
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Phenyl acrylate is a versatile monomer for the synthesis of acrylic diblock copolymer nano-objects via polymerization-induced self-assembly†
S. L.
Canning
 ,
V. J.
Cunningham
,
L. P. D.
Ratcliffe
and
S. P.
Armes
*
,
V. J.
Cunningham
,
L. P. D.
Ratcliffe
and
S. P.
Armes
*
Department of Chemistry, University of Sheffield, Dainton Building, Brook Hill, Sheffield, South Yorkshire S3 7HF, UK. E-mail: s.p.armes@sheffield.ac.uk
First published on 25th July 2017
Abstract
Over the last decade or so, polymerization-induced self-assembly (PISA) has become widely recognized as a versatile technique for the rational synthesis of diblock copolymer nano-objects in the form of concentrated dispersions. However, there are relatively few examples of acrylic-based PISA formulations in the literature, partly because such copolymers typically possess relatively low glass transition temperatures (Tg) that preclude morphological characterization by transmission electron microscopy. To address this problem, we have selected phenyl acrylate (PhA) as a model monomer to generate the solvophobic block in three PISA formulations using reversible addition–fragmentation chain transfer (RAFT) polymerization. Thus, a poly(dimethyl acrylamide)-based chain transfer agent (CTA) is chain-extended using PhA via RAFT aqueous emulsion polymerization to produce a series of well-defined sterically-stabilized spheres whose mean diameter can be readily adjusted from 38 nm to 188 nm by varying the target degree of polymerization (DP). In contrast, RAFT alcoholic dispersion polymerization of PhA using a poly(acrylic acid) CTA leads to an evolution of copolymer morphology from spheres to worms to lamellae and finally vesicles as the target DP of the structure-directing PPhA block is increased. Similarly, RAFT dispersion polymerization of PhA in n-heptane also produces spheres, worms or vesicles depending on the target DP of the PPhA block. 1H NMR studies indicate that >98% PhA conversion is achieved in all cases, while GPC analysis indicates high blocking efficiencies. However, relatively broad molecular weight distributions are observed (Mw/Mn = 1.37 to 2.48), which suggests extensive chain transfer to polymer in such PISA syntheses, particularly in the case of the RAFT aqueous emulsion polymerization formulation. Nevertheless, the relatively high Tg of PPhA (50 °C) enables characterization of the various copolymer morphologies using conventional TEM.
Introduction
AB diblock copolymers are well-known to self-assemble into well-defined micelles in a solvent that is selective for one of the two blocks.1–9 In principle, a wide range of morphologies can be obtained depending on the relative volume fractions of each block.10,11 Traditionally, this self-assembly involves some form of post-polymerization processing (e.g. a pH switch, a solvent switch or thin film rehydration) that is conducted at high dilution and typically at rather low final copolymer concentrations (<1% w/w). With the advent of controlled radical polymerization (CRP) techniques such as nitroxide-mediated polymerization (NMP),12 atom transfer radical polymerization (ATRP)13,14 and reversible addition–fragmentation chain transfer (RAFT) polymerization,15 polymerization-induced self-assembly (PISA) has recently become established as a versatile platform technology for the rational synthesis of diblock copolymer nano-objects at high solids, which eliminates the requirement for further processing steps.16–19In practice, most of the PISA formulations reported in the literature are based on RAFT polymerization.16–22 Robust RAFT dispersion polymerization formulations have been developed for water,16,19,20,23–32 lower alcohols17,33–40 and non-polar solvents17,41–45 while RAFT aqueous emulsion polymerization has also been thoroughly explored.46–52 However, the vast majority of PISA syntheses reported in the literature involve polymerization of methacrylic or styrenic monomers rather than acrylic monomers, despite the commercial importance of the latter monomer class. Acrylic polymers typically exhibit glass transition temperatures (Tg) well below ambient temperature, which makes them very useful for paints and coatings53 and also adhesion applications.54 Unfortunately, this makes the examination of copolymer morphology via conventional transmission electron microscopy (TEM) rather problematic, and such characterization is usually an important component of most PISA studies. In principle, cryo-TEM can be utilized instead, but in practice this technique is not widely available and relatively expensive; moreover, appropriate sample preparation protocols are not well-developed for imaging non-aqueous formulations.
Chain transfer to polymer is well known for the polymerization of acrylic monomers.55–57 This side reaction leads to branching and hence significant broadening of the molecular weight distribution (MWD), although this can be mitigated to some extent by using CRP techniques.58,59 This problem may explain why relatively few acrylic-based PISA formulations have been reported in the literature.45,60–67 These include syntheses conducted in either water,60,61,64,65n-alkanes45,63,66 or ethanol/water mixtures.62,67 For example, Ratcliffe et al. recently chain-extended a poly(lauryl acrylate) (PLA) macromolecular chain transfer agent (macro-CTA) using benzyl acrylate (BzA) to produce PLA-PBzA diblock copolymer nano-objects in either n-heptane, n-dodecane or iso-hexadecane.66 By varying the target DP of the PBzA core-forming block, pure phases of spheres, worms and vesicles could be produced. However, relatively broad molecular weight distributions were observed, with Mw/Mn values ranging from 1.53 to 1.67 when targeting PBzA DPs of 55–90 at 25% w/w solids. Moreover, the relatively low Tg of PBzA (6 °C) prevented characterization of the nano-object morphology by conventional TEM, so cryo-TEM was utilized instead.66
In principle, one solution to this imaging problem is to simply select an acrylic polymer with a relatively high Tg. In practice, very few acrylic monomers are suitable in this regard. Nevertheless, this approach was recently reported by Tan et al., who polymerized isobornyl acrylate (IBOA; Tg of PIBOA = 94 °C) using a poly(ethylene glycol)-based macro-CTA.67 This PISA synthesis was conducted in an 85/15 ethanol/water mixture at 40 °C using visible light to initiate the polymerization. High conversions (>95%) were obtained within 30 min and relatively narrow molecular weight distributions were reported (e.g. Mw/Mn = 1.26 when targeting a PIBOA DP of 150). However, diblock copolymers were typically contaminated with approximately 10–20% unreacted macro-CTA. Nevertheless, either spheres, worms or vesicles could be produced depending on the PISA synthesis conditions, as judged by conventional TEM studies.
In the present work, we have chosen to study phenyl acrylate (PhA) as a model acrylic monomer in the context of three PISA formulations. Poly(phenyl acrylate) (PPhA) has a relatively high Tg of 50 °C,68 which should be sufficient to enable copolymer morphology studies using conventional TEM. To evaluate the versatility of this new monomer, we studied its use in RAFT aqueous emulsion polymerization, RAFT alcoholic dispersion polymerization and RAFT dispersion polymerization in n-alkanes using suitable macro-CTAs as the steric stabilizer block.
Experimental section
Materials
Lauryl acrylate (LA, 98%) was purchased from Tokyo Chemical Industry UK Ltd. Phenyl acrylate (PhA, 97%) was purchased from Alfa Aesar (UK). Both monomers were used as received. 2-(Dodecylthiocarbonothioylthio)-2-methylpropionic acid (DDMAT, 98%), dimethyl acrylamide (DMAC, 99%), acrylic acid (AA, 99%), potassium persulfate (KPS, 99%), L-ascorbic acid (AscAc, 99%) and azoisobutyronitrile (AIBN, 98%) were purchased from Sigma Aldrich (UK) and were used as received. tert-Butyl peroxy-2-ethylhexanoate (T21S) was purchased from AkzoNobel (The Netherlands). Dimethyl sulfoxide-d6, CDCl3 and DMF-d7 were purchased from Goss Scientific Instruments Ltd (Cheshire, UK). All other solvents were purchased from Fisher Scientific (Loughborough, UK) and were used as received. Deionized water was used for all aqueous PISA formulations.Synthesis of PDMAC53 macro-CTA by RAFT solution polymerization
A typical protocol for the synthesis of a PDMAC53 macro-CTA was as follows. DMAC monomer (0.141 mol, 14.021 g), DDMAT RAFT agent (2.57 mmol, 0.9376 g) and dioxane (0.397 mol, 35.00 g, corresponding to a 30% w/w solution) were added to a round-bottomed flask in order to target a mean DP of 55. AIBN initiator (0.257 mmol, 0.0422 g, CTA/AIBN molar ratio = 10.0) was added to this mixture, and the resulting yellow solution was cooled by immersion in an ice bath while sparging with N2 gas for 30 min, before the sealed flask was placed in an oil bath set at 70 °C. After 35 min, the polymerization was quenched by immersion of the reaction flask in ice, followed by exposure to air. The final DMAC conversion was determined to be 92% by 1H NMR spectroscopy. The crude PDMAC macro-CTA was then precipitated twice into a ten-fold excess of diethyl ether to remove unreacted DMAC and other residual reagents before being dried in a vacuum oven overnight at 20 °C. 1H NMR analysis indicated a mean DP of 53 for the final purified PDMAC macro-CTA, which suggests a CTA efficiency of 95%. THF GPC analysis of PDMAC53 macro-CTA indicated Mn and Mw/Mn values of 5400 g mol−1 and 1.08, respectively.Synthesis of PDMAC53-PPhAx diblock copolymer nano-objects by RAFT aqueous emulsion polymerization
A typical protocol for the synthesis of PDMAC53-PPhA100 diblock copolymer was as follows: PDMAC53 macro-CTA (0.200 g, 0.0356 mmol), KPS (1.90 mg, 0.007 mmol; CTA/initiator molar ratio = 5.0) and water (2.192 g, corresponding to a 25% w/w aqueous solution) were weighed into a 25 mL round-bottom flask and purged with nitrogen for 30 min. Ascorbic acid (2.0 mg, 16 μmol, CTA/initiator molar ratio = 5.0; prepared as a 10% w/w aqueous solution) was degassed separately with nitrogen for 30 min prior to addition to the aqueous solution in the reaction flask. Phenyl acrylate (0.527 g, 3.56 mmol) was also degassed separately with nitrogen for 30 min and added last to the reaction flask, which was then sealed and immersed in an oil bath set at 30 °C. The reaction mixture was stirred for 4.5 h to ensure high monomer conversion (>99% by 1H NMR analysis) and subsequently quenched by cooling to 20 °C, followed by exposure to air. DMF GPC analysis of PDMAC53-PPhA100 indicated Mn and Mw/Mn values of 18![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 400 g mol−1 and 2.10, respectively.
400 g mol−1 and 2.10, respectively.
Synthesis of PAA26 macro-CTA
A typical protocol for the synthesis of a PAA26 macro-CTA was as follows. AA monomer (0.080 mol, 6.000 g), DDMAT RAFT agent (2.78 mmol, 1.011 g), and ethanol (0.081 mol, 7.102 g, corresponding to a 50% w/w solution) were added to a round-bottomed flask in order to target a mean DP of 30. AIBN initiator (0.56 mmol, 0.091 g, CTA/AIBN molar ratio = 5.0) was added to this mixture, and the resulting yellow solution was cooled in an ice bath while sparging with N2 gas for 30 min, before the sealed flask was immersed in an oil bath set at 60 °C. After 150 min, the polymerization was quenched by immersion of the reaction flask in an ice bath, followed by exposure to air. 1H NMR spectroscopy indicated a final AA conversion of 85%. The crude polymer was then precipitated twice into a ten-fold excess of diethyl ether before being dried in a vacuum oven at 20 °C. 1H NMR analysis indicated a mean DP of 26 for this PAA macro-CTA, which suggests a CTA efficiency of 98%. THF GPC analysis of this PAA26 macro-CTA indicated Mn and Mw/Mn values of 3400 g mol−1 and 1.07, respectively.Synthesis of PAA26-PPhAx diblock copolymers by RAFT alcoholic dispersion polymerization
A typical protocol for the synthesis of PAA26-PPhA50 diblock copolymer nano-objects was as follows: PAA26 macro-CTA (0.080 g, 0.035 mmol) was added to a glass vial, followed by PhA monomer (0.265 g, 1.787 mmol) and ethanol (1.038 g, corresponding to a 25% w/w solution). AIBN initiator was then added (1.20 mg, 7.15 μmol, CTA/AIBN molar ratio = 5.0). This flask was cooled using an ice bath, and the solution was sparged with N2 gas for 25 min. The flask was then sealed and immersed in an oil bath set at 70 °C. The reaction mixture was stirred for 16 h to ensure high monomer conversion (>99% by 1H NMR analysis) and subsequently quenched by cooling followed by exposure to air. THF GPC analysis of PAA26-PPhA50 indicated Mn and Mw/Mn values of 8100 g mol−1 and 1.68, respectively.Synthesis of PLA14 macro-CTA
A typical protocol for the synthesis of a PLA14 macro-CTA was as follows. LA monomer (0.32 mol, 76.92 g), DDMAT RAFT agent (12.8 mmol, 4.67 g), and acetone (1.41 mol, 82.0 g) were added to a round-bottomed flask in order to target a mean DP of 25. AIBN initiator (2.56 mmol, 0.420 g, CTA/AIBN molar ratio = 5.0) was added to this mixture, and the resulting yellow solution was cooled in an ice bath while sparging with N2 gas for 30 min, before the sealed flask was immersed in an oil bath set at 70 °C. After 125 min, the polymerization was quenched by immersion of the reaction flask in ice, followed by exposure to air. A final LA conversion of 55% was determined by 1H NMR spectroscopy. This crude polymer solution was then precipitated twice into a ten-fold excess of cold methanol and washed in further methanol before being dried under vacuum for 72 h at 20 °C. 1H NMR analysis indicated a mean DP of 14 for this PLA macro-CTA, which suggests a CTA efficiency of 98%. THF GPC analysis indicated Mn and Mw/Mn values of 3700 g mol−1 and 1.12, respectively.Synthesis of PLA14-PPhAx diblock copolymers by RAFT dispersion polymerization in n-alkanes
A typical protocol for the synthesis of PLA14-PPhA80 diblock copolymer nano-objects was as follows: PLA14 macro-CTA (0.180 g, 0.05 mmol) was added to a 14 mL glass vial, followed by PhA monomer (0.572 g, 3.86 mmol) and n-heptane (2.05 g). T21S initiator was then added (2.08 mg, 0.01 mmol; CTA/T21S molar ratio = 5.0), as a 1.0% solution in n-heptane to make up a 25% w/w solution. This flask was cooled using an ice bath, and the solution was sparged with N2 gas for 20 min. The flask was then sealed and immersed in an oil bath set at 80 °C. The reaction mixture was stirred for 16 h to ensure high monomer conversion (99% according to 1H NMR analysis) and subsequently quenched by cooling to 20 °C, followed by exposure to air. THF GPC analysis of PLA14-PPhA80 indicated Mn and Mw/Mn values of 13800 g mol−1 and 1.44, respectively.
Copolymer characterization
1H NMR spectroscopy
All 1H NMR spectra were recorded on a 400 MHz Bruker Avance-400 spectrometer using DMF-d7 (PDMAC53-PPhAx), dimethyl sulfoxide-d6 (PAA26-PPhAx) and CDCl3 (PLA14-PPhAx) as deuterated solvents.Gel permeation chromatography (GPC)
Molecular weight distributions of the PAA26-PPhAx and PLA14-PPhAx diblock copolymers were assessed by GPC using THF eluent. For the former series of copolymers, such analysis required exhaustive methylation of the carboxylic acid groups in the PAA block with excess trimethylsilyldiazomethane to prevent column adsorption. The GPC set-up consisted of two 5 μm Mixed C columns (30 cm) and a WellChrom K-2301 refractive index detector operating at 950 ± 30 nm. The mobile phase contained 2.0% v/v triethylamine and 0.05% w/v butylhydroxytoluene (BHT) with a toluene flow rate marker and the flow rate was fixed at 1.0 mL min−1. Copolymer solutions (1.0% w/v) were prepared in THF. A series of ten near-monodisperse poly(methyl methacrylate) standards (Mp values ranging from 1280 to 330000 g mol−1) were used for calibration. Data were analyzed using Agilent GPC/SEC software (version 1.2). The molecular weights and dispersities of the PDMAC53-PPhAx diblock copolymers were determined by GPC using DMF eluent at 60 °C. The GPC set-up consisted of two Polymer Laboratories PL gel 5 μm Mixed C columns connected in series to an Agilent 1260 Infinity GPC/SEC system with refractive index and variable wavelength detectors and autosampler. The mobile phase was HPLC-grade DMF containing 10 mM LiBr with a flow rate of 1.0 mL min−1. Copolymer solutions (1.0% w/v) were prepared in DMF. Ten near-monodisperse PMMA standards (Mn = 625 to 618000 g mol−1) were used for calibration. Data were analyzed using Agilent GPC/SEC software (version 1.2).
Dynamic light scattering (DLS)
The intensity-average hydrodynamic diameter of each batch of diblock copolymer nanoparticles was determined from the Stokes–Einstein equation using a Malvern Instruments Zetasizer NanoZS instrument. Dilute (0.20% w/w) copolymer dispersions were analyzed and data were averaged over three consecutive runs. Measurements were made at 25 °C and the scattered light was detected at an angle of 173°.Transmission electron microscopy (TEM)
Copper/palladium TEM grids (Agar Scientific, UK) were coated in-house to yield a thin film of amorphous carbon. For the PDMAC53-PPhAx and PAA26-PPhAx samples, the grids were then subjected to a glow discharge for 30 s to create a hydrophilic surface. Individual samples (0.20% w/w dispersion, 10.0 μL) were placed on the freshly-treated grids for 1 min and then blotted with filter paper to remove excess solution. To stain the colloidal aggregates, uranyl formate (9.0 μL of a 0.75% w/w solution) was absorbed onto the sample-loaded grid for 20 s and then carefully blotted to remove excess stain. The grids were then dried using a vacuum hose. For the PLA14-PPhAx samples, each dispersion (0.20% w/w, 11 μL) was placed on a grid for 1 min and then blotted with filter paper to remove excess solution. To stain the deposited nanoparticles, each grid was exposed to ruthenium(IV) oxide vapor for 7 min at 20 °C prior to analysis.41 This heavy metal compound acted as a positive stain to improve electron contrast. The ruthenium(IV) oxide was prepared as follows: ruthenium(II) oxide (0.30 g) was added to water (50 g) to form a black slurry; addition of sodium periodate (2.0 g) with stirring produced a yellow solution of ruthenium(IV) oxide within 1 min. Imaging of all grids was performed using a Philips CM100 instrument operating at 100 kV equipped with a Gatan 1 k CCD camera.Results and discussion
One major obstacle to the more widespread use of acrylates in PISA formulations are the relatively low Tg values exhibited by such polymers. In contrast, PPhA has a Tg of 50 °C, which is comparable to that of poly(2-hydroxypropyl methacrylate) or poly(benzyl methacrylate).69 Many high-quality TEM images have been reported for PISA formulations based on such methacrylic core-forming blocks,33,34,46,70 so it was considered likely that similar results could be obtained for PPhA. To exemplify the versatility offered by the use of phenyl acrylate, we devised three new acrylic-based RAFT-mediated PISA formulations based on this monomer (see Fig. 1). Thus RAFT aqueous emulsion polymerization of PhA utilized a PDMAC53 macro-CTA, while RAFT dispersion polymerization of PhA in ethanol used PAA26 as a stabilizer block. Finally, RAFT dispersion polymerizations conducted in n-heptane were conducted using a PLA14 macro-CTA. Each of these three PISA formulations is discussed in turn below.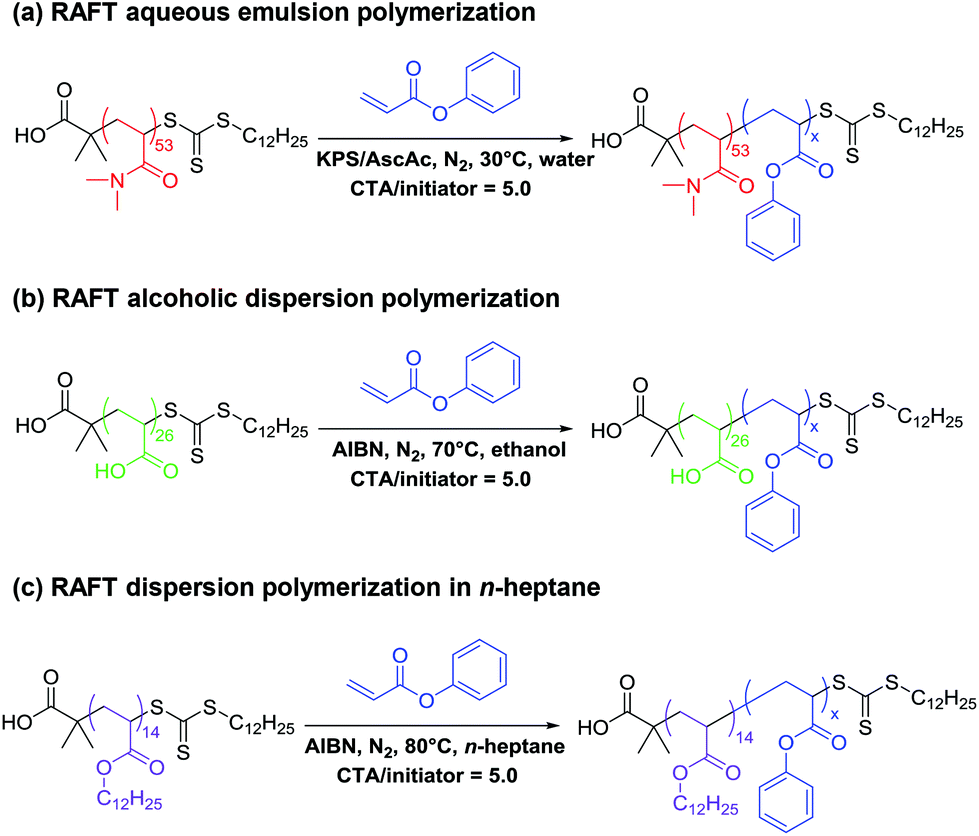 | ||
| Fig. 1 Polymerization of phenyl acrylate via three different PISA formulations: (a) RAFT aqueous emulsion polymerization at 30 °C, (b) RAFT alcoholic dispersion polymerization in ethanol at 70 °C and (c) RAFT dispersion polymerization in n-heptane at 80 °C. | ||
RAFT aqueous emulsion polymerization of phenyl acrylate using PDMAC53 stabilizer
The PDMAC53 macro-CTA was synthesized by RAFT solution polymerization in dioxane using 2-(dodecylthiocarbonothioylthio)-2-methylpropionic acid (DDMAT) as the RAFT agent, as this CTA is well-suited for the controlled polymerization of both acrylic and acrylamide-based monomers.30,66 A low-temperature synthesis protocol was selected for the synthesis of PDMAC53-PPhAx diblock copolymer nanoparticles via RAFT aqueous emulsion polymerization in an attempt to minimize chain transfer to polymer, which is well-known for the polymerization of acrylic monomers.55–59 This problem leads to broadening of the molecular weight distribution via long-chain branching. Thus a redox initiator system comprising KPS and ascorbic acid was selected to enable polymerizations to be conducted at 30 °C.65A series of PDMAC53-PPhAx copolymers was prepared with a target PPhA DP (x) ranging from 50 to 500, see Table 1. High conversions were obtained in all cases (>98% for x = 50–500; entries 1–7). DMF GPC analysis indicated relatively high blocking efficiencies, with the copolymer molecular weight increasing monotonically with target core-forming block DP, as expected. However, Mw/Mn values ranged from 1.85 to 2.48 with no apparent trend between DP and dispersity (Fig. 2). This is perhaps surprising given the relatively low reaction temperature selected for these PISA syntheses, and this point will be considered later.
 | ||
| Fig. 2 DMF GPC curves for PDMAC53-PPhAx diblock copolymers prepared at 25% w/w solids via RAFT aqueous emulsion polymerization of PhA at 30 °C. Mn values are expressed relative to a series of near-monodisperse poly(methyl methacrylate) calibration standards. | ||
| Entry no. | Target diblock composition | Conversionb/% | DMF GPCc | DLS diameterd/nm | TEM morphology | |
|---|---|---|---|---|---|---|
| M n | M w/Mn | |||||
|
a Conditions: [macro-CTA]:[initiator] molar ratio = 5.0; copolymer concentration = 25% w/w.
b Monomer conversion determined by 1H NMR spectroscopy in DMF-d7.
c Refractive index detector, calibrated with a series of near-monodisperse poly(methyl methacrylate) standards.
d DLS: intensity-average diameters were calculated using cumulants analysis software provided by the manufacturer (Malvern Instruments, UK). The numbers in brackets denote the polydispersity in the particle diameter.
|
||||||
| 1 | PDMAC53-PPhA50 | >99 | 12400 |
1.85 | 38 (0.27) | Spheres |
| 2 | PDMAC53-PPhA100 | >99 | 18400 |
2.10 | 54 (0.12) | Spheres |
| 3 | PDMAC53-PPhA150 | >99 | 24700 |
2.19 | 68 (0.12) | Spheres |
| 4 | PDMAC53-PPhA200 | >99 | 30600 |
2.22 | 79 (0.07) | Spheres |
| 5 | PDMAC53-PPhA300 | >99 | 41200 |
2.48 | 105 (0.08) | Spheres |
| 6 | PDMAC53-PPhA400 | 98 | 53300 |
2.43 | 133 (0.08) | Spheres |
| 7 | PDMAC53-PPhA500 | 99 | 61400 |
2.06 | 188 (0.21) | Spheres |
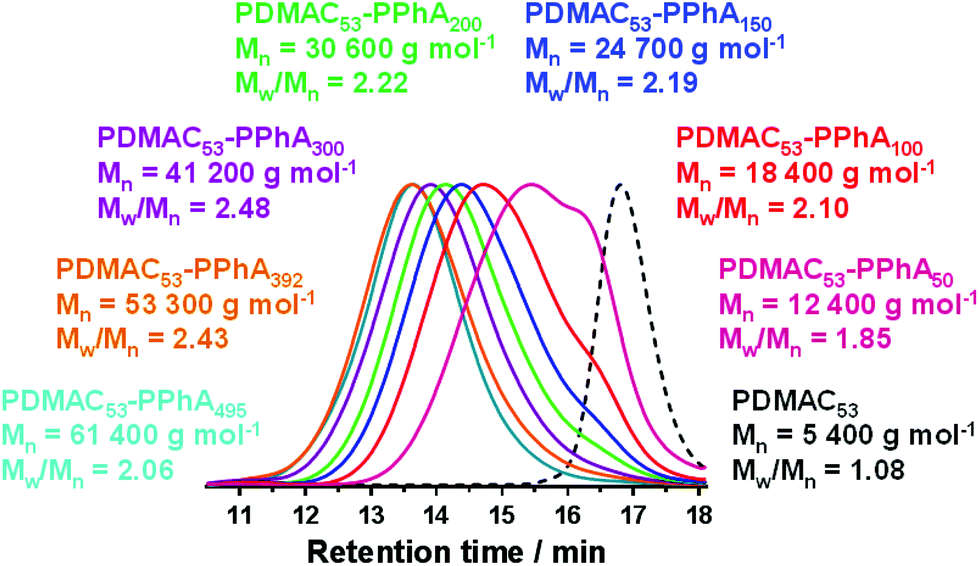
Spherical nanoparticles were produced in all cases, as confirmed by both DLS analysis and TEM (Fig. 3). This kinetically-trapped morphology has been reported for many RAFT aqueous emulsion polymerization syntheses,46,48,61,71–76 but this phenomenon is not properly understood. However, it is perhaps noteworthy that the relatively few such PISA formulations yielding non-spherical nano-objects employed either (i) statistical copolymer macro-CTAs comprising a 1:1 molar ratio of (meth)acrylic acid and oligo(ethylene glycol) (meth)acrylate,51,52,77,78 (ii) copolymer macro-CTAs comprising a poly(N-acryloylmorpholine) (PNAM) block with a few poly(ethylene glycol) acrylate (PEGA) units with varying position within the polymer chain or (iii) a monomer exhibiting a relatively high aqueous solubility (e.g. 4-hydroxybutyl methacrylate).47 Nevertheless, in the present study the PISA mechanism provided good control over the particle size, with a monotonic increase in the intensity-average particle diameter being observed when targeting core-forming DPs ranging from 50 to 500 (see Fig. 3).
 | ||
| Fig. 3 A series of PDMAC53-PPhAx diblock copolymers prepared at 25% w/w solids via RAFT aqueous emulsion polymerization of PhA conducted at 30 °C using a PDMAC53 macro-CTA. (a) Selected TEM images showing well-defined spherical nanoparticles; (b) corresponding DLS intensity-average size distributions and (c) the relationship between the DP of the core-forming PPhA block and the intensity-average DLS diameter, where α is the scaling factor. | ||
RAFT dispersion polymerization of phenyl acrylate in ethanol
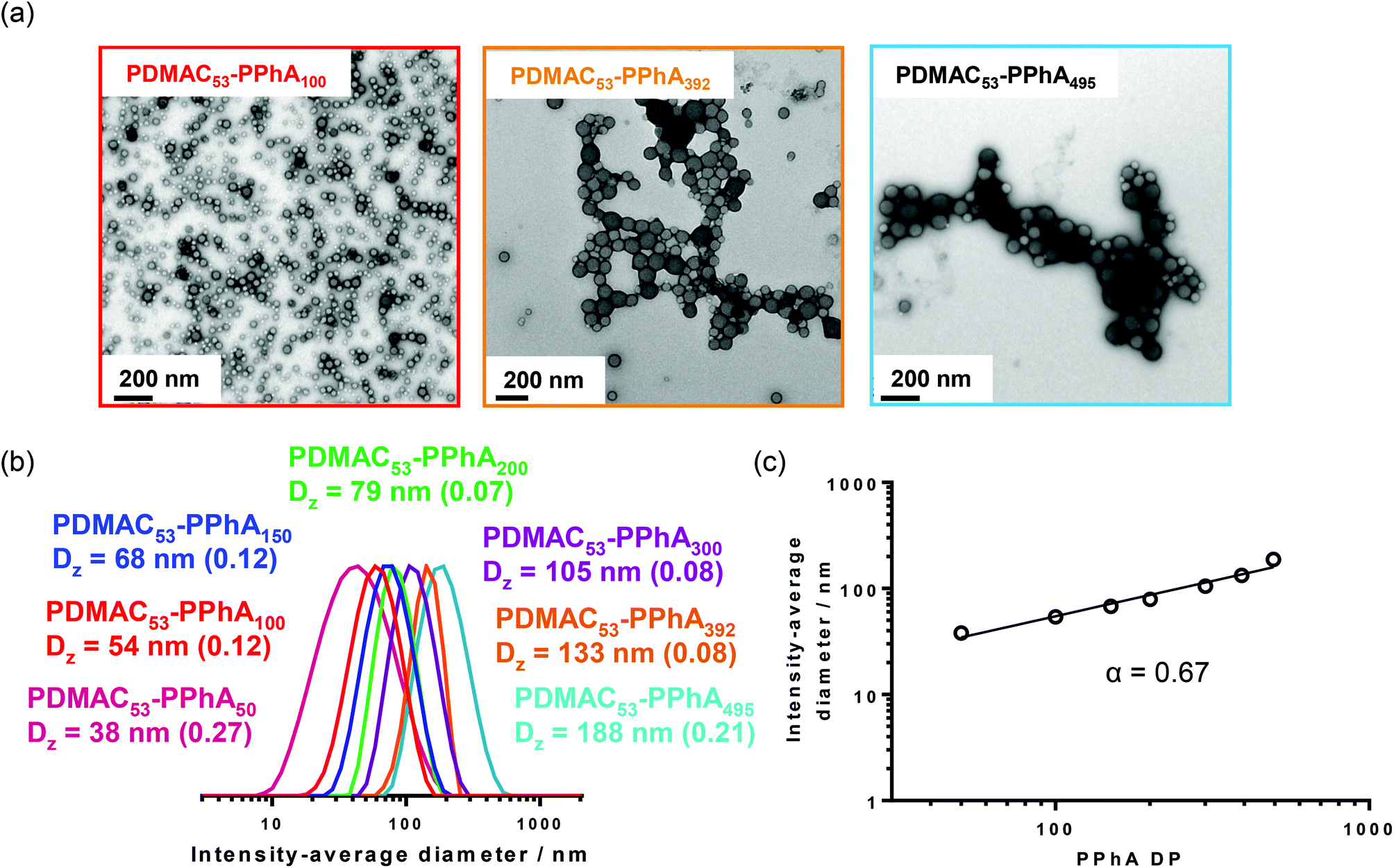
It is now well-established that RAFT alcoholic dispersion polymerization normally enables access to worms or vesicles when targeting longer solvophobic blocks, provided that the steric stabilizer block is sufficiently short to allow efficient sphere-sphere fusion to occur on the time scale of the PISA synthesis.16–18,20,23,25,28,79–81 Accordingly, for the RAFT dispersion polymerization of PhA in ethanol, a PAA macro-CTA with a mean DP of 26 was synthesized via RAFT solution polymerization in ethanol using DDMAT as the RAFT agent. Following exhaustive methylation with excess trimethylsilyldiazomethane, THF GPC analysis indicated an Mw/Mn of 1.07, suggesting good RAFT control. This PAA26 macro-CTA was then used for the polymerization of PhA in ethanol at 70 °C using AIBN initiator. For such PISA formulations, higher copolymer concentrations (e.g. 25% w/w solids, as used herein) favor the production of pure (rather than mixed) phases.Table 2 summarizes the target PAA26-PPhAx compositions, with x ranging from 30 to 300. Analysis by 1H NMR indicated that the final monomer conversion was high (>98%) in all cases. Following exhaustive methylation of the carboxylic acid residues in the PAA block, THF GPC analysis indicated marginally narrower molecular weight distributions compared to those obtained for the series of PDMAC53-PPhAx diblock copolymers prepared by RAFT aqueous emulsion polymerization (compare Tables 1 and 2). Although relatively high blocking efficiencies were obtained, the molecular weight distributions were again rather broad compared to those typically obtained for PISA syntheses based on methacrylic diblock copolymers.16–22 For this second series of all-acrylic copolymers, increasing the target DP of the core-forming block led to a gradual increase in dispersity (see Fig. 4). The weak high molecular weight shoulder is similar to that previously reported by Ratcliffe and co-workers66 for PLA14-PBzAx syntheses conducted via PISA in non-polar solvents; this feature is postulated to be either the result of chain transfer to polymer or possibly owing to termination by combination.
 | ||
| Fig. 4 THF GPC curves obtained for PAA26-PPhAx diblock copolymers prepared at 25% w/w solids via RAFT alcoholic dispersion polymerization of PhA at 70 °C. Molecular weight data are expressed relative to a series of near-monodisperse poly(methyl methacrylate) calibration standards. | ||
| Entry no. | Target diblock composition | Monomer conversionb/% | GPC datac | DLS diameterd/nm | TEM morphology | |
|---|---|---|---|---|---|---|
| M n | M w/Mn | |||||
|
a Conditions: [macro-CTA]:[initiator] molar ratio = 5.0; copolymer concentration = 25% w/w.
b Monomer conversion determined by 1H NMR spectroscopy in DMSO-d6.
c Determined by THF GPC (refractive index detector, calibrated using a series of poly(methyl methacrylate) standards).
d Intensity-average diameters were calculated using cumulants analysis software provided by the manufacturer (Malvern Instruments, UK). The numbers in brackets denote the polydispersity in the particle diameter.
|
||||||
| 1 | PAA26-PPhA30 | 98 | 7200 | 1.45 | 24 (0.22) | Spheres |
| 2 | PAA26-PPhA50 | >99 | 8100 | 1.68 | 231 (0.38) | Worms |
| 3 | PAA26-PPhA75 | 99 | 14500 |
1.50 | 2800 (0.88) | Lamellae |
| 4 | PAA26-PPhA100 | 98 | 12300 |
1.77 | 1300 (0.28) | Lamellae + vesicles |
| 5 | PAA26-PPhA150 | 98 | 12200 |
2.13 | 970 (0.09) | Vesicles |
| 6 | PAA26-PPhA300 | 98 | 41200 |
2.48 | 3400 (0.95) | Precipitate |
An evolution in copolymer morphology occurs when targeting higher PPhA DPs for this PISA system. Thus a relatively transparent dispersion is obtained at a PPhA DP of 30, with DLS and TEM studies indicating the presence of relatively small spheres with an intensity-average diameter of 24 nm. As the PPhA DP is increased to 50, a pure worm phase is obtained in the form of a transparent soft gel. At a PPhA DP of 74, a transparent brittle gel was produced with DLS studies indicating a large sphere-equivalent diameter (>2 μm) and relatively high polydispersity (0.88). TEM studies suggest that this is actually a lamellar phase, with large stacked ‘sheets’. Similar lamellae have been reported by Yang et al.,26,82 who also utilized a RAFT dispersion polymerization formulation comprising ethanol plus a co-solvent (either dioxane or methyl ethyl ketone). In these two earlier studies the formation of such a lamellar morphology was rationalized in terms of the relatively high Tg (219 °C) of the poly(styrene-alt-N-phenylmaleimide) core-forming block impeding vesicle formation. However, the present study suggests that other as-yet-unidentified factors may be responsible for the formation of such lamellar morphologies.
Increasing the PPhA DP up to 98 produced a mixed phase comprising lamellae and vesicles, suggesting that the lamellar sheets are sufficiently deformable to enable vesicle formation to occur via ‘wrap up’. The visual appearance of PAA26-PPhA147 (entry 6 in Table 2) was a turbid free-flowing dispersion, which is characteristic of vesicles in the PISA literature.66,81,83,84 TEM studies confirmed this morphology, with some evidence for the presence of oligolamellar vesicles (see Fig. 5). Finally, a precipitate phase comprising polydisperse particles of approximately 6 μm was observed when targeting PAA26-PPhA300 (see entry 6 in Table 2 and Fig. 5). Given the highly asymmetric nature of these latter diblock copolymer chains, it seems likely that ‘vesicle death’ has occurred in this case. As reported by Warren et al.,27 the overall vesicle diameter is conserved during PISA. Thus, as the membrane thickens, the volume of the vesicle lumen must shrink. This leads to sterically congested stabilizer chains within the inner leaflet, which ultimately leads to vesicle destabilization. Strong evidence to support this upper limit constraint on vesicle growth was recently confirmed by Derry et al.85via in situ synchrotron SAXS studies. Given that ‘vesicle death’ has been observed for PISA syntheses conducted in both water and mineral oil, this suggests a generic mechanism that is hence also likely to apply to the current RAFT alcoholic dispersion polymerization formulation.
 | ||
| Fig. 5 PAA26-PPhAx diblock copolymers prepared at 25% w/w solids by RAFT alcoholic dispersion polymerization of PhA at 70 °C: selected TEM images showing the evolution in copolymer morphology to form (a) spheres, (b) worms, (c) lamellae, (d) a mixed phase of lamellae and vesicles, (e) vesicles or (f) precipitate on increasing the PPhA target DP. | ||
RAFT dispersion polymerization of phenyl acrylate in n-heptane
A wide range of copolymer morphologies could also be accessed via RAFT dispersion polymerization of PhA in n-heptane at 80 °C. These syntheses were conducted using a PLA14 macro-CTA at 25% w/w solids to enable direct comparison with the other two PISA formulations. Again, a series of PLA14-PPhAx diblock copolymers were produced by systematically varying the PPhA target DP (see Table 3). According to 1H NMR studies, high conversions (>98%) were obtained in each case.| Entry no. | Target diblock composition | Monomer conversionb/% | GPC datac | DLS diameterd/nm | TEM morphology | |
|---|---|---|---|---|---|---|
| M n | M w/Mn | |||||
|
a Conditions: [macro-CTA]:[initiator] molar ratio = 5.0; copolymer concentration = 25% w/w.
b Monomer conversion determined by 1H NMR spectroscopy in CDCl3.
c Determined by THF GPC (refractive index detector, calibrated with poly(methyl methacrylate) standards).
d Intensity-average diameters were calculated using cumulants analysis software provided by the manufacturer (Malvern Instruments, UK). The numbers in brackets denote the polydispersity in the particle diameter.
|
||||||
| 1 | PLA14-PPhA55 | >99 | 11300 |
1.42 | 32 (0.10) | Spheres |
| 2 | PLA14-PPhA65 | >99 | 13000 |
1.40 | 75 (0.15) | Spheres + worms |
| 3 | PLA14-PPhA80 | >99 | 14600 |
1.37 | 381 (0.49) | Worms |
| 4 | PLA14-PPhA120 | 99 | 18300 |
1.40 | 904 (0.94) | Worms + vesicles |
| 5 | PLA14-PPhA130 | 98 | 20800 |
1.53 | 244 (0.32) | Vesicles |
As expected, the apparent GPC Mn for this series of diblock copolymers increased with increasing PPhA DP. Moreover, THF GPC analysis suggested high blocking efficiencies, with this formulation providing significantly better control over the molecular weight distribution; Mw/Mn values range from 1.37 to 1.53 in all cases. Nevertheless, the GPC curve for each copolymer contained a high molecular weight shoulder (see Fig. 6). DLS and TEM studies confirmed that the expected evolution in copolymer morphology occurred on varying the target DP for the structure-directing PPhA block. Thus, the PLA14-PPhA55 dispersion was a transparent free-flowing fluid. This is consistent with the formation of spheres, which were found to be approximately 32 nm and relatively uniform in size, as judged by DLS and TEM studies respectively (see Fig. 7a).
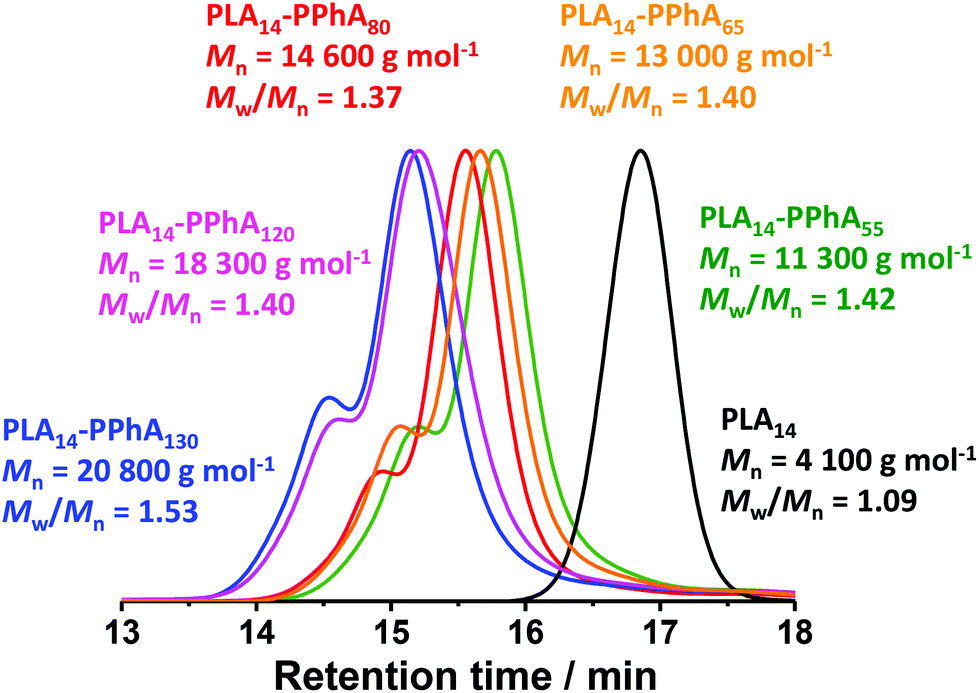 | ||
| Fig. 6 THF GPC curves obtained for a series of PLA14-PPhAx diblock copolymers prepared at 25% w/w solids via RAFT dispersion polymerization of PhA in n-heptane at 80 °C (see Table 3). Molecular weight data are expressed relative to a series of near-monodisperse poly(methyl methacrylate) calibration standards. | ||
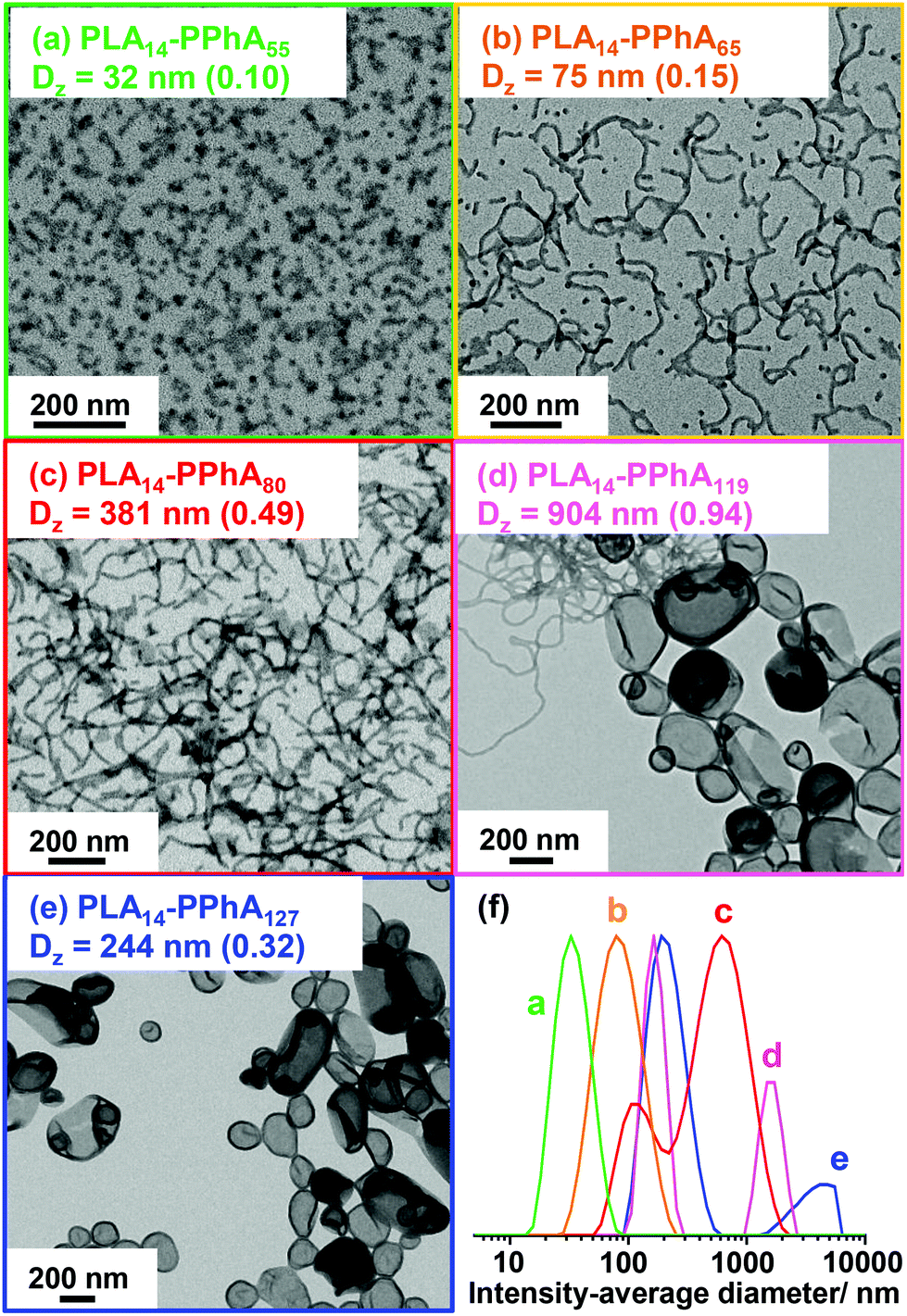 | ||
| Fig. 7 TEM images and corresponding DLS intensity-average diameters (polydispersity index shown in parentheses) obtained for a series of PLA14-PPhAx diblock copolymers prepared at 25% w/w solids via RAFT dispersion polymerization of phenyl acrylate in n-heptane at 80 °C. Increasing the target DP of the structure-directing PPhA block leads to a gradual evolution in copolymer morphology from (a) pure spheres, (b) spheres and worms, (c) worms, (d) worms and vesicles and (e) pure vesicles. (f) The corresponding DLS intensity-average size distributions obtained for these copolymer dispersions. | ||
An increase in PPhA DP of only ten units produced a turbid free-flowing liquid comprising a mixed phase of spheres and worms (see Fig. 7b). A free-standing transparent gel comprising a pure worm phase was obtained when targeting a PPhA DP of 80 (see Fig. 7c). Further increasing the core-forming block DP produced a mixed phase of worms and vesicles for PLA14-PPhA119 (see Fig. 7d), while TEM studies confirmed the existence of a pure vesicle phase for a PPhA DP of 127 (see Fig. 7e).
Comparing these three PhA-based PISA formulations (see Tables 1–3), significantly higher dispersities are obtained for RAFT aqueous emulsion polymerization at 30 °C compared to the two RAFT dispersion polymerizations conducted at 70 or 80 °C. Similarly broad molecular weight distributions have been reported by other workers for all-acrylic RAFT aqueous emulsion formulation.60,61 In contrast, significantly lower dispersities (Mw/Mn < 1.30) have been achieved for certain low-temperature RAFT dispersion polymerization formulations conducted in either water65 or ethanol/water mixtures.67 We postulate that the relatively high dispersities associated with RAFT aqueous emulsion polymerization may be related to stronger partitioning of unreacted monomer within the growing sterically-stabilized nanoparticles. This higher local monomer concentration is consistent with the significantly faster rate of polymerization observed for such PISA formulations relative to RAFT dispersion polymerizations. According to Lovell and co-workers, such a rate enhancement is likely to favor a higher degree of branching via chain transfer to polymer.58 To examine this hypothesis, the following control experiments were performed. The PDMAC53 macro-CTA was used to polymerize phenyl acrylate via RAFT solution polymerization in DMF at 30 °C (see ESI† for the precise synthetic protocol and associated characterization data). Three different DPs were targeted for the PPhA core-forming block: 50, 100 and 150. Thus these diblock compositions correspond to entries 1–3 in Table 1. In each case, high PhA conversions (>96%) were achieved after approximately 16 h at 30 °C. DMF GPC analyses indicated high blocking efficiencies and significantly narrower MWDs (Mw/Mn ≤ 1.26) for all three diblock copolymers. These additional experiments confirm that the PDMAC53 macro-CTA has a relatively high end-group fidelity and hence indicate that the problem of high dispersity is instead intimately related to the heterogeneous nature of these PISA formulations. This is consistent with our hypothesis that chain transfer to polymer becomes much more likely within monomer-swollen particles.
Conclusions
Phenyl acrylate is a useful model monomer for conducting fundamental studies focused on acrylic-based PISA formulations. Unlike the vast majority of acrylic polymers, the Tg of poly(phenyl acrylate) is sufficiently high (50 °C) to allow morphological studies to be conducted using conventional TEM, rather than cryo-TEM. Accordingly, phenyl acrylate has been employed to generate the structure-directing solvophobic block in RAFT aqueous emulsion polymerization, RAFT alcoholic dispersion polymerization and RAFT n-alkane dispersion polymerization. In the former case, only kinetically-trapped sterically-stabilized spheres could be obtained; this morphological limitation is often reported in the literature and remains poorly understood. In contrast, RAFT dispersion polymerization in either ethanol or n-heptane yielded the expected range of copolymer morphologies, depending on the relative volume fractions of the soluble and insoluble blocks. In each case high-quality images could be obtained by TEM. Final monomer conversions were invariably high (>98%) for these PISA syntheses, but GPC analyses indicated relatively broad molecular weight distributions, particularly for RAFT aqueous emulsion polymerization. This is believed to be the result of chain transfer to polymer, which is a well-known side reaction for the polymerization of acrylic monomers when employing either conventional free radical polymerization or pseudo-living radical polymerization techniques such as RAFT. Nevertheless, such branching is no barrier to the convenient formation of well-defined sterically-stabilized diblock copolymer nanoparticles at high solids. In summary, phenyl acrylate is a versatile monomer in the context of fundamental PISA research, which has been hitherto mainly focused on methacrylic monomers or styrene.Conflict of interest
The authors declare no competing financial interest.Acknowledgements
EPSRC is thanked for a Platform grant (EP/J007846) and the European Research Council is acknowledged for a five-year Advanced Investigator grant (PISA 320372).Notes and references
- B. M. Discher, Y. Won, D. S. Ege, J. C.-M. Lee, F. S. Bates, D. E. Discher and D. A. Hammer, Science, 1999, 284, 1143–1146 CrossRef CAS PubMed.
- D. E. Discher and A. Eisenberg, Science, 2002, 297, 967–973 CrossRef CAS PubMed.
- L. Zhang and A. Eisenberg, Science, 1995, 268, 1728–1731 CAS.
- L. Zhang and A. Eisenberg, J. Am. Chem. Soc., 1996, 118, 3168–3181 CrossRef CAS.
- Y. Mai and A. Eisenberg, Chem. Soc. Rev., 2012, 41, 5969 RSC.
- S. Jain and F. S. Bates, Macromolecules, 2004, 37, 1511–1523 CrossRef CAS.
- R. Xu, M. A. Winnik, F. R. Hallett, G. Riess and M. D. Croucher, Macromolecules, 1991, 24, 87–93 CrossRef CAS.
- Y. Won, Science, 1999, 283, 960–963 CrossRef CAS PubMed.
- C. Booth and D. Attwood, Macromol. Rapid Commun., 2000, 21, 501–527 CrossRef CAS.
- J. N. Israelachvili, D. J. Mitchell and B. W. Ninham, J. Chem. Soc., Faraday Trans. 2, 1976, 72, 1525–1568 RSC.
- A. Blanazs, S. P. Armes and A. J. Ryan, Macromol. Rapid Commun., 2009, 30, 267–277 CrossRef CAS PubMed.
- C. J. Hawker, A. W. Bosman and E. Harth, Chem. Rev., 2001, 101, 3661–3688 CrossRef CAS PubMed.
- M. Kato, M. Kamigaito, M. Sawamoto and T. Higashimura, Macromolecules, 1996, 28, 1721–1723 CrossRef.
- J. S. Wang and K. Matyjaszewski, J. Am. Chem. Soc., 1995, 117, 5614–5615 CrossRef CAS.
- J. Chiefari, Y. K. B. Chong, F. Ercole, J. Krstina, J. Jeffery, T. P. T. Le, R. T. A. Mayadunne, G. F. Meijs, C. L. Moad, G. Moad, E. Rizzardo and S. H. Thang, Macromolecules, 1998, 31, 5559–5562 CrossRef CAS.
- N. J. Warren and S. P. Armes, J. Am. Chem. Soc., 2014, 136, 10174–10185 CrossRef CAS PubMed.
- M. J. Derry, L. A. Fielding and S. P. Armes, Prog. Polym. Sci., 2016, 52, 1–18 CrossRef CAS.
- S. L. Canning, G. N. Smith and S. P. Armes, Macromolecules, 2016, 49, 1985–2001 CrossRef CAS PubMed.
- B. Charleux, G. Delaittre, J. Rieger and F. D'Agosto, Macromolecules, 2012, 45, 6753–6765 CrossRef CAS.
- J. Rieger, Macromol. Rapid Commun., 2015, 36, 1458–1471 CrossRef CAS PubMed.
- J.-T. Sun, C.-Y. Hong and C.-Y. Pan, Polym. Chem., 2013, 4, 873–881 RSC.
- J.-T. Sun, C.-Y. Hong and C.-Y. Pan, Soft Matter, 2012, 8, 7753–7767 RSC.
- N. J. Warren, O. O. Mykhaylyk, D. Mahmood, A. J. Ryan and S. P. Armes, J. Am. Chem. Soc., 2014, 136, 1023–1033 CrossRef CAS PubMed.
- S. Sugihara, A. Blanazs, S. P. Armes, A. J. Ryan and A. L. Lewis, J. Am. Chem. Soc., 2011, 133, 15707–15713 CrossRef CAS PubMed.
- A. Blanazs, A. J. Ryan and S. P. Armes, Macromolecules, 2012, 45, 5099–5107 CrossRef CAS.
- P. Yang, L. P. D. Ratcliffe and S. P. Armes, Macromolecules, 2013, 46, 8545–8556 CrossRef CAS.
- N. J. Warren, O. O. Mykhaylyk, A. J. Ryan, M. Williams, T. Doussineau, P. Dugourd, R. Antoine, G. Portale and S. P. Armes, J. Am. Chem. Soc., 2015, 137, 1929–1937 CrossRef CAS PubMed.
- L. P. D. Ratcliffe, A. J. Ryan and S. P. Armes, Macromolecules, 2013, 46, 769–777 CrossRef CAS.
- M. Williams, N. J. W. Penfold, J. R. Lovett, N. J. Warren, C. W. I. Douglas, N. Doroshenko, P. Verstraete, J. Smets and S. P. Armes, Polym. Chem., 2016, 7, 3864–3873 RSC.
- S. J. Byard, M. Williams, B. E. Mckenzie, A. Blanazs and S. P. Armes, Macromolecules, 2017, 50, 1482–1493 CrossRef CAS PubMed.
- W. Zhou, Q. Qu, Y. Xu and Z. An, ACS Macro Lett., 2015, 4, 495–499 CrossRef CAS.
- J. Tan, H. Sun, M. Yu, B. S. Sumerlin and L. Zhang, ACS Macro Lett., 2015, 4, 1249–1253 CrossRef CAS.
- M. Semsarilar, E. R. Jones, A. Blanazs and S. P. Armes, Adv. Mater., 2012, 24, 3378–3382 CrossRef CAS PubMed.
- E. R. Jones, M. Semsarilar, A. Blanazs and S. P. Armes, Macromolecules, 2012, 45, 5091–5098 CrossRef CAS.
- M. Semsarilar, V. Ladmiral, A. Blanazs and S. P. Armes, Polym. Chem., 2014, 5, 3466–3475 RSC.
- M. Semsarilar, N. J. W. Penfold, E. R. Jones and S. P. Armes, Polym. Chem., 2015, 6, 1751–1757 RSC.
- W. Zhao, G. Gody, S. Dong, P. B. Zetterlund and S. Perrier, Polym. Chem., 2014, 5, 6990–7003 RSC.
- X. Zhang, J. Rieger and B. Charleux, Polym. Chem., 2012, 3, 1502–1509 RSC.
- W. M. Wan and C. Y. Pan, Macromolecules, 2010, 43, 2672–2675 CrossRef CAS.
- W. M. Wan, X. L. Sun and C. Y. Pan, Macromol. Rapid Commun., 2010, 31, 399–404 CrossRef CAS PubMed.
- L. A. Fielding, M. J. Derry, V. Ladmiral, J. Rosselgong, A. M. Rodrigues, L. P. D. Ratcliffe, S. Sugihara and S. P. Armes, Chem. Sci., 2013, 4, 2081–2087 RSC.
- M. J. Derry, L. A. Fielding and S. P. Armes, Polym. Chem., 2015, 6, 3054–3062 RSC.
- A. P. Lopez-Oliva, N. J. Warren, A. Rajkumar, O. O. Mykhaylyk, M. J. Derry, K. E. B. Doncom, M. J. Rymaruk and S. P. Armes, Macromolecules, 2015, 48, 3547–3555 CrossRef CAS.
- L. A. Fielding, J. A. Lane, M. J. Derry, O. O. Mykhaylyk and S. P. Armes, J. Am. Chem. Soc., 2014, 136, 5790–5798 CrossRef CAS PubMed.
- L. Houillot, C. Bui, M. Save, B. Charleux, C. Farcet, C. Moire, J.-A. Raust and I. Rodriguez, Macromolecules, 2007, 40, 6500–6509 CrossRef CAS.
- V. J. Cunningham, A. M. Alswieleh, K. L. Thompson, M. Williams, G. J. Leggett, S. P. Armes and O. M. Musa, Macromolecules, 2014, 47, 5613–5623 CrossRef CAS.
- A. A. Cockram, T. J. Neal, M. J. Derry, O. O. Mykhaylyk, N. S. J. Williams, M. W. Murray, S. N. Emmett and S. P. Armes, Macromolecules, 2017, 50, 796–802 CrossRef CAS PubMed.
- J. Rieger, W. Zhang, F. Stoffelbach and B. Charleux, Macromolecules, 2010, 43, 6302–6310 CrossRef CAS.
- W. Zhang, F. D'Agosto, O. Boyron, J. Rieger and B. Charleux, Macromolecules, 2011, 44, 7584–7593 CrossRef CAS.
- S. Boissé, J. Rieger, G. Pembouong, P. Beaunier and B. Charleux, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 3346–3354 CrossRef.
- W. Zhang, F. D'Agosto, O. Boyron, J. Rieger and B. Charleux, Macromolecules, 2012, 45, 4075–4084 CrossRef CAS.
- W. Zhang, F. D'Agosto, P. Y. Dugas, J. Rieger and B. Charleux, Polym., 2013, 54, 2011–2019 CrossRef CAS.
- P. A. Steward, J. Hearn and M. C. Wilkinson, Adv. Colloid Interface Sci., 2000, 86, 195–267 CrossRef CAS PubMed.
- Z. Czech, Polym. Int., 2003, 52, 347–357 CrossRef CAS.
- N. M. Ahmad, D. J. Britton, F. Heatley and P. A. Lovell, Macromol. Symp., 1999, 143, 231–241 CrossRef CAS.
- F. Heatley, P. A. Lovell and T. Yamashita, Macromolecules, 1998, 31, 2828–2837 CrossRef.
- C. Plessis, G. Arzamendi, J. M. Alberdi, M. Agnely, J. R. Leiza and J. M. Asua, Macromolecules, 2001, 34, 6138–6143 CrossRef CAS.
- N. M. Ahmad, B. Charleux, C. Farcet, C. J. Ferguson, S. G. Gaynor, B. S. Hawkett, F. Heatley, B. Klumperman, D. Konkolewicz, P. A. Lovell, K. Matyjaszewski and R. Venkatesh, Macromol. Rapid Commun., 2009, 30, 2002–2021 CrossRef CAS PubMed.
- Y. Reyes and J. M. Asua, Macromol. Rapid Commun., 2011, 32, 63–67 CrossRef CAS PubMed.
- M. Chenal, L. Bouteiller and J. Rieger, Polym. Chem., 2013, 4, 752–762 RSC.
- C. J. Ferguson, R. J. Hughes, D. Nguyen, B. T. T. Pham, R. G. Gilbert, A. K. Serelis, C. H. Such and B. S. Hawkett, Macromolecules, 2005, 38, 2191–2204 CrossRef CAS.
- X. Zhang, S. Boissé, C. Bui, P.-A. Albouy, A. Brûlet, M.-H. Li, J. Rieger and B. Charleux, Soft Matter, 2012, 8, 1130–1141 RSC.
- L. Houillot, C. Bui, C. Farcet, C. Moire, J. A. Raust, H. Pasch, M. Save and B. Charleux, ACS Appl. Mater. Interfaces, 2010, 2, 434–442 CAS.
- J. Zhou, R. He and J. Ma, Polymers, 2016, 8, 207 CrossRef.
- G. Liu, Q. Qiu, W. Shen and Z. An, Macromolecules, 2011, 44, 5237–5245 CrossRef CAS.
- L. P. D. Ratcliffe, B. E. McKenzie, G. M. D. Le Bouëdec, C. N. Williams, S. L. Brown and S. P. Armes, Macromolecules, 2015, 48, 8594–8607 CrossRef CAS.
- J. Tan, C. Huang, D. Liu, X. Zhang, Y. Bai and L. Zhang, ACS Macro Lett., 2016, 5, 894–899 CrossRef CAS.
- R. Diaz-Calleja, E. Riande and J. San Román, Macromolecules, 1991, 24, 1854–1858 CrossRef CAS.
- Polymer Handbook, ed. J. Brandrup and E. H. Immergut, Wiley & Sons, 1975 Search PubMed.
- A. Blanazs, J. Madsen, G. Battaglia, A. J. Ryan and S. P. Armes, J. Am. Chem. Soc., 2011, 133, 16581–16587 CrossRef CAS PubMed.
- C. J. Ferguson, R. J. Hughes, B. T. T. Pham, B. S. Hawkett, R. G. Gilbert, A. K. Serelis and C. H. Such, Macromolecules, 2002, 35, 9243–9245 CrossRef CAS.
- N. P. Truong, M. V. Dussert, M. R. Whittaker, J. F. Quinn and T. P. Davis, Polym. Chem., 2015, 6, 3865–3874 RSC.
- L. Carlsson, A. Fall, I. Chaduc, L. Wagberg, B. Charleux, E. Malmstrom, F. D'Agosto, M. Lansalot and A. Carlmark, Polym. Chem., 2014, 5, 6076–6086 RSC.
- Y. Ning, L. A. Fielding, T. S. Andrews, D. Growney and S. P. Armes, Nanoscale, 2015, 7, 6691–6702 RSC.
- L. Guo, Y. Jiang, T. Qiu, Y. Meng and X. Li, Polym., 2014, 55, 4601–4610 CrossRef CAS.
- J. Rieger, G. Osterwinter, C. Bui, F. Stoffelbach and B. Charleux, Macromolecules, 2009, 42, 5518–5525 CrossRef CAS.
- X. Zhang, S. Boissé, W. Zhang, P. Beaunier, F. D'Agosto, J. Rieger and B. Charleux, Macromolecules, 2011, 44, 4149–4158 CrossRef CAS.
- S. Boissé, J. Rieger, K. Belal, A. Di-Cicco, P. Beaunier, M.-H. Li and B. Charleux, Chem. Commun., 2010, 46, 1950–1952 RSC.
- M. Semsarilar, V. Ladmiral, A. Blanazs and S. P. Armes, Langmuir, 2013, 29, 7416–7424 CrossRef CAS PubMed.
- M. Semsarilar, V. Ladmiral, A. Blanazs and S. P. Armes, Langmuir, 2012, 28, 914–922 CrossRef CAS PubMed.
- K. E. B. Doncom, N. J. Warren and S. P. Armes, Polym. Chem., 2015, 6, 7264–7273 RSC.
- P. Yang, O. O. Mykhaylyk, E. R. Jones and S. P. Armes, Macromolecules, 2016, 49, 6731–6742 CrossRef CAS PubMed.
- W. Cai, W. Wan, C. Hong, C. Huang and C. Pan, Soft Matter, 2010, 6, 5554–5561 RSC.
- J. R. Lovett, N. J. Warren, L. P. D. Ratcliffe, M. K. Kocik and S. P. Armes, Angew. Chem., 2015, 127, 1295–1299 CrossRef.
- M. J. Derry, L. A. Fielding, N. J. Warren, C. J. Mable, A. J. Smith, O. O. Mykhaylyk and S. P. Armes, Chem. Sci., 2016, 7, 5078–5090 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Assigned 1H NMR spectra of the macro-CTA and diblock copolymer in each of the three solvent systems. See DOI: 10.1039/c7py01161j |
| This journal is © The Royal Society of Chemistry 2017 |