
Rational skeletal rigidity of conjugated microporous polythiophenes for gas uptake†
 *b
*b
Abstract
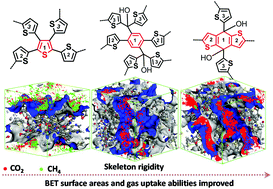
Three microporous polythiophenes, P-TTT, P-THIDT and P-DTBDT, with increasing rigidity of their cores were designed and prepared by FeCl3 catalyzed polymerization. The step by step strengthening of the rigidity of these polymers, stacking morphologies (planar, three-dimensional mesh pattern, and spherical), and Brunauer–Emmett–Teller (BET) surface areas have been extensively improved (thienyl core is 58 m2 g−1, phenyl core is 869 m2 g−1 and BDT fused ring is 1140 m2 g−1). At the same time, the carbon dioxide uptake capacity has been enhanced from 2.1 wt% to 12.1 wt% at 1.1 bar and 273 K temperature, accompanied by an increase of the heat adsorption from 17 to 37 kJ mol−1. Molecular-level models were developed that showed the same trend, which was similar to the experimental results. The rational skeletal rigidity can counteract the nanostructural packing and avoid the nanopores collapsing in organic microporous polymers.
 Please wait while we load your content...
Please wait while we load your content...

