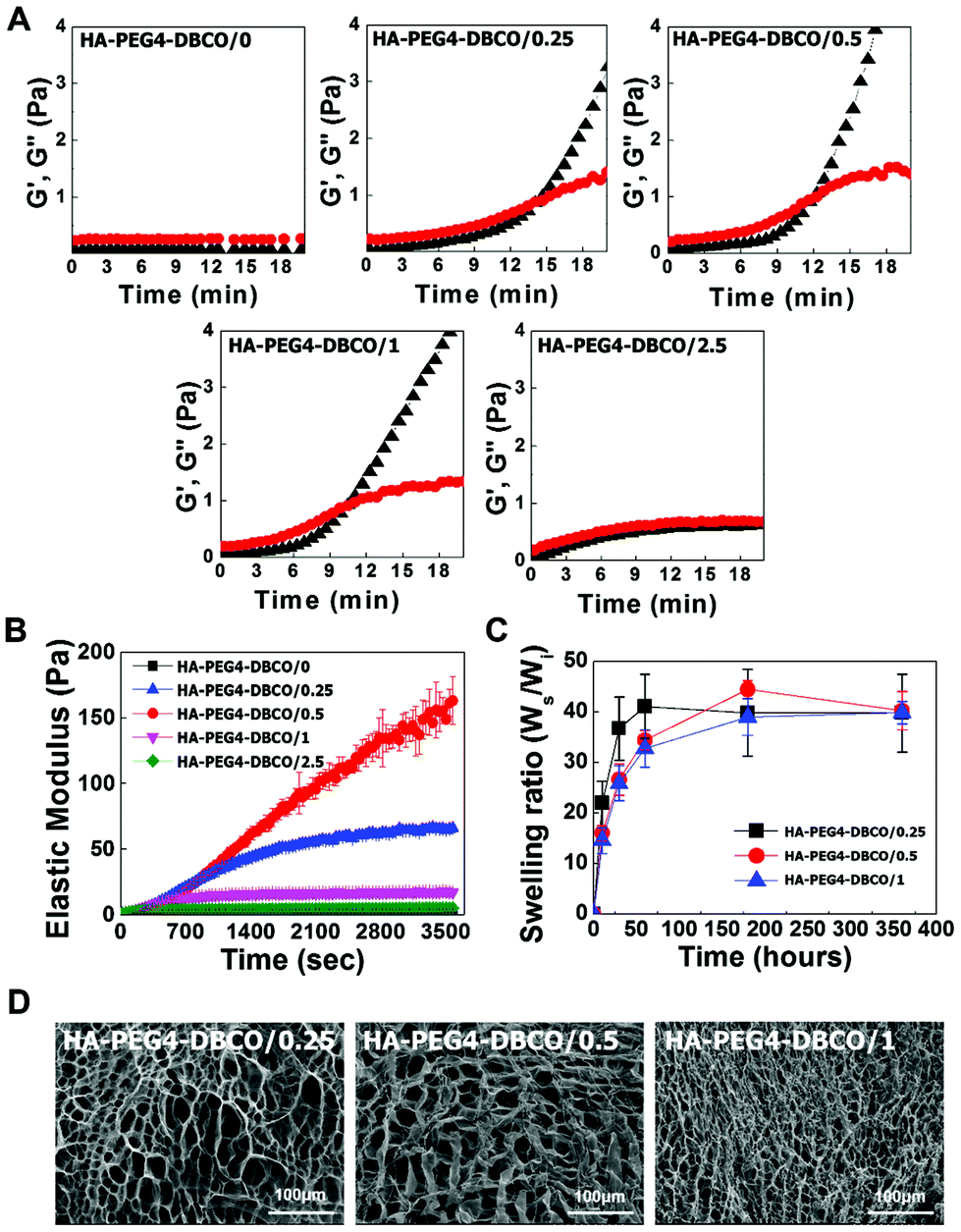 | ||
| Fig. 3 Physical characterization of HA-DBCO hydrogels with different 4-arm PEG azide concentrations. (A) Gelation times of the HA-DBCO hydrogels. Storage (G′, black triangles) and loss (G′′, red circles) moduli of the hydrogels were obtained with time after mixing HA-PEG4-DBCO with various concentrations of 4-arm PEG amine. The gelation time is the intersection of G′ and G′′. (B) Elastic moduli of the HA-DBCO hydrogels. HA-PEG4-DBCO and 4-arm PEG azide were mixed for 20 min, and the elasticity of the hydrogels was obtained at a constant stress rate of 40 mN min−1 up to 20% strain. (C) The swelling ratio of the HA-DBCO hydrogels. The freeze-dried hydrogels are incubated in PBS at 37 °C until equilibrium is reached, and the weights of the swollen hydrogels were measured with time. The hydrogel swelling ratio was calculated from the equation (Ws − Wd)/Wd, where Ws and Wd represent the weights of the swollen hydrogel and the dried hydrogel. (D) SEM images of HA-DBCO hydrogels. Scale bar: 100 μm. | ||
The elastic modulus of the hydrogels was measured using a dynamic mechanical analysis method (Fig. 3B). As the concentration of 4-arm PEG azide was increased from 0 mM to 0.5 mM, the elastic modulus of the hydrogels improved correspondingly. The HA-PEG4-DBCO/0.5 hydrogel had a significantly larger elastic modulus than the HA-PEG4-DBCO/1 hydrogel (p < 0.01).
Fig. 3C indicates the swelling ratio of freeze-dried hydrogels determined in phosphate-buffered saline (PBS) (pH = 7.4). The swelling ratio was not measured for dried HA-PEG4-DBCO/2.5, which became water-soluble after 6 h of incubation due to poor cross-linking. The swelling ratio of HA-PEG4-DBCO/0.25 in PBS was slightly higher than those of HA-PEG4-DBCO/0.5 and HA-PEG4-DBCO/1 for 72 h, whereas no difference was found in any hydrogels after 360 h. The equilibrium swelling value was 40.3 ± 3.8.
SEM images were obtained to characterize the microstructure morphologies of three freeze-dried HA-PEG4-DBCO hydrogels. According to the cross-sectional SEM images, all the hydrogel samples showed a highly porous three-dimensional structure, but the pore diameters in the hydrogel were different (Fig. 3D). The HA-PEG4-DBCO/0.25 hydrogel presented approximately spherical pores below 100 μm. The HA-PEG4-DBCO/1 hydrogel also showed spherical pores, but have a smaller pore size (10 μm) compared to the HA-PEG4-DBCO/0.25 hydrogel.
In vitro cell viability
Cross-linkers for hydrogel formation in situ should be non-cytotoxic and should not induce adverse effects on any function of the cells. In this study, the cytotoxicity of 4-arm PEG azide as a cross-linker was determined by the MTT assay as shown in Fig. 4A. Chondrocytes were selected to evaluate the cytotoxicity of the cross-linker and hydrogels given the possible uses of the injectable scaffold for cartilage tissue engineering. The 4-arm PEG azide induced low cytotoxicity of chondrocytes at various concentrations. The cytotoxicity of 4-arm PEG azide at 2.5 mM slightly increased. At concentrations of 0.25, 0.5, 1, and 2.5 mM, the cell viabilities were 96.9 ± 2.6, 97.6 ± 3.7, 93.9 ± 2.8 and 90.2 ± 3.5%, respectively. The cytotoxicity tests on the in situ cross-linkable HA-PEG4-DBCO hydrogels were performed using live/dead assays. The chondrocytes encapsulated within the HA-PEG4-DBCO hydrogels were observed by confocal microscopy after 3D culture for 1 and 5 days (Fig. 4B). Round chondrocytes were uniformly distributed in the HA-PEG4-DBCO hydrogels. Most of the encapsulated chondrocytes survived in the HA-PEG4-DBCO hydrogels after the cross-linking process using 4-arm PEG azide at concentrations of 0.25, 0.5, and 1 mM. In addition, cell aggregation was observed at 5 days of culture in the HA-PEG4-DBCO/0.25 and HA-PEG4-DBCO/1 hydrogels. This result may be due to the differences in the elastic moduli of the HA-PEG4-DBCO hydrogels.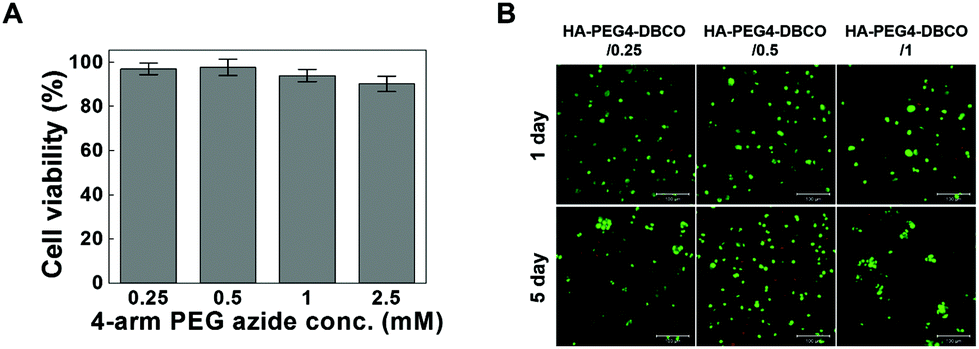 | ||
| Fig. 4 (A) In vitro analysis of cytotoxicity and biocompatibility of HA-DBCO hydrogels. The cells (5,000 cells per well in 96-well plates) were cultured with DMEM/F-12 medium containing various concentrations of 4-arm PEG azide. After 24 h of incubation, cell viability was measured by MTT assays. (B) Live/dead staining images (green: live cells, red: dead cells) of chondrocytes that were encapsulated in various HA-DBCO hydrogels after 1 and 5 days in culture. The cells were encapsulated in HA-DBCO hydrogels at a cell density of 1.0 × 107 cells per ml. Scale bar: 100 μm. | ||
To determine whether hydrogel injections can maintain new tissue formation in vivo, HA-PEG4-DBCO/0.5 hydrogels were injected into the subcutaneous dorsum of mice (Fig. 5). In all animals, subcutaneous mounds were created at the sites of injection of the hydrogel. There was no evidence of complication, including erythema or inflammation, around any of the implants at various time points (Fig. 5A). The approximate shapes of the implants were maintained over the entire implantation period, but changes in the volume were observed over 5 weeks (Fig. 5B). The volume of the implants was increased by 2.3-fold the original volume after 1 week, which may be due to the swelling of the hydrogel. Thereafter, the volumes of the implants formed by the injection slowly decreased after 2 weeks in vivo. The volume of the implants decreased to 30% of the 7-day volume at 5 weeks, which may be caused by the removal of the solution that induced the initial swelling and degradation of the hydrogel. These results confirmed that our HA hydrogels based on azide-DBCO click chemistry can be used as in situ cross-linkable hydrogels and have potential applications as scaffolds for tissue engineering.
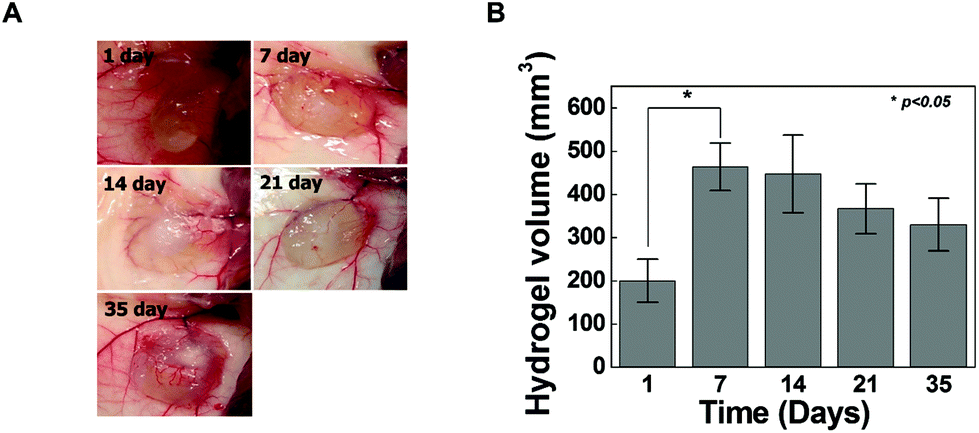 | ||
| Fig. 5 In vivo injectable HA-DBCO hydrogel formation (A) and analysis of change in volume (B) at different intervals 1, 7, 14, 21 and 35 days. HA-DBCO hydrogels were subcutaneously injected into Balb-c mice, and injection sites were opened to observe the state of the hydrogels and to measure the volume changes of hydrogels after the specified times. | ||
Cartilage regeneration using HA-PEG4-DBCO hydrogels
To determine whether the HA-PEG4-DBCO hydrogel as an injectable scaffold is able to regenerate cartilaginous tissue in vivo, cultured chondrocytes were encapsulated with HA-PEG4-DBCO hydrogels and immediately injected into the subcutaneous dorsum of athymic mice. Five weeks after transplantation, the chondrocytes encapsulated within HA-PEG4-DBCO formed solid and milky-white tissue (Fig. 6A). Before harvesting, the constructs were easily visualized under the dorsal skin of the athymic mice. Histological analysis by H&E staining of implants retrieved at 5 weeks showed that the chondrocytes encapsulated within the HA-PEG4-DBCO hydrogels regenerated cartilaginous tissue, as evidenced by chondrocytes in lacunae. Neocartilage in the HA-PEG4-DBCO/0.25 hydrogel showed that more chondrocytes formed a lacunae structure compared with neocartilage in the HA-PEG4-DBCO/1 hydrogel. In the HA-PEG4-DBCO/1 hydrogel, the host cells from the surrounding tissues migrated to the injected hydrogel and formed new hybrid tissue constructs (Fig. 6B).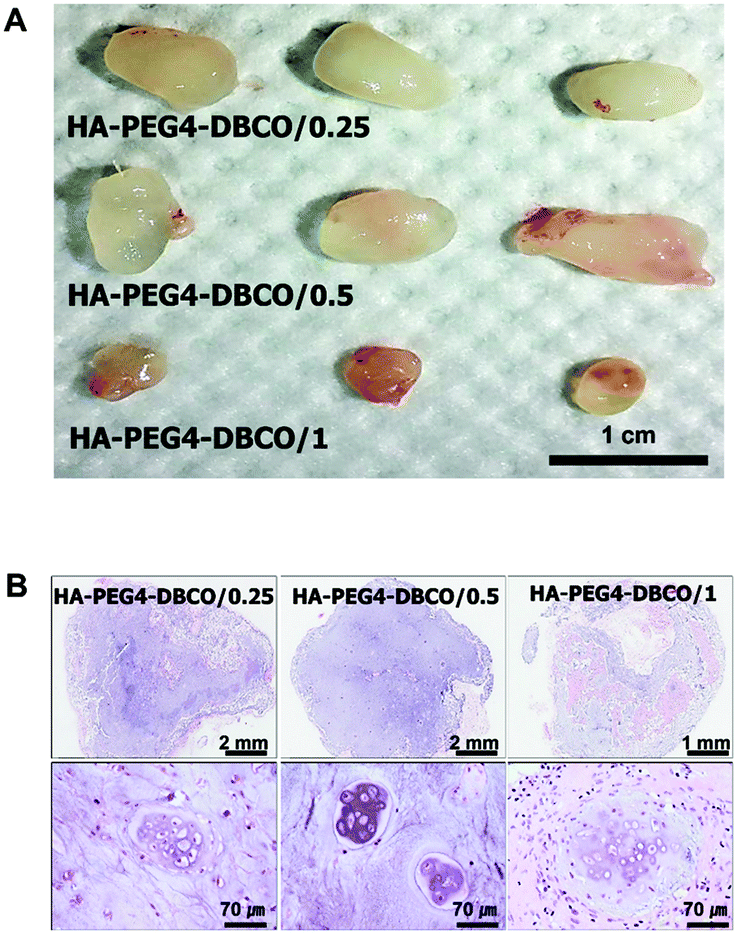 | ||
| Fig. 6 In vivo cartilage regeneration after injection of various HA-DBCO hydrogels that encapsulated chondrocytes at 35 days. The cells were cultured in DMEM/F-12 medium and encapsulated in HA-DBCO hydrogels at a density of 3 × 106 cells per 300 μl. (A) HA-DBCO hydrogels removed from the mice. Both HA-DBCO/0.25 and HA-DBCO/0.5 hydrogels with chondrocytes formed milky-white solid tissues similar to cartilage tissue. (B) Histological analysis of HA-DBCO hydrogels using H&E staining. | ||
The injectable hydrogels have emerged as ideal biomaterials for in situ tissue repair. Although to fabricate the injectable hydrogels, various cross-linking methods including photoinduced cross-linking, Michael addition reaction, Schiff base reaction and copper-catalyzed azide–alkyne cycloaddition reaction were developed, these chemical reactions commonly caused side effects.15 Recently, bioorthogonal click reactions such as Diels–Alder and strain-promoted cyclooctyne–azide cycloaddition reaction have been introduced to fabricate the injectable hydrogels.30,34 A lot of studies demonstrated that this bioorthogonal cross-linking has allowed for rapid gelation and highly specific chemical reactions in live cells without harmful effects.25,35 Fan et al. have shown the in vivo analysis after the injection of the only chitosan/hyaluronic acid hydrogels without the cells in subcutaneous tissues, which was focused on the biocompatibility and inflammation of hydrogels.26 In another study, the regeneration of cartilage tissue using injectable dextran hydrogels was shown, which was analyzed by spheroid consisting of hydrogels and chondrocytes under in vitro conditions.30 Thus, we developed an in situ cross-linkable HA hydrogel using bioorthogonal copper-free click chemistry, and we have directly demonstrated the potential of our injectable hydrogel for cartilage tissue regeneration using the in vivo model.
One of the most notable benefits of our HA hydrogel is that our cross-linking method using bioorthogonal copper-free click chemistry provides efficient and biocompatible cross-linking for the fabrication of HA hydrogels as injectable scaffolds. To this end, HA-PEG4-DBCO derivatives were prepared in a one-step reaction by coupling DBCO-PEG4-amine to HA. This process uses a biocompatible solution and very mild conditions that allow the physical properties of natural HA to be preserved.36 For gelation, we chose 4-arm PEG azide as a cross-linker due to its well-defined symmetric structure, high hydrophilicity, and commercial availability.37 In addition, click reactions between azide groups and DBCO proceed quickly in cell culture media with biocompatible pH and temperature.38 In this study, the cytotoxicity of 4-arm PEG azide to chondrocytes, as measured by MTT assays, was low at various concentrations (Fig. 4A). To study the toxicity of the cross-linker during the cross-linking process, chondrocytes were encapsulated with HA-PEG4-DBCO followed by mixing with various concentrations of 4-arm PEG azide for cross-linking. To evaluate the toxicity of 4-arm PEG azide during the cross-linking process, a live/dead assay was carefully performed (Fig. 4B). Most of the encapsulated chondrocytes in HA-PEG4-DBCO survived through 4-arm PEG azide treatments at concentrations of 0.25, 0.5, and 1 mM. Taken together, no obvious indication of severe toxicity due to the use of 4-arm PEG azide as a cross-linker appeared in the chondrocytes during the cross-linking process. In addition, chondrocytes encapsulated within the HA-PEG4-DBCO hydrogel generated new cartilage after transplantation into the subcutaneous dorsum, as evidenced by the presence of chondrocytes in lacunae without signs of inflammation.
In this study, the extent of cartilage formation was low, and the regenerated cartilage did not distribute homogeneously (Fig. 6B). The efficacy of cartilage regeneration is affected by many different factors, including hydrogel stiffness and cell concentration.39 Previous studies reported that new cartilage formation was more extensive at a higher concentration (1 × 107 cells per ml) treatment groups compared to the low concentration (1 × 106 cells per ml) treatment groups.40 However, we deliberately used a low concentration (1 × 106 cells per ml) treatment group to avoid complicating factors, such as cell concentration, in order to ensure a more controlled comparison of the three types of HA-PEG4-DBCO hydrogels with different mechanical properties. This resulted in a smaller area of cartilage formation compared with the results of other studies. We found that the HA-PEG4-DBCO/0.25 hydrogel (medium stiffness) exhibited superior neocartilage formation in vivo compared to the other HA-PEG4-DBCO hydrogels with different stiffnesses. In addition, for the low-stiffness HA-PEG4-DBCO/1 hydrogel, host cells surrounding the hydrogel migrated within the degraded hydrogel after injection. These results indicate that the stiffness of the hydrogel plays an important role in cell proliferation and hydrogel degradation for cartilage regeneration. To confirm the efficacy of medium-stiffness HA-PEG4-DBCO for promoting cartilage regeneration in vivo, we also injected the HA-PEG4-DBCO hydrogel with a high cell concentration (1 × 107 cells per ml) and observed the histological changes by alcian blue and safranin O staining (Fig. S1†). More extensively and homogeneously distributed mature cartilage structures with lacuna and clusters consisting of two or three cells were observed in the high concentration treatment group.
For hydrogels applied to induce regeneration of a target tissue, hydrogels with controllable physical properties could provide additional benefits. Various cells, such as mesenchymal stem cells, hepatocytes, and cardiomyocytes, have the potential to regenerate target tissues and secrete bioactive molecules to surrounding cells.41,42 The therapeutic efficacy of these cells is regulated by different stiffnesses of the extracellular matrix.43,44 In this study, the stiffness of HA-PEG4-DBCO hydrogels was readily controlled by the concentration of the cross-linker. Thus, we propose that HA-PEG4-DBCO could also be utilized for designing hydrogels that mimic the physiological environment of various tissues for developing advanced tissue engineering therapies.
Experimental section
Materials
Sodium hyaluronic acid (HA, 2 × 103 kDa) was purchased from Lifecore Biomedical (Chaska, USA) and confirmed by size exclusion chromatography-multi-angle laser light scattering (SEC-MALLS). N-Hydroxysuccinimide (NHS), dimethylsulfoxide (DMSO), 1-ethyl-3(3-dimethylaminopropyl)carbodiimide (EDC), deuterium oxide (D, 99.9%), dimethylsulfoxide-d6 (D, 99.9%), and a live/dead assay kit were purchased from Sigma-Aldrich Co (St Louis, MO, USA). Dibenzocyclooctyl-PEG-amine (DBCO-PEG-NH2) was purchased from Click Chemistry Tools (Scottsdale, AZ, USA). 4-Arm-PEG azide (MW 2 kDa) was purchased from Creative PEGWorks (Chapel Hill, NC, USA). All chemicals were analytical grade and used without further purification.Preparation of HA-PEG4-DBCO hydrogels
HA and DBCO-PEG-NH2 were chemically conjugated to synthesize HA-PEG4-DBCO. HA sodium (1 g) was dissolved in DMSO/distilled water (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v, 200 ml), and EDC (23.4 mg) and NHS (14.1 mg) were added to activate the carboxylic groups of HA. DBCO-PEG-NH2 (43 mg) dissolved in DMSO (1 ml) was added dropwise to the HA solution, and the mixture was vigorously stirred for 3 days at room temperature. And then, purification was conducted by dialysis from DMSO and DMSO/distilled water (1:1 v/v). The resulting solution was lyophilized to obtain the dry product of HA-PEG4-DBCO. To fabricate the HA-PEG4-DBCO-based hydrogel, 100 mg HA-PEG4-DBCO was dissolved in 10 ml distilled water. 4-Arm-PEG azide was added to the HA-PEG4-DBCO solution as an in situ cross-linker, and the mixture was incubated at 37 °C for 30 min. The dose of 4-arm-PEG azide was varied from 0 to 2.5 mM to optimize the physical properties of the resulting hydrogel.
:1 v/v, 200 ml), and EDC (23.4 mg) and NHS (14.1 mg) were added to activate the carboxylic groups of HA. DBCO-PEG-NH2 (43 mg) dissolved in DMSO (1 ml) was added dropwise to the HA solution, and the mixture was vigorously stirred for 3 days at room temperature. And then, purification was conducted by dialysis from DMSO and DMSO/distilled water (1:1 v/v). The resulting solution was lyophilized to obtain the dry product of HA-PEG4-DBCO. To fabricate the HA-PEG4-DBCO-based hydrogel, 100 mg HA-PEG4-DBCO was dissolved in 10 ml distilled water. 4-Arm-PEG azide was added to the HA-PEG4-DBCO solution as an in situ cross-linker, and the mixture was incubated at 37 °C for 30 min. The dose of 4-arm-PEG azide was varied from 0 to 2.5 mM to optimize the physical properties of the resulting hydrogel.
Analysis of the physicochemical properties of the HA-PEG4-DBCO hydrogel
The chemical composition and structure of the polymers were analyzed by 1H NMR (UnityPlus300, Varian, CA, USA) and FT-IR (Bruker Optic GmbH, Germany) spectroscopy. HA and HA-PEG4-DBCO were dissolved in D2O for 1H NMR analysis, and the 1H NMR spectra were recorded at 300 MHz. The FT-IR spectra of HA, HA-PEG4-DBCO, and HA-PEG4-DBCO hydrogels were determined by attenuated total reflectance (ATR) FT-IR, and each FT-IR spectrum was recorded with 16 scans in the range of 600–4000 cm−1. Rheological characterization of the HA-PEG4-DBCO hydrogels was performed using a dynamic mechanical analyzer (ELF3200, Endura TEC, Minnetonka, MN, USA). To confirm the gelation time of the hydrogels, the HA-PEG4-DBCO aqueous solution was prepared and gelated by adding various concentrations of 4-arm PEG azide. The storage modulus (G′) and loss modulus (G′′) were measured at a stress of 25 Pa at room temperature, and the gelation time was determined at the intersection point of the G′ and G′′ curves. To measure the elasticity of the hydrogels, HA-PEG4-DBCO aqueous solution and 4-arm PEG azide were mixed in 12-well tissue culture plates for 20 min to prepare 6 mm-thick hydrogels, and the compressive modulus of elasticity was tested at a constant stress rate of 40 mN min−1 up to 20% strain at room temperature. To examine the swelling properties of the HA-PEG4-DBCO hydrogels, known weights of freeze-dried hydrogels were immersed in phosphate buffered saline (PBS) and kept at 37 °C until the swelling reached equilibrium. At an appropriate time, the swollen hydrogels were removed and immediately weighed using a microbalance after any excess water remaining on the surface was absorbed with filter paper. The equilibrium swelling ratio (ESR) was calculated using the following equation: ESR = (Ws − Wd)/Wd, where Ws and Wd represent the weights of the swollen hydrogel and dried hydrogel, respectively. The cross-sectional morphologies of the hydrogels were characterized by scanning electron microscopy (SEM, JSM-6330F, Peabody, MA, USA) operating at an accelerating voltage of 10 kV after gelation. For SEM analysis, the HA-PEG4-DBCO hydrogels (HA-PEG4-DBCO/0.25, HA-PEG4-DBCO/0.5 and HA-PEG4-DBCO/1) were fabricated and freeze-dried overnight. The samples were placed on carbon tape and then cross-sectioned and coated with a thin layer of platinum.In vitro culture of chondrocytes and cytotoxicity of HA-PEG4-DBCO hydrogels
Rabbit articular chondrocytes were isolated from the knee joints of New Zealand white rabbits (n = 5, Jung-Ang Lab Animal, Seoul, Korea) using a sterile scalpel.45 The cartilage fragments were chopped, washed with PBS (pH 7.4), and digested with 0.5% collagenase type II (Sigma) containing Dulbecco's modified Eagle's medium/nutrient mixture F-12 (DMEM/F12, Gibco, Grand Island, NY, USA) for 10 h. The culture media were supplemented with 10% (v/v) fetal bovine serum (FBS, Gibco) and penicillin/streptomycin combined antibiotics (100 units per ml). Recovered chondrocytes were washed with PBS and cultured in a humidified 5% CO2 incubator using DMEM/F12 media. Adherent chondrocytes were expanded for a period of 7 days, and the medium was changed every 3 days.Cytotoxicity of 4-arm PEG azide and HA-PEG4-DBCO hydrogels
We confirmed the cytotoxicity of 4-arm PEG azide as a cross-linker using MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) (Sigma) assays. Expanded chondrocytes (passage number 2) were collected with 0.05% trypsin-EDTA (Gibco) and plated in a 96-well culture plate (Corning) at a cell density of 5,000 cells per well. After 24 h of incubation, the medium was replaced with fresh medium that contained various concentrations of 4-arm PEG azide (0.25, 0.5, 1, and 2.5 mM). On the 1st day after adding 4-arm PEG azide, 20 μl MTT solution (5 mg ml−1 in PBS) was added to each well and incubated for 2 h at 37 °C. The formed formazan crystal was solubilized by dimethylsulfoxide (DMSO), and the absorbance was measured at 540 nm using a reader (SpectraMax M3, Molecular Devices, Sunnyvale, CA, USA). To evaluate the cytotoxicity of the hydrogels, chondrocytes were suspended in HA-PEG4-DBCO (1.0 × 106 cells per ml). The chondrocytes in the HA-PEG4-DBCO were encapsulated using the in situ cross-linker, 4-arm-PEG azide (final concentration of 0.25 to 1 mM). The HA-PEG4-DBCO and 4-arm-PEG azide mixture (n = 9 and 200 mm3) was injected into a 24-well culture plate and incubated at 37 °C for 20 min to form hydrogels. DMEM/F12 (2 ml per well) was added to the in vitro culture. The cells in the hydrogels were stained using a Live/Dead assay kit (Abcam) after 1 and 5 days of in vitro culture. In addition, the distribution of chondrocytes in the hydrogels was observed. Stained chondrocytes were observed using confocal laser scanning microscopy (CLSM, Olympus IX 81).Application of the hydrogels for the transplantation of chondrocytes in vivo
All animals received care according to the guidelines for the care and use laboratory animals of the Korea Institute of Toxicology (KIT). The study was approved by the Institutional Animal Care and Use Committee of KIT (IACUC 1607-0235). Prior to chondrocyte transplantation using hydrogels, the chondrocyte-free hydrogel was subcutaneously injected into Balb-c mice to monitor the potential immune responses and in vivo changes of the optimized hydrogel. To evaluate the biodegradation of the hydrogel, injected hydrogels (n = 3/each time-point) were collected to measure their masses and volumes at 1, 7, 14, 21, and 35 days post-injection. At each time-point, gross lesions of the injection sites were carefully observed. To transplant, the hydrogel with chondrocytes, Balb-c mice (5.5 weeks old; NARA Biotech., Korea) were anesthetized by isoflurane (2.5–5%) inhalation. The HA-PEG4-DBCO containing chondrocytes (1 × 106 cells per ml) were mixed with 0.25, 0.5, or 1 mM 4-arm-PEG azide, and 1 ml of the mixture was immediately subcutaneously injected into the mice using 24-gauge needles. After 35 days, all mice were euthanized, and the injected HA-PEG4-DBCO hydrogels (n = 3/each group) were excised for histology. The excised specimens were fixed in 10% neutral buffered formalin and embedded in paraffin. Tissue (5 μm) slides were primarily stained with H&E for evaluating tissue responsiveness and chondrogenesis in the cell-seeded hydrogels. Alcian blue and safranin-O staining was additionally performed to measure cartilage regeneration in the hydrogels.Statistical analysis
Statistical analysis was performed using an Origin pro version 8 software package (OriginLab Corp, MA, USA) to determine the statistical differences. The experimental data are presented as the mean ± standard deviation and were performed with one-way analysis of variance (One Way ANOVA). A value of p < 0.05 was considered statistically significant.Conclusions
In this study, we synthesized in situ cross-linkable and injectable HA-PEG4-DBCO hydrogels for tissue engineering using a simple one-step bioorthogonal click reaction. The cross-linker is non-toxic, and our HA-PEG4-DBCO hydrogel has good biocompatibility during in situ physical gelation. The elastic modulus can be efficiently modulated by changing the concentration of the cross-linker. In addition, cartilage regeneration using our hydrogel revealed the successful development of cartilage lacunae. Therefore, we demonstrated the feasibility of our hydrogel for use as prototype cell delivery vehicles, which will be further refined in the future for particular biomedical applications in tissue engineering and other industries.Conflicts of interest
There are no conflicts to declareAcknowledgements
This study was supported by the INNOPOLIS Foundation grant (2016-02-DD-016), the Bio & Medical Technology Development Program of the National Research Foundation (NRF-2016 M3A9B4919616) funded by the Korean government (Ministry of Science, ICT & Future Planning). We thank Eun Hee Han of the Korea Basic Science Institute (Daejeon, Republic of Korea) for her technical support in the confocal microscopy analysis.References
- B. P. Toole, Nat. Rev. Cancer, 2004, 4, 528–539 CrossRef CAS PubMed.
- D. Jiang, J. Liang and P. W. Noble, Annu. Rev. Cell Dev. Biol., 2007, 23, 435–461 CrossRef CAS PubMed.
- J. A. Burdick and G. D. Prestwich, Adv. Mater., 2011, 23, H41–H56 CrossRef CAS PubMed.
- E. Caló and V. V. Khutoryanskiy, Eur. Polym. J., 2015, 65, 252–267 CrossRef.
- J.-P. Chen and T.-H. Cheng, Polymer, 2009, 50, 107–116 CrossRef CAS.
- W. Y. Su, Y. C. Chen and F. H. Lin, Acta Biomater., 2010, 6, 3044–3055 CrossRef CAS PubMed.
- R. M. A. Domingues, M. Silva, P. Gershovich, S. Betta, P. Babo, S. G. Caridade, J. F. Mano, A. Motta, R. L. Reis and M. E. Gomes, Bioconjugate Chem., 2015, 26, 1571–1581 CrossRef CAS PubMed.
- K. H. Bae, L.-S. Wang and M. Kurisawa, J. Mater. Chem. B, 2013, 1, 5371–5388 RSC.
- M. K. Nguyen and D. S. Lee, Macromol. Biosci., 2010, 10, 563–579 CrossRef CAS PubMed.
- Y. Li, J. Rodrigues and H. Tomas, Chem. Soc. Rev., 2012, 41, 2193–2221 RSC.
- V. Crescenzi, A. Francescangeli, A. Taglienti, D. Capitani and L. Mannina, Biomacromolecules, 2003, 4, 1045–1054 CrossRef CAS PubMed.
- S. Ibrahim, Q. K. Kang and A. Ramamurthi, J. Biomed. Mater. Res., Part A, 2010, 94, 355–370 Search PubMed.
- P. L. Lu, J. Y. Lai, D. H. Ma and G. H. Hsiue, J. Biomater. Sci., Polym. Ed., 2008, 19, 1–18 CrossRef CAS PubMed.
- E. M. Ahmed, J. Adv. Res., 2015, 6, 105–121 CrossRef CAS PubMed.
- P. M. Kharkar, K. L. Kiick and A. M. Kloxin, Chem. Soc. Rev., 2013, 42, 7335–7372 RSC.
- J. Baier Leach, K. A. Bivens, C. W. Patrick Jr. and C. E. Schmidt, Biotechnol. Bioeng., 2003, 82, 578–589 CrossRef PubMed.
- J. B. Leach, K. A. Bivens, C. N. Collins and C. E. Schmidt, J. Biomed. Mater. Res., Part A, 2004, 70, 74–82 CrossRef PubMed.
- R. Jin, L. S. Moreira Teixeira, A. Krouwels, P. J. Dijkstra, C. A. van Blitterswijk, M. Karperien and J. Feijen, Acta Biomater., 2010, 6, 1968–1977 CrossRef CAS PubMed.
- H. Tan, J. P. Rubin and K. G. Marra, Organogenesis, 2010, 6, 173–180 CrossRef PubMed.
- K. Xu, K. Narayanan, F. Lee, K. H. Bae, S. Gao and M. Kurisawa, Acta Biomater., 2015, 24, 159–171 CrossRef CAS PubMed.
- X. Hu, D. Li, F. Zhou and C. Gao, Acta Biomater., 2011, 7, 1618–1626 CrossRef CAS PubMed.
- S. Piluso, B. Hiebl, S. N. Gorb, A. Kovalev, A. Lendlein and A. T. Neffe, Int. J. Artif. Organs, 2011, 34, 192–197 CrossRef CAS PubMed.
- M. Schrag, C. Mueller, U. Oyoyo and W. M. Kirsch, Prog. Neurobiol., 2011, 94, 296–306 CrossRef CAS PubMed.
- R. Hatano, M. Ebara, H. Fukuda, M. Yoshikawa, N. Sugiura, F. Kondo, M. Yukawa and H. Saisho, J. Gastroenterol. Hepatol., 2000, 15, 786–791 CrossRef CAS PubMed.
- A. Takahashi, Y. Suzuki, T. Suhara, K. Omichi, A. Shimizu, K. Hasegawa, N. Kokudo, S. Ohta and T. Ito, Biomacromolecules, 2013, 14, 3581–3588 CrossRef CAS PubMed.
- M. Fan, Y. Ma, J. Mao, Z. Zhang and H. Tan, Acta Biomater., 2015, 20, 60–68 CrossRef CAS PubMed.
- N. J. Agard, J. A. Prescher and C. R. Bertozzi, J. Am. Chem. Soc., 2004, 126, 15046–15047 CrossRef CAS PubMed.
- K. Lang and J. W. Chin, ACS Chem. Biol., 2014, 9, 16–20 CrossRef CAS PubMed.
- D. Steinhilber, T. Rossow, S. Wedepohl, F. Paulus, S. Seiffert and R. Haag, Angew. Chem., Int. Ed., 2013, 52, 13538–13543 CrossRef CAS PubMed.
- X. Wang, Z. Li, T. Shi, P. Zhao, K. An, C. Lin and H. Liu, Mater. Sci. Eng., C, 2017, 73, 21–30 CrossRef CAS PubMed.
- G. Huerta-Angeles, M. Nemcova, E. Prikopova, D. Smejkalova, M. Pravda, L. Kucera and V. Velebny, Carbohydr. Polym., 2012, 90, 1704–1711 CrossRef CAS PubMed.
- D. A. Ossipov, X. Yang, O. Varghese, S. Kootala and J. Hilborn, Chem. Commun., 2010, 46, 8368–8370 RSC.
- H. Koo, S. Lee, J. H. Na, S. H. Kim, S. K. Hahn, K. Choi, I. C. Kwon, S. Y. Jeong and K. Kim, Angew. Chem., Int. Ed., 2012, 51, 11836–11840 CrossRef CAS PubMed.
- C. M. Nimmo, S. C. Owen and M. S. Shoichet, Biomacromolecules, 2011, 12, 824–830 CrossRef CAS PubMed.
- H. Jiang, S. Qin, H. Dong, Q. Lei, X. Su, R. Zhuo and Z. Zhong, Soft Matter, 2015, 11, 6029–6036 RSC.
- J. Zhu and R. E. Marchant, Expert Rev. Med. Devices, 2011, 8, 607–626 CrossRef CAS PubMed.
- J. Xu, E. Feng and J. Song, J. Am. Chem. Soc., 2014, 136, 4105–4108 CrossRef CAS PubMed.
- M. Pretze, D. Pietzsch and C. Mamat, Molecules, 2013, 18, 8618–8665 CrossRef CAS PubMed.
- K. L. Spiller, S. A. Maher and A. M. Lowman, Tissue Eng., Part B, 2011, 17, 281–299 CrossRef CAS PubMed.
- Y.-B. Park, C.-W. Ha, J.-A. Kim, J.-H. Rhim, Y.-G. Park, J. Y. Chung and H.-J. Lee, PLoS One, 2016, 11, e0165446 Search PubMed.
- T. J. Myers, F. Granero-Molto, L. Longobardi, T. Li, Y. Yan and A. Spagnoli, Expert Opin. Biol. Ther., 2010, 10, 1663–1679 CrossRef CAS PubMed.
- M. T. Alrefai, D. Murali, A. Paul, K. M. Ridwan, J. M. Connell and D. Shum-Tim, Stem Cells Cloning: Adv. Appl., 2015, 8, 81–101 Search PubMed.
- F. Gattazzo, A. Urciuolo and P. Bonaldo, Biochim. Biophys. Acta, 2014, 1840, 2506–2519 CrossRef CAS PubMed.
- J. S. Park, J. S. Chu, A. D. Tsou, R. Diop, Z. Tang, A. Wang and S. Li, Biomaterials, 2011, 32, 3921–3930 CrossRef CAS PubMed.
- S. W. Kang, S. M. Son, J. S. Lee, E. S. Lee, K. Y. Lee, S. G. Park, J. H. Park and B. S. Kim, J. Biomed. Mater. Res., Part A, 2006, 78, 659–671 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7py01654a |
| ‡ These authors contributed equally to this work as first author. |
| This journal is © The Royal Society of Chemistry 2018 |