
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis of well-defined epoxy-functional spherical nanoparticles by RAFT aqueous emulsion polymerization†
Fiona L.
Hatton
 *,
Joseph R.
Lovett
and
Steven P.
Armes
*
*,
Joseph R.
Lovett
and
Steven P.
Armes
*
Dainton Building, Department of Chemistry, University of Sheffield, Brook Hill, Sheffield, South Yorkshire S3 7HF, UK. E-mail: f.l.hatton@sheffield.ac.uk; s.p.armes@sheffield.ac.uk
First published on 27th July 2017
Abstract
The environmentally-friendly synthesis of epoxy-functional spherical nanoparticles has been achieved using polymerization-induced self-assembly (PISA) in aqueous solution. Firstly, a non-ionic hydrophilic stabilizer block, poly(glycerol monomethacrylate) (PGMA), was prepared by reversible addition–fragmentation chain transfer (RAFT) solution polymerization in ethanol. This water-soluble precursor was subsequently chain-extended via RAFT aqueous emulsion polymerization of glycidyl methacrylate (GlyMA) at 50 °C and neutral pH to ensure maximum retention of the epoxy functionality. PISA leads to the formation of well-defined PGMA-PGlyMA spherical diblock copolymer nanoparticles at up to 35% w/w solids and 1H NMR spectroscopy studies indicated that virtually all of the epoxy groups survive such relatively mild conditions. DMF GPC studies confirmed that relatively low dispersities (Mw/Mn < 1.30) were obtained if the mean degree of polymerization of the core-forming PGlyMA block remained below 100. Well-defined triblock copolymer nanoparticles could also be prepared via seeded RAFT emulsion polymerization of n-butyl methacrylate, with DMF GPC analysis indicating a relatively narrow molecular weight distribution (Mw/Mn < 1.20). The epoxy groups within the nanoparticle cores were ring-opened by adding sodium azide to a 10% w/w aqueous copolymer dispersion at 50 °C, as confirmed by FT-IR spectroscopy. PGMA45-PGlyMA100 diblock copolymer nanoparticles could be conveniently converted into cationic nanogels by utilizing water-soluble diamines as crosslinkers. These nanogels were characterized by DLS and aqueous electrophoresis and remained intact when dispersed in DMF; in contrast, the corresponding linear precursor nanoparticles dissociated to form molecularly-dissolved copolymer chains under the same conditions.
Introduction
Homopolymerization or copolymerization of glycidyl methacrylate (GlyMA) has been widely reported using atom transfer radical polymerization,1 nitroxide-mediated polymerization2 or reversible addition–fragmentation chain transfer (RAFT) polymerization3–5 to form well-defined statistical or diblock copolymers with high degrees of epoxy functionality. This approach enables a wide range of post-polymerization derivatizations with nucleophiles such as amines, hydroxyl groups, thiols, carboxylic acids or sodium azide. For example, the reaction of PGlyMA homopolymer with various secondary amines, sodium azide, or a thiol has been studied in DMSO, with high degrees of functionality being achieved within 2 h in most cases.5 Direct thiolation, bromination, iodination and phosphorylation of PGlyMA-based triblock copolymers has also been reported, although reaction times of up to 24 h were often required.6 Potential applications suggested for such derivatized GlyMA-based copolymers include gene and drug delivery,7 biocatalysis,8 cell imaging,9 inkjet printing,10 chromatographic media,11 denitrogenation of petroleum feedstock,12 heavy metal absorbents13,14 and pressure-sensitive adhesives.15GlyMA has also been utilized as a comonomer to prepare various types of epoxy-functional polymer colloids such as latexes,16,17 microspheres,18–20 anisotropic particles21 and microgels.22,23 For example, surfactant-free emulsion polymerization was used to prepare both poly(styrene-co-GlyMA) latexes containing surface epoxy groups and also core–shell latexes comprising polystyrene cores and poly(GlyMA-co-methyl methacrylate) shells.16 Surfactant-free emulsion polymerization was also utilized to prepare crosslinked poly(styrene-co-GlyMA) particles, whose distinctive framboidal morphology was attributed to phase separation.24 Mesoporous silica nanoparticles have been modified via surface-initiated polymerization of GlyMA: post-polymerization crosslinking with cystamine afforded hybrid nanocarriers that could release encapsulated cargo through external stimuli such as a pH switch or the addition of glutathione.23 The self-assembly of GlyMA-based copolymers has also been reported, including the formation of micelles and reverse micelles, polymersomes (vesicles), organic–inorganic hybrid materials and polyion complexes.7 Surface-initiated polymerization has been used to prepare PGlyMA brushes with subsequent derivatization with octylamine.25 In related work, Klok and coworkers found that the presence of the tertiary amine methacrylate residues in P(GlyMA-co-2-(diethylamino)ethyl methacrylate) brushes resulted in enhanced rates of ring-opening when reacted with various amines.26
Organic nanoparticles prepared via block copolymer self-assembly have received growing attention.27 In particular, polymerization-induced self-assembly (PISA) has become recognized as a highly versatile technique that enables the efficient formation of a wide range of functional diblock copolymer nano-objects in various media utilizing many types of vinyl monomers.28–32 PISA involves chain extension of a soluble homopolymer with a second monomer that, once polymerized, becomes insoluble in the reaction media, thereby driving in situ self-assembly to form diblock copolymer nano-objects. First reported in 2002 by Hawkett and coworkers,33 PISA was initially developed in the context of reversible addition–fragmentation chain transfer (RAFT) aqueous emulsion polymerization to prepare poly(acrylic acid)-poly(n-butyl acrylate) latexes34 and poly(acrylic acid)-polystyrene latexes.35 Subsequently, PISA has been extended to include various dispersion polymerization formulations conducted in either aqueous,36 alcoholic or non-polar media,32 while RAFT aqueous emulsion polymerization has continued to be explored by various groups.28,30,31,37–40 PISA formulations based on dispersion polymerization usually allow facile access to many nanoparticle morphologies, including spheres, worms and vesicles.36,41 In contrast, RAFT aqueous emulsion polymerizations often result in the formation of kinetically-trapped spheres, particularly for methacrylic core-forming blocks based on methyl methacrylate,42–45n-butyl methacrylate,46,47 benzyl methacrylate48 or 2,2,2-trifluoroethyl methacrylate.49,50 Nevertheless, the latter PISA formulations offer a potentially important route for the preparation of functional diblock copolymer nano-objects at up to 50% w/w solids, which represents a substantial improvement over traditional post-polymerization self-assembly techniques.30
We have previously reported using poly(glycerol monomethacrylate) (PGMA) as a steric stabilizer block for the preparation of diblock copolymer nano-objects via RAFT aqueous dispersion polymerization,30 RAFT alcoholic dispersion polymerization,36 and RAFT aqueous emulsion polymerization.48,49 In principle, PGMA can be obtained via reaction of PGlyMA with one equivalent of water, but this derivatization suffers from a cross-linking side reaction and in practice GMA is commercially available as a specialty monomer. However, the conversion of water-immiscible GlyMA into water-soluble GMA can be readily achieved simply by heating an initial aqueous GlyMA emulsion at 80 °C for 8–9 h.51 Remarkably, if this reaction is conducted at around neutral pH in the presence of air then no background polymerization occurs and no hydrolysis of the ester bond is detected. We have also reported using GlyMA as a comonomer in PISA syntheses to facilitate core cross-linking of block copolymer vesicles in order to improve their surfactant tolerance.52 A similar approach also works for cationic block copolymer worms, which were recently demonstrated to be ‘superflocculants’ for micrometer-sized silica particles.53 In both cases, the PISA synthesis involved statistical copolymerization of relatively low levels of GlyMA with 2-hydroxypropyl methacrylate. Subsequent crosslinking was achieved by adding either a diamine52 or 3-aminopropyltriethoxysilane (APTES).53,54 Of particular relevance to the present work, Tan et al. recently reported the photoinitiated RAFT dispersion polymerization of GlyMA in ethanol–water mixtures to form various copolymer morphologies.55 Other research teams have also incorporated relatively low amounts of GlyMA during PISA to introduce desired functionality. For example, Esser et al. conjugated a gadolinium chelate to an epoxy-functional stabilizer block for MRI imaging.56
Herein we report the highly convenient and environmentally-friendly PISA synthesis of epoxy-functional spherical diblock copolymer nanoparticles via RAFT aqueous emulsion polymerization of GlyMA. Systematic variation of the radical initiator type, reaction temperature and solution pH enabled optimum reaction conditions to be identified that preserved virtually all of the original epoxy groups on the time scale of the PISA synthesis. The block copolymer chains were characterized by 1H NMR spectroscopy and GPC, while TEM and DLS were used to characterize the particle size and morphology of the resulting nanoparticles. The epoxy groups were subsequently reacted with either sodium azide or model diamines. The latter reagents produced highly crosslinked primary amine-based nanogels with appreciable cationic character, as confirmed by aqueous electrophoresis studies.
Experimental
Materials
Glycerol monomethacrylate (GMA; >99%) was donated by GEO Specialty Chemicals (Hythe, UK) and used without further purification. Glycidyl methacrylate (GlyMA; 97%), n-butyl methacrylate (BuMA; 99%), 4,4′-azobis(4-cyanopentanoic acid) (ACVA; 99%), sodium azide (≥99.5%), ethylenediamine (EDA; 99%), bis(3-aminopropyl)terminated poly(ethylene oxide) (PEG31DA; Mn ≈ 1500 g mol−1) and tert-butyl hydroperoxide (TBH; 70% H2O solution) were purchased from Sigma-Aldrich UK. 2,2′-Azobis[2-(2-imidazolin-2-yl)propane]dihydrochloride (VA-044; ≥97%) was purchased from Wako Chemicals GmBH. Sodium sulphite (Na2SO3; 98%) was purchased from Alfa Aesar UK and 2-cyano-2-propyl dithiobenzoate (CPDB) was purchased from STREM Chemicals Ltd (Cambridge, UK). Ammonium chloride (≥99%), hydrochloric acid (35%) and sodium hydroxide (98%) were obtained from VWR Chemicals UK. All chemicals were used as received, unless otherwise stated. d6-DMSO was purchased from Goss Scientific Instruments Ltd (Cheshire, UK). All other solvents were purchased from Fisher Scientific (Loughborough, UK) and used as received. Deionized water was used for all aqueous experiments. Dialysis tubing used in this study was standard-grade regenerated cellulose with molecular weight cut-off values of 1 and 50 kDa, purchased from Spectrum Laboratories, Inc. (Rancho Dominguez CA, USA).Characterisation
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1). Chromatograms were analyzed using Agilent GPC/SEC software.
000 g mol−1). Chromatograms were analyzed using Agilent GPC/SEC software.
Synthesis of PGMA45 macro-CTA by RAFT solution polymerization in ethanol
A PGMA45 macro-CTA was prepared as previously described.57 Briefly, CPDB (1.21 g, 4.37 mmol), GMA (50.0 g, 0.312 mol) and ethanol (78.6 g, 60% w/w) were weighed into a round-bottomed flask. ACVA initiator (245 mg, 0.874 mmol, CPDB/ACVA = 5.0) was added and the reaction mixture was degassed with N2 in an ice bath for 40 min prior to placing the flask in an oil bath set at 70 °C. The polymerization was quenched after 160 min after the GMA conversion had reached 63%. The crude macro-CTA was diluted with methanol and precipitated into excess dichloromethane (twice) to remove unreacted GMA monomer and other impurities. The mean DP of the purified PGMA45 macro-CTA was confirmed to be 45 by 1H NMR end-group analysis by comparing the integrated aromatic protons at 7.4–7.9 ppm assigned to the dithiobenzoate chain-ends to the GMA pendant proton signals (–C![[H with combining low line]](https://www.rsc.org/images/entities/i_char_0048_0332.gif) 2–C(OH)–C2OH) at 3.4–4.3 ppm and methacrylate backbone signals (–C2–C(C3)–) at 0.7–2.5 ppm.
2–C(OH)–C2OH) at 3.4–4.3 ppm and methacrylate backbone signals (–C2–C(C3)–) at 0.7–2.5 ppm.
Synthesis of PGMA45-PGlyMAn nanoparticles by RAFT aqueous emulsion polymerization
The synthesis of PGMA45-PGlyMA100 is representative of the general protocol. PGMA45 macro-CTA (1.20 g, 0.162 mmol), and deionized water (31.58 g) were weighed into a 100 mL round-bottomed flask to target a final solids content of 10% w/w. VA-044 initiator (13.0 mg, 0.0404 mmol, macro-CTA/VA-044 = 4.0) was added and the solution pH was adjusted to pH 7.0–7.5 by addition of either 0.1 or 0.01 M NaOH. GlyMA (2.30 g, 16.2 mmol) was added and the reaction mixture was degassed with N2 in an ice bath for 30 min, before placing in an oil bath at 50 °C. The GlyMA polymerization was quenched by removal from the oil bath after 1 h, followed by exposure to air. The copolymer chains were analyzed by 1H NMR and GPC while the copolymer nanoparticles were characterized by TEM, DLS and aqueous electrophoresis.Synthesis of PGMA45-PGlyMA25-PBuMA75 triblock copolymer by seeded RAFT aqueous emulsion polymerization
The PGMA45-PGlyMA25-PBuMA75 triblock copolymer was prepared by in situ chain extension of PGMA45-PGlyMA25 nanoparticles using BuMA. PGMA45 macro-CTA (0.15 g, 20.2 μmol), and deionized water (2.01 g) were weighed into a vial to give a final target solids content of 10% w/w. VA-044 initiator (0.16 mg, 5.05 μmol, macro-CTA/VA-044 = 4.0) was added using a freshly prepared stock solution (163 μL, 0.03 M VA-044 stock solution) and the solution pH was adjusted to pH 7.0–7.5 using 0.01 M NaOH. GlyMA (0.072 g, 0.50 mmol) was added to the vial prior to degassing the reaction mixture with N2 in an ice bath for 30 min, after which the vial was placed in an oil bath at 50 °C. Aliquots were extracted for 1H NMR and GPC analyses after 1 h, then degassed BuMA (0.215 g, 1.50 mmol) was added via syringe and the aqueous dispersion was stirred at 50 °C for a further 3 h. The BuMA polymerization was quenched by removing the reaction vessel from the oil bath followed by exposure to air. The copolymer chains were analyzed by 1H NMR and GPC while the copolymer nanoparticles were characterized by TEM, DLS and aqueous electrophoresis. For the preparation of the corresponding statistical diblock copolymer, the BuMA (0.215 g, 1.50 mmol) was added at the same time as the GlyMA (0.072 g, 0.50 mmol) followed by degassing of the comonomer mixture using nitrogen.Derivatization of PGMA45-PGlyMA100 nanoparticles and dissolved chains using sodium azide
PGMA45-PGlyMA100 nanoparticles were prepared at 20% w/w solids and diluted to 10% w/w solids using deionized water. For the DMF scoping experiment, the PGMA45-PGlyMA100 nanoparticle dispersion was freeze-dried and then redissolved in DMF at a copolymer concentration of 3.5% w/w. For an aqueous PGMA45-PGlyMA100 nanoparticle dispersion (2.0 mL, 10% w/w, containing 1.15 mmol epoxy groups), NaN3 (150 mg, 2.30 mmol) and NH4Cl (123 mg, 2.30 mmol) were added and the ring-opening reaction was allowed to proceed for 24 h at either 20 °C or at 50 °C using an oil bath. Subsequently, the aqueous dispersions or DMF solution were purified by dialysis (molecular weight cut-off = 1 kDa) against deionized water and freeze-dried for FT-IR spectroscopy studies and elemental microanalysis. Such copolymers were also redissolved in d6-DMSO for 1H NMR analysis. A control experiment was also performed in the absence of NH4Cl in the case of the aqueous copolymer dispersion derivatization.Diamine crosslinking of PGMA45-PGlyMA100 nanoparticles
A 20% w/w aqueous dispersion of PGMA45-PGlyMA100 nanoparticles was diluted to 10% w/w solids using deionized water. Each diamine was weighed into a vial and a known volume of the copolymer dispersion (2.0 mL, 10% w/w, 1.15 mmol epoxy groups) was added, following which each sample vial was placed on a roller mixer for 24 h at 20 °C. Each copolymer dispersion was then dialyzed against deionized water using regenerated cellulose dialysis tubing (molecular weight cut-off = 50 kDa) to remove any free unreacted diamine. Following dialysis, 1.0–2.0 mL of the purified copolymer dispersions were freeze-dried for elemental microanalyses and FT-IR spectroscopy studies.Results and discussion
First, a PGMA45 macro-CTA was synthesized by RAFT solution polymerization of GMA in ethanol at 70 °C, as previously described.57 The resulting purified PGMA45 macro-CTA was subsequently chain-extended via RAFT aqueous emulsion polymerization of GlyMA. The synthesis parameters employed for this PISA formulation were systematically varied while targeting a constant diblock copolymer composition (PGMA45-PGlyMA100) in order to identify the optimum reaction conditions required to preserve the epoxy functionality, see Table 1. In principle, epoxy groups can be ring-opened via nucleophilic attack by either water or a hydroxyl group (which may be located on the PGMA stabilizer chains or formed in situ after initial reaction of GlyMA residues with water). In the latter case, intermolecular reaction can result in chain branching and hence broadening of the molecular weight distribution. In principle, conducting the RAFT aqueous emulsion polymerization of GlyMA at lower temperatures, for shorter reaction times, and at neutral pH should suppress ring-opening side-reactions. The two azo initiators investigated in this initial set of experiments, ACVA and VA-044, have 10 h half-lives of 69 and 44 °C respectively in water. In addition, a tert-butyl hydroperoxide-sodium sulfite (TBH-Na2SO3) redox couple was also examined to enable these PISA syntheses to be conducted at 30 °C. Indeed, GlyMA polymerizations conducted using ACVA at 70 °C resulted in relatively broad molecular weight distributions (Mw/Mn = 1.34–1.50) (Table 1, entries 1–4). Perhaps surprisingly, syntheses conducted at 40 °C using VA-044 (Table 1, entries 5 and 6) yielded similar results, while the redox-initiated polymerizations performed at 30 °C actually gave the highest dispersities (Table 1, entries 11 and 12).| Entry number | Initiator typea | Temperature (°C) | Solution pH | Reaction timed (h) |
M
ne (g mol−1) |
M
we (g mol−1) |
M
w/Mne |
|---|---|---|---|---|---|---|---|
| a PGMA45 CTA/initiator molar ratio = 4.0, unless otherwise stated. b PGMA45 CTA/TBH-Na2SO3 molar ratio = 10. c PGMA45 CTA/TBH-Na2SO3 molar ratio = 5. d Monomer conversion reached more than 98% for all entries, as determined by 1H NMR analysis in d6-DMSO. e Determined by gel permeation chromatography using DMF containing 10 mM LiBr as eluent and calibrated using a series of near-monodisperse poly(methyl methacrylate) standards. | |||||||
| 1 | ACVA | 70 | 5.2 | 1.0 | 28300 |
37900 |
1.34 |
| 2 | ACVA | 70 | 5.2 | 2.0 | 29800 |
41500 |
1.39 |
| 3 | ACVA | 70 | 7.1 | 1.0 | 27600 |
38000 |
1.38 |
| 4 | ACVA | 70 | 7.1 | 2.0 | 29900 |
44800 |
1.50 |
| 5 | VA-044 | 40 | 4.5 | 1.0 | 23400 |
36700 |
1.57 |
| 6 | VA-044 | 40 | 7.2 | 1.0 | 24000 |
37500 |
1.56 |
| 7 | VA-044 | 50 | 4.2 | 1.0 | 23900 |
29200 |
1.22 |
| 8 | VA-044 | 50 | 4.2 | 2.0 | 25100 |
32700 |
1.30 |
| 9 | VA-044 | 50 | 7.1 | 1.0 | 25100 |
31200 |
1.24 |
| 10 | VA-044 | 50 | 7.1 | 2.0 | 24600 |
32400 |
1.32 |
| 11 |
tBH/Na2SO3b |
30 | 3.1 | 2.5 | 24500 |
48800 |
1.99 |
| 12 |
tBH/Na2SO3c |
30 | 3.0 | 2.5 | 25200 |
46700 |
1.85 |
Notably, employing VA-044 at 50 °C produced relatively narrow molecular weight distributions at both pH 4 and pH 7, particularly for shorter reaction times (entries 7–10). In all cases, monomer conversion exceeded 98% within 1.0 h as determined by 1H NMR analysis, with longer reaction times merely leading to broader molecular weight distributions. Thus, intermolecular side-reactions can be suppressed simply by minimizing the reaction time without having any deleterious effect on the monomer conversion. Henceforth, all further PISA syntheses were conducted for just 1.0 h at 50 °C using VA-044 (PGMA45 CTA/VA-044 = 4.0) at a solution pH of approximately 7.0, see Scheme 1. Under such conditions, the potential loss of epoxy functionality during GlyMA polymerization was investigated by 1H NMR spectroscopy, employing 3-(trimethylsilyl) propionic acid (TMSP) as an internal standard (see Fig. S1†). Comparison of the integrated epoxy proton signals at 2.7 and 2.8 ppm with the TMSP signals located at 0.0 ppm indicated that minimal (<1%) ring-opening occurred under the above optimized conditions when targeting PGMA45-PGlyMA100.
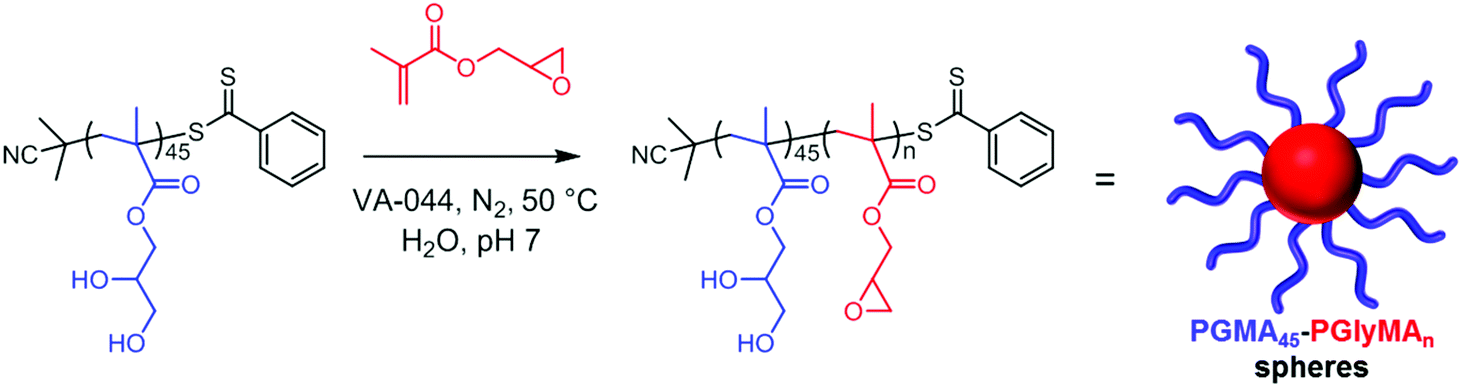 | ||
| Scheme 1 Schematic representation of the chain extension of a PGMA45 macro-CTA via RAFT aqueous emulsion polymerization of GlyMA. | ||
Following these initial studies, the kinetics of the RAFT aqueous emulsion polymerization of GlyMA was investigated when targeting PGMA45-PGlyMA100, see Fig. 1. The reaction solution was periodically sampled throughout the GlyMA polymerization and aliquots were diluted using d6-DMSO for 1H NMR analysis and DMF for GPC analysis; this ensures molecular dissolution of the copolymer chains in each case. The PGMA45 CTA/VA-044 molar ratio was varied from 2.0 to 8.0 at a fixed PGMA45 CTA concentration. As expected, faster polymerizations were observed when using a higher VA-044 concentration, with more than 99% conversion being attained within 30 min at 50 °C for a PGMA45 CTA/VA-044 molar ratio of 2.0. DMF GPC analysis revealed a linear increase in number-average molecular weight (Mn) with monomer conversion, while dispersities (Mw/Mn) remained below 1.35 in all cases. Perhaps surprisingly, using a PGMA45 CTA/VA-044 molar ratio of 4.0 resulted in the lowest final Mw/Mn of 1.26. Previous studies have shown that increasing the CTA/initiator molar ratio usually reduces the dispersity.48 Presumably, the slightly higher final dispersity (Mw/Mn = 1.32) obtained when employing a PGMA45 CTA/VA-044 molar ratio of 8.0 indicates that some degree of latent branching via epoxy ring-opening occurs over the longer time scale required for approximately full conversion (80 min vs. 40 min; see Fig. 1A).
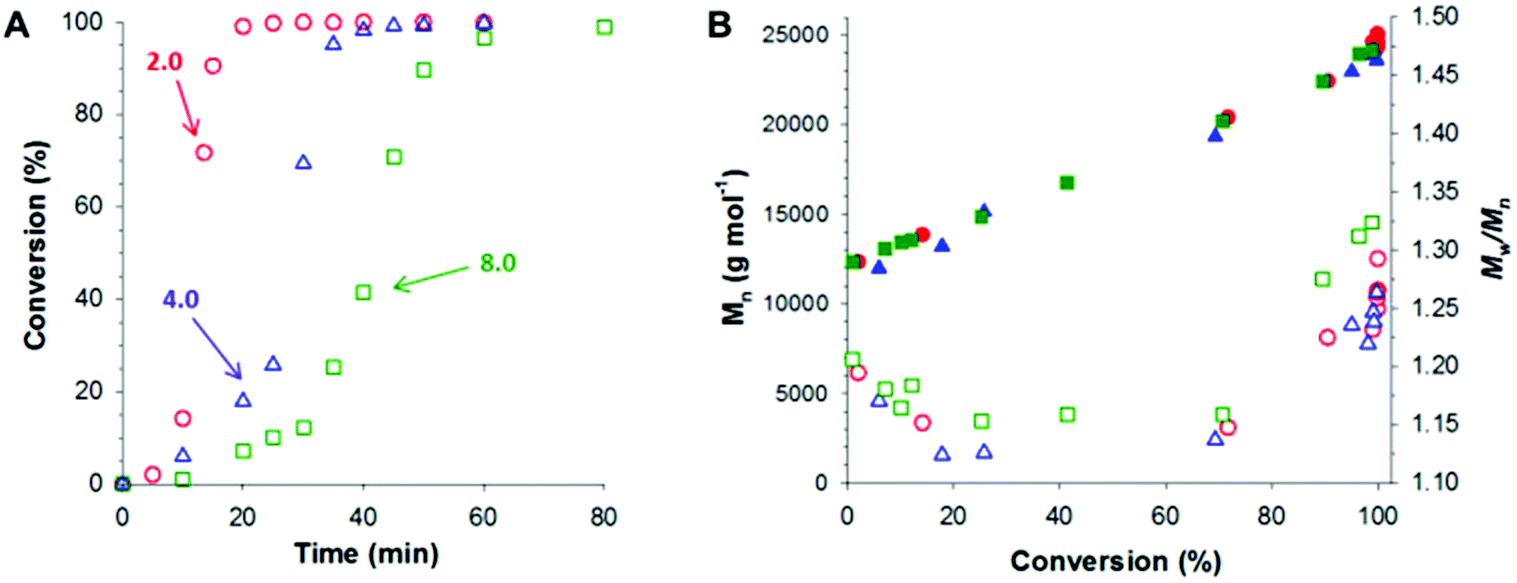 | ||
| Fig. 1 Kinetics of the RAFT aqueous emulsion polymerization of GlyMA at 50 °C. These PISA syntheses were conducted at 10% w/w solids and a solution pH of 7.0–7.5. The PGMA45 macro-CTA/VA-044 molar ratio was varied from 2.0 (red circles) to 4.0 (blue triangles) to 8.0 (green squares). (A) Conversion vs. time curves obtained from 1H NMR studies. (B) Evolution of Mn (closed symbols) and Mw/Mn (open symbols) with monomer conversion. The target copolymer composition was PGMA45-PGlyMA100 in each case. | ||
Varying the target degree of polymerization (DP) of the core-forming PGlyMA block from 35 to 300 resulted in the formation of a series of sterically-stabilized nanoparticles of increasing size, as judged by dynamic light scattering (DLS) and transmission electron microscopy (TEM) studies, see Fig. 2 and Table S1.† For relatively short core-forming block DPs (≤100), the former technique indicated narrow particle size distributions (PDI < 0.10), with mean particle diameters ranging from 20 to 40 nm. Targeting longer PGlyMA blocks (100 < DP < 300) produced less uniform nanoparticles with hydrodynamic diameters of up to 462 nm. TEM studies confirmed an exclusively spherical morphology in all cases. However, this technique revealed the presence of much smaller particles than those indicated by DLS, suggesting incipient aggregation. This is consistent with the much broader size distributions observed when targeting higher PGlyMA DPs (e.g. DP = 300). This is illustrated in the data set shown in Fig. 2A, where the error bars represent the width of each particle size distribution, rather than the experimental error. There is also a discernible change in gradient at a PGlyMA DP of approximately 100, which appears to correspond to the onset of flocculation. Further TEM images of other PGMA45-PGlyMAn nanoparticles support this hypothesis, see Fig. S2.† Furthermore, targeting PGlyMA DPs of more than 400 led to visible signs of macroscopic precipitation. Subsequent analysis of such failed PISA syntheses confirmed the presence of large aggregates comprising spherical nanoparticles, see Fig. S3.† We do not have a satisfactory explanation for this unexpectedly low upper limit DP: we have been able to target much longer core-forming blocks for other aqueous PISA syntheses using PGMA stabilizer blocks with similar DPs without significant nanoparticle aggregation.48,58,59
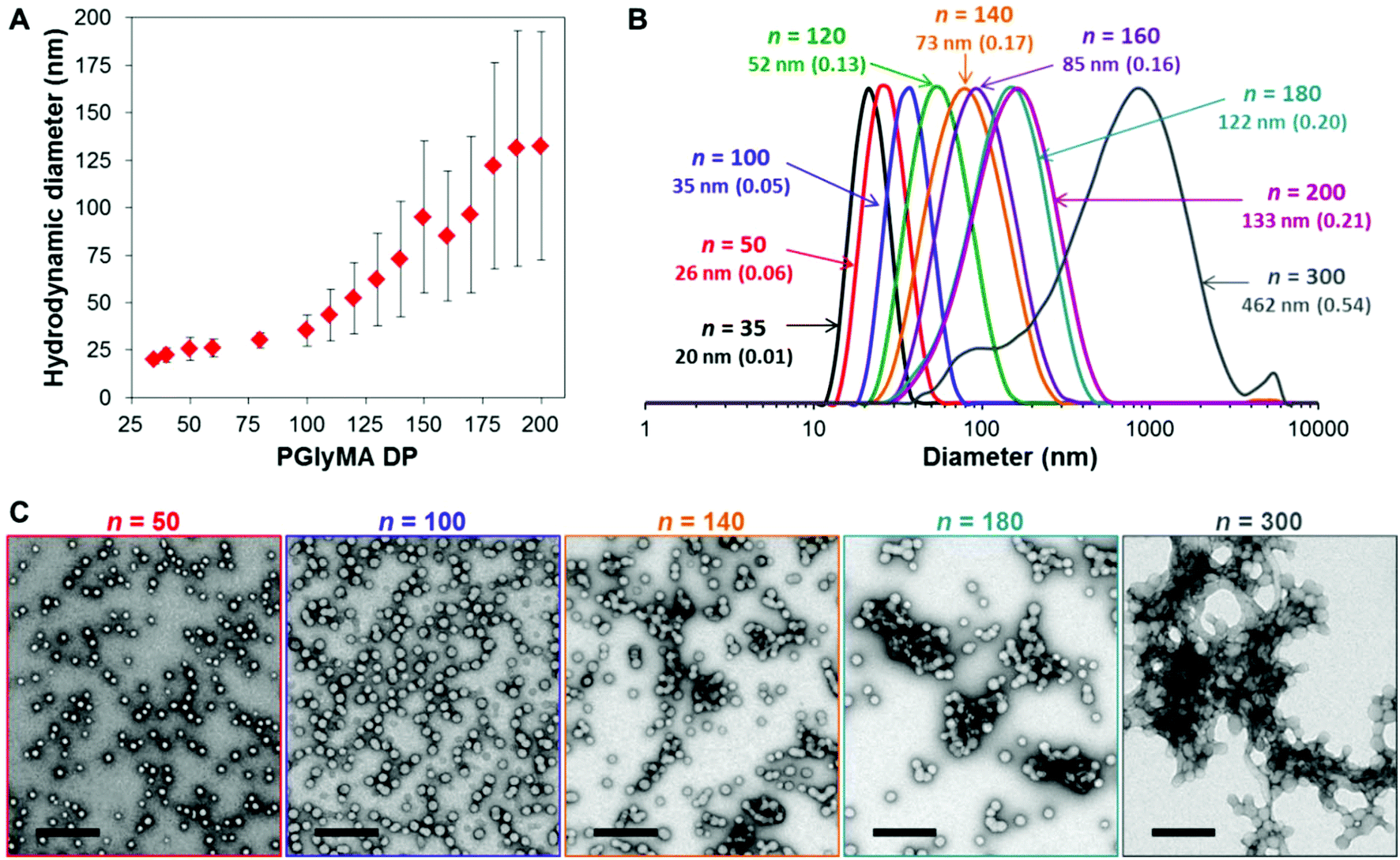 | ||
| Fig. 2 (A) Relationship between DLS diameter (Dh) and target PGlyMA DP for a series of PGMA45-PGlyMAn diblock copolymer nanoparticles prepared by RAFT aqueous emulsion polymerization of GlyMA at 10% w/w solids; error bars indicate the standard deviations for each particle size distribution, rather than the experimental error. (B) Selected DLS intensity-average size distributions, with Dh and PDI indicated, and (C) TEM images of representative samples, where n indicates the mean DP of the core-forming PGlyMA block. Scale bars represent 200 nm in all five images. | ||
DMF GPC analyses indicate that relatively low dispersities (Mw/Mn < 1.30) were obtained when targeting PGlyMA DPs of up to 100 (Fig. 3 and Table S1†). Given the well-known propensity of GlyMA residues to undergo intermolecular crosslinking reactions in the presence of water and/or other hydroxyl-functional polymers such as PGMA,52 we were pleasantly surprised by these observations. However, increasing the core-forming block DP above 100 led to a dramatic broadening in the molecular weight distribution (Mw/Mn = 1.44 to 1.85). Inspecting the relevant chromatograms reveals substantial tailing to higher molecular weight, which is most obvious for the PGMA45-PGlyMA400 diblock copolymer (Fig. 3 and also Table S1†). This feature is consistent with increasing levels of light branching.60,61 Nevertheless, blocking efficiencies were high in all cases, with little or no evidence for unreacted PGMA45 macro-CTA. The pseudo-living character of such RAFT aqueous emulsion polymerizations was confirmed by a ‘self-blocking’ experiment, see Fig. S4.†
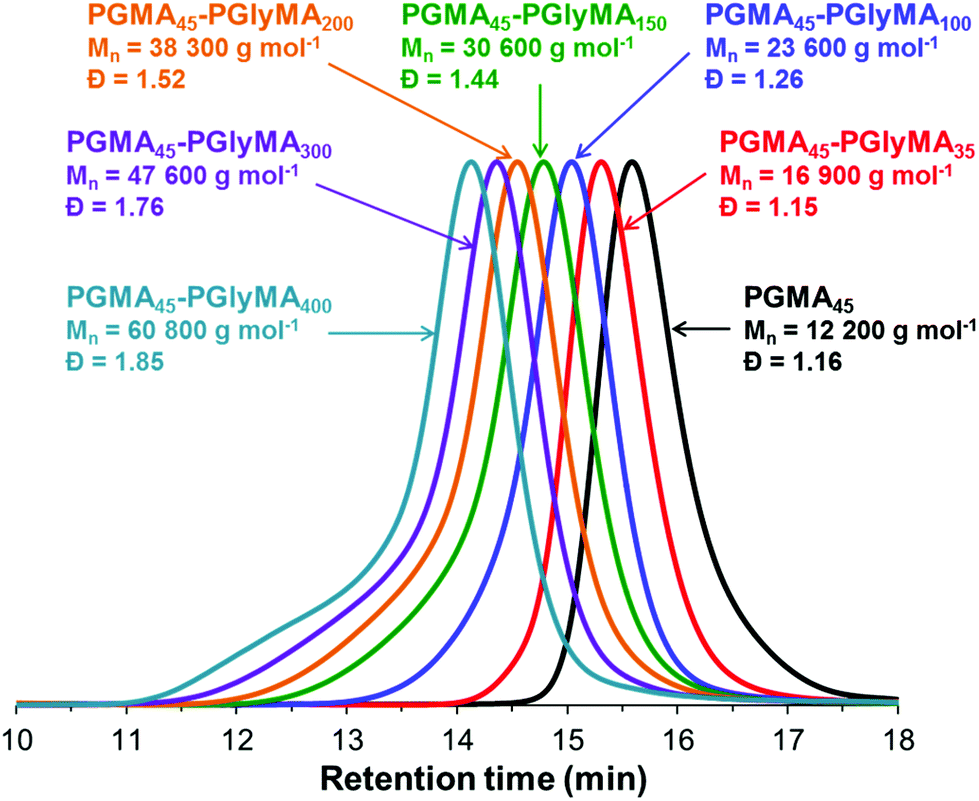 | ||
| Fig. 3 Overlaid GPC chromatograms obtained for PGMA45-PGlyMAn diblock copolymers prepared by aqueous RAFT emulsion polymerization of GlyMA at 50 °C and 10% w/w solids, where the mean DP for the core-forming PGlyMA block (n) = 35, 100, 150, 200, 300 or 400. | ||
Thus a just-nucleated PGMA45-PGlyMA25 precursor prepared by RAFT aqueous emulsion polymerization was used as a seed and chain-extended by a further 75 GlyMA units. This second-stage polymerization proceeded to more than 99%, yielding PGMA45-PGlyMA100 with a unimodal molecular weight distribution and a final Mw/Mn of 1.20 (Fig. S4†). For RAFT aqueous dispersion polymerizations, Blanazs et al. have shown that a relatively high copolymer concentration is an important parameter for the formation of higher order morphologies.57 However, with only a few exceptions,62–68 RAFT aqueous emulsion polymerization formulations usually result in kinetically-trapped spheres.40,42–50,69,70 Nevertheless, it was considered prudent to explore higher copolymer concentrations, if only to establish the upper limit for the production of well-defined epoxy-functional spheres. Thus PGMA45-PGlyMA100 syntheses were conducted at 50 °C at up to 40% w/w solids, see Table 2. In each case, uniform spherical nanoparticles were obtained with remarkably similar diameters and polydispersities reported by DLS (Table 2). Moreover, the molecular weight distributions obtained for these five diblock copolymers were almost identical, which suggests that relatively high copolymer concentrations are not detrimental to the preservation of the epoxy groups. However, a thin film was formed at the surface of the relatively viscous reaction mixture for the PISA synthesis conducted at 40% w/w solids. In principle, more efficient stirring might alleviate this problem, but this possibility was not explored in the present study. It is perhaps noteworthy that other aqueous and non-aqueous PISA formulations have also been reported to have effective upper limit copolymer concentrations of around 35–40% w/w solids.48,71
| Target composition | Solidsa (% w/w) | Conversionb (%) |
M
nc (g mol−1) |
M
wc (g mol−1) |
M
w/Mnc |
D h (nm) | PDI |
|---|---|---|---|---|---|---|---|
| a The amounts of monomer and macro-CTA were both increased in order to increase the solids content. b Determined by 1H NMR analysis in d6-DMSO after a reaction time of 1.0 h. c Determined by gel permeation chromatography analysis using DMF eluent containing 10 mM LiBr and calibrated with a series of near-monodisperse poly(methyl methacrylate) standards. d Reaction mixture became viscous and a thin film formed on its surface. | |||||||
| PGMA45-PGlyMA100 | 10 | >99 | 25100 |
31200 |
1.24 | 35 | 0.05 |
| PGMA45-PGlyMA100 | 20 | >99 | 25400 |
31400 |
1.24 | 35 | 0.03 |
| PGMA45-PGlyMA100 | 30 | >99 | 23900 |
30000 |
1.26 | 37 | 0.06 |
| PGMA45-PGlyMA100 | 35 | >99 | 25000 |
31000 |
1.24 | 37 | 0.05 |
| PGMA45-PGlyMA100 | 40d | >99 | 24900 |
29900 |
1.20 | 38 | 0.06 |
Preparation of ABC triblock copolymers via seeded RAFT aqueous emulsion polymerization
In principle, ABC triblock copolymer nanoparticles can also be prepared via seeded RAFT aqueous emulsion polymerization.34,45 This approach could either lead to the generation of higher order morphologies (i.e. worms or framboidal vesicles)72–74 or produce onion-type nanostructures if the final copolymer morphology remains confined to kinetically-trapped spheres.75 Accordingly, PGMA45-PGlyMA25 spheres were chain-extended with n-butyl methacrylate (BuMA). PGMA45-PGlyMA25 was selected for the initial diblock composition in this experiment because a PGlyMA DP of 25 is sufficient to induce nucleation but not so high as to cause incipient flocculation. 1H NMR confirmed essentially full GlyMA conversion after 1.0 h at 50 °C, and BuMA was then added via syringe, targeting a DP of 75 for the third block. After a further 3.0 h at 50 °C, the polymerization was quenched and analyzed by 1H NMR spectroscopy and high BuMA conversion (>99%) was achieved. In a control experiment targeting the same overall DP of 100, the corresponding statistical diblock copolymer nanoparticles were also prepared whereby the core-forming block comprised GlyMA and BuMA. DMF GPC data for the resulting PGMA45-PGlyMA25-PBuMA75 triblock copolymer and PGMA45-[PGlyMA25-co-BuMA75] statistical diblock copolymer are shown in Fig. 4A, with the GPC curve for the PGMA45 macro-CTA precursor included as a reference. The two copolymers have essentially identical unimodal molecular weight distributions and relatively low dispersities (Mw/Mn < 1.20), which suggests that high blocking efficiencies and good living character were achieved in these syntheses. DLS particle size distribution curves for the same two copolymers are shown in Fig. 4B, with the PGMA45-PGlyMA25 seed nanoparticles included for reference.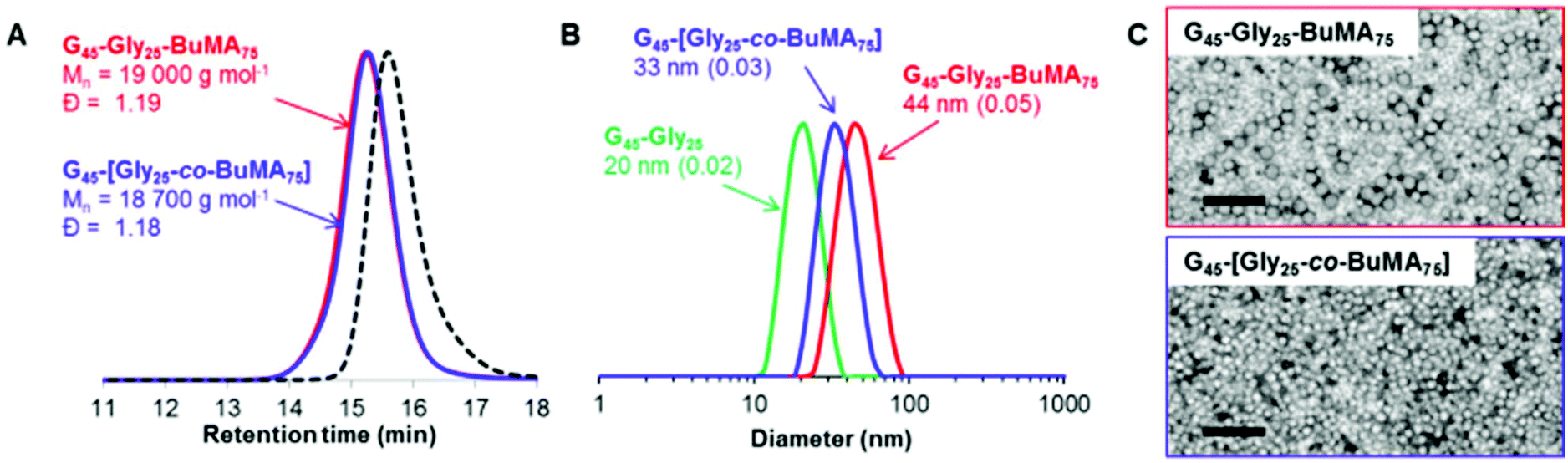 | ||
| Fig. 4 (A) Overlaid GPC chromatograms recorded for PGMA45-PGlyMA25-PBuMA75 triblock copolymer prepared by seeded RAFT aqueous emulsion polymerization at 10% w/w solids. In a control experiment, the corresponding PGMA45-[GlyMA25-co-PBuMA75] statistical diblock copolymer was also prepared by RAFT aqueous emulsion copolymerization. The original PGMA45 CTA precursor (black dotted lines; Mn = 12200 g mol−1, Mw/Mn = 1.16) is shown as a reference. (B) DLS intensity-average size distributions, with Dh and PDI indicated, and (C) TEM images obtained for the PGMA45-PGlyMA25-PBuMA75 and PGMA45-[GlyMA25-co-PBuMA75] nanoparticles. Scale bars represent 200 nm in both images. PGMA and PGlyMA are denoted by ‘G’ and ‘Gly’ for brevity. | ||
Colloidally stable nanoparticles with rather narrow size distributions are obtained in both cases, but their hydrodynamic diameters are 44 nm and 33 nm, respectively. This unexpected difference is also apparent in TEM images recorded for these two dispersions, see Fig. 4C. Clearly, these preliminary results are very encouraging, but higher DPs will most likely be required to drive microphase separation within such triblock copolymer spheres.
Long-term stability of epoxy groups for aqueous dispersions of PGMA45-PGlyMA100 nanoparticles
In order to assess whether epoxy ring-opening occurs during long-term storage at ambient temperature and pH 7, a 10% w/w aqueous dispersion of PGMA45-PGlyMA100 nanoparticles was periodically sampled for 1H NMR, GPC, and DLS analyses over twelve weeks. The DLS diameter and polydispersity remained constant over this time period (Fig. 5A), which indicates no change in the colloidal stability of the nanoparticles. However, DMF GPC analysis (Fig. 5B) provided strong evidence for intermolecular reactions occurring during long-term storage: gradual evolution of a high molecular weight shoulder led to a significant increase in both Mw and Mw/Mn. 1H NMR analysis corroborated the concomitant loss of epoxy functionality (Fig. 5C). Ring-opening causes a reduction in the intensity of the two non-equivalent CH2 protons in the epoxy ring at 2.6 and 2.8 ppm, and a concomitant increase in the signal corresponding to the PGMA oxymethylene protons observed at 3.4 ppm. This spectral shift enables the loss of epoxy functionality to be conveniently monitored by comparing these integrated signals to those of the methacrylate backbone protons, since the latter remain constant. After six weeks storage at 20 °C, the epoxy functionality was reduced by 15%, and after 12 weeks only 73% of the original epoxy groups remained intact. This hydrolytic instability may be related to the relatively high local concentration of epoxy groups within the nanoparticle cores. Since the partially-reacted epoxy groups only lead to light branching rather than highly crosslinked nanoparticles, this suggests that the majority of the ring-opening events actually involve intramolecular cyclization, rather than intermolecular reactions.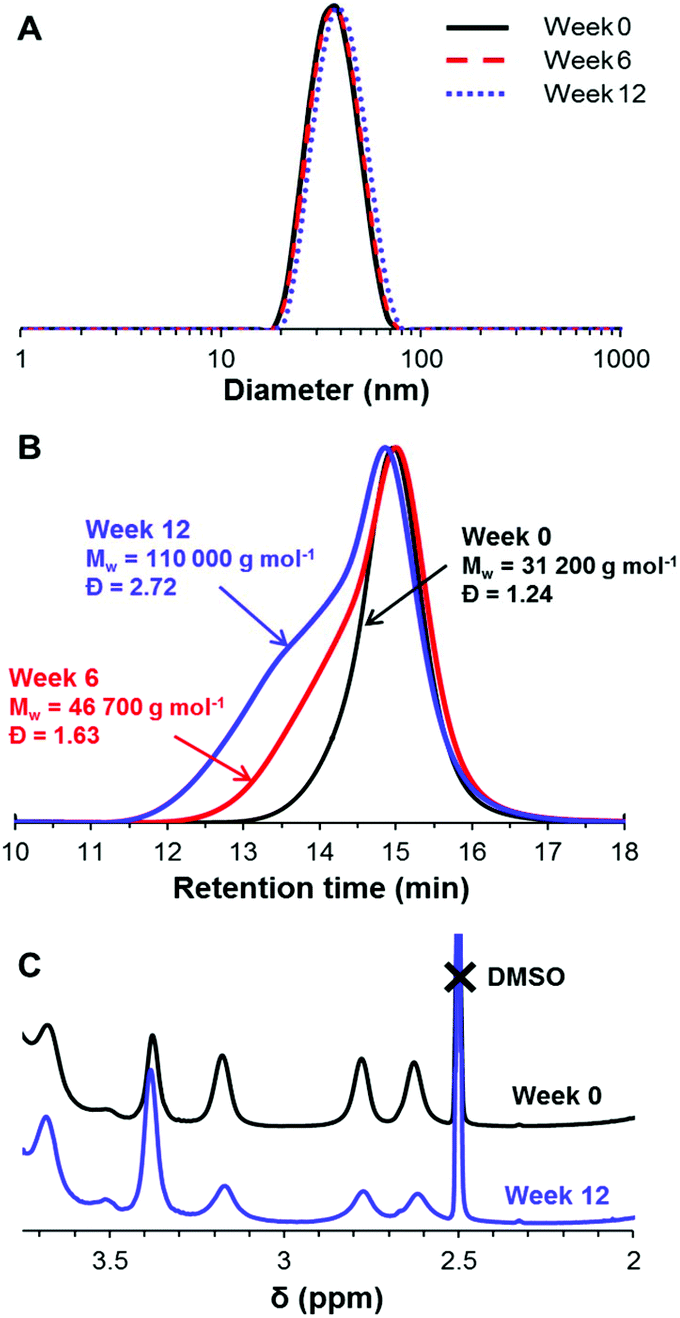 | ||
| Fig. 5 Evaluation of the colloidal and chemical stability of a 10% w/w aqueous dispersion of PGMA45-PGlyMA100 nanoparticles during long-term storage at 20 °C and pH 7. (A) Intensity-average DLS particle size distributions and (B) DMF GPC chromatograms recorded for diluted copolymer dispersions at various time points. The former indicates excellent colloidal stability, whereas the latter suggests light branching caused by intermolecular ring-opening reactions. (C) Partial 1H NMR spectra confirming the gradual loss of epoxy group functionality over time as a result of ring-opening. | ||
Most likely, a small fraction of epoxy groups first react with water to form cis-diol units, which can then react with neighboring epoxy groups either on the same chain or on adjacent chains. In view of these observations, all of the following derivatization reactions were conducted on fresh aqueous dispersions of PGMA45-PGlyMAn nanoparticles to ensure maximum epoxy functionality.
Preparation of azide-functional nanoparticles
It is well-known that epoxy groups can be ring-opened by reaction with sodium azide:76 the resulting azide-functional copolymers can be further reacted with various alkynes via Huisgen cycloaddition, a so-called ‘click’ reaction.77 Moreover, this chemistry is known to be highly orthogonal, hence azide derivatization significantly broadens the scope for designing bespoke functional nanoparticles for potential applications. In an initial scoping experiment, PGMA45-PGlyMA100 was molecularly dissolved in DMF to afford a 3.5% w/w solution and its epoxy groups were reacted with excess sodium azide at 20 °C in the presence of NH4Cl, which acts as a coordinating salt.56This model reaction was successful: the 1H NMR spectrum recorded in d6-DMSO for the product indicated complete loss of epoxy signals (CH2 at 2.6 and 2.8 ppm and CH at 4.3 ppm) while a characteristic azide stretch appeared at 2100 cm−1 in the corresponding FT-IR spectrum (Fig. 6). Subsequently, the same azide ring-opening reaction was performed on 10% w/w aqueous dispersions of PGMA45-PGlyMA100 nanoparticles at either 20 °C or 50 °C. At ambient temperature, epoxy signals (at 2.6, 2.8 and 4.3 ppm) were still discernible in the 1H NMR spectrum even after 24 h. However, no epoxy signals could be detected after the same time period when this reaction was run at 50 °C. Moreover, such derivatizations did not require the addition of NH4Cl. Visual inspection and 1H NMR end-group analysis also confirmed that cleavage of the dithiobenzoate RAFT end-groups occurred under these conditions, as indicated by the change in color (from pink to white) of the dispersion and complete loss of the aromatic proton signals at 7.9–7.5 ppm (data not shown).
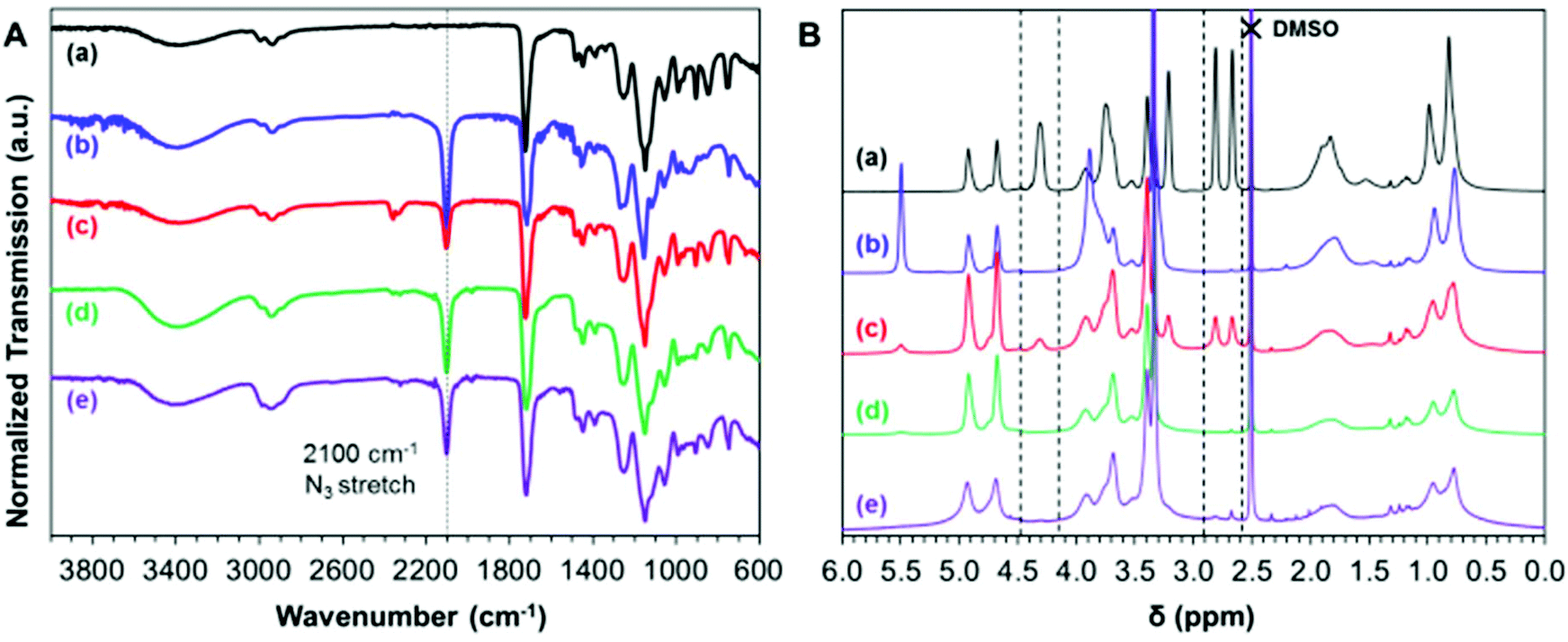 | ||
| Fig. 6 (A) ATR-FTIR spectra and (B) 1H NMR spectra (d6-DMSO) recorded for PGMA45-PGlyMA100 nanoparticles/chains after their reaction with excess sodium azide: (a) PGMA45-PGlyMA100 precursor; and (b)–(d) after reaction with 2 eq. NaN3 in the presence of 2 eq. NH4Cl, (b) in DMF at 20 °C; (c) for a 10% w/w aqueous dispersion at 20 °C; (d) for a 10% w/w aqueous dispersion at 50 °C; and (e) under the same conditions as (d), but in the absence of any NH4Cl. | ||
Core crosslinking of PGMA45-PGlyMA100 nanoparticles using various diamines
Given the high density of epoxy groups within the nanoparticle cores, diamine crosslinking should be readily achieved, see Scheme 2. Indeed, nucleophilic reaction of GlyMA residues with diamines has been utilized to covalently stabilize diblock copolymer vesicles prepared via PISA.52 Bearing in mind the long-term hydrolytic instability of the GlyMA residues discussed above, all derivatizations were conducted within 1–2 weeks of the PISA synthesis of the precursor PGMA45-PGlyMA100 nanoparticles to ensure minimal loss of epoxy functionality. Two water-soluble diamines were investigated in this study: ethylenediamine (EDA) and bis(3-aminopropyl)-terminated poly(ethylene oxide) (PEG31DA). In the following experiments, the amount of diamine crosslinker added to the PGMA45-PGlyMA100 nanoparticles is expressed in terms of an amine/epoxy molar ratio. Thus an amine/epoxy molar ratio of 2.0 corresponds to a diamine/epoxy molar ratio of 1.0. The desired diamine was added directly to a 10% w/w aqueous dispersion of the nanoparticles. Initially, DLS studies were conducted in DMF to examine the extent of crosslinking.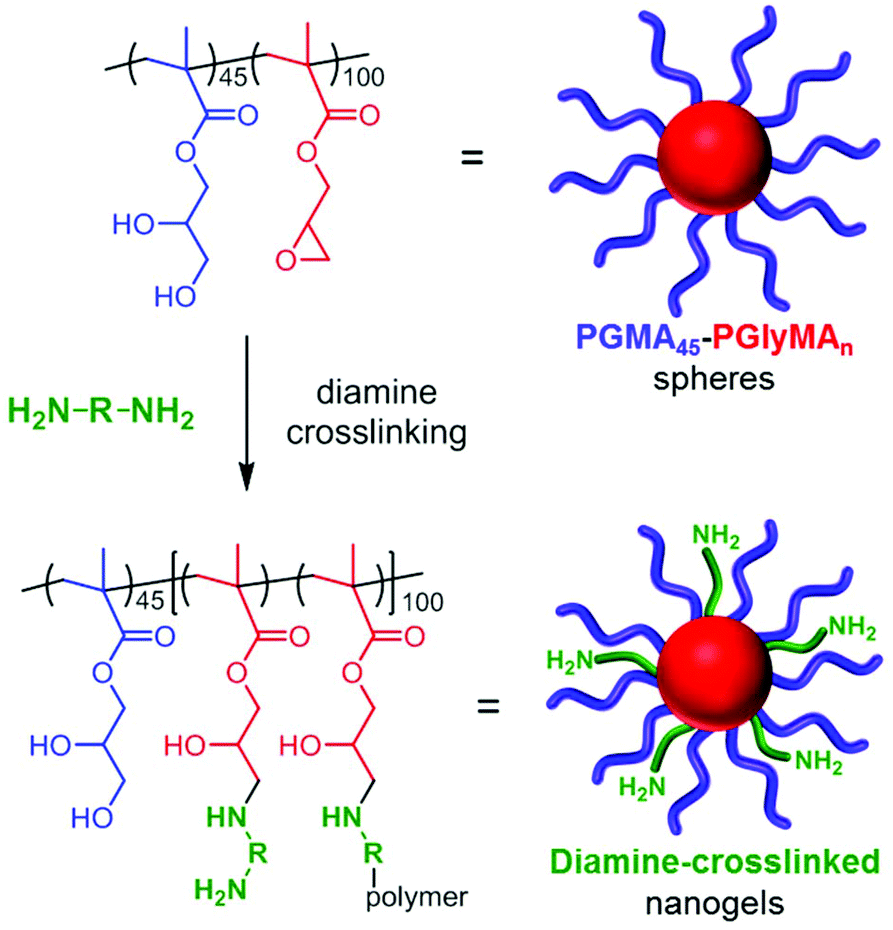 | ||
| Scheme 2 Schematic representation of diamine crosslinking for 10% w/w aqueous dispersions of the epoxy-functional PGMA45-PGlyMA100 nanoparticles. | ||
DMF is a good solvent for both PGMA and PGlyMA, so the linear precursor nanoparticles are molecularly dissolved in this solvent, as confirmed by GPC analysis (Fig. 3) and also the very weak light scattering observed by DLS. In contrast, successful diamine crosslinking produces appreciably swollen nanogels that cannot be dissolved in DMF (Fig. 7). The same nanogels are significantly less swollen in aqueous media, as confirmed by DLS studies conducted at pH 3–10, and the nanogel diameters did not vary significantly when adjusting the solution pH. EDA crosslinking at 10% w/w solids gave nanogels of around 37 nm diameter in water and 42–44 nm diameter in DMF, respectively. Similarly, PEG31DA crosslinked nanogels exhibited DLS diameters ranging from 50 to 59 nm when dispersed in DMF but only approximately 38–42 nm when analysed in dilute aqueous solution. Following crosslinking at 10% w/w solids, nanogels were purified by dialysis against deionized water to remove any unreacted diamine, followed by dilution with d6-DMSO for 1H NMR analysis of the residual epoxy protons at 2.6 and 2.8 ppm. No epoxy signals could be detected in any of the EDA-crosslinked nanogels (see Fig. S5†), indicating the formation of densely crosslinked nanogels even when using relatively low amine/epoxy molar ratios. However, residual epoxy signals could be detected for the PEG31DA-crosslinked nanogels when using amine/epoxy molar ratios of 0.1 or 0.25 (Fig. S6†). Moreover, using higher PEG31DA concentrations increases the extent of crosslinking of the nanogels.
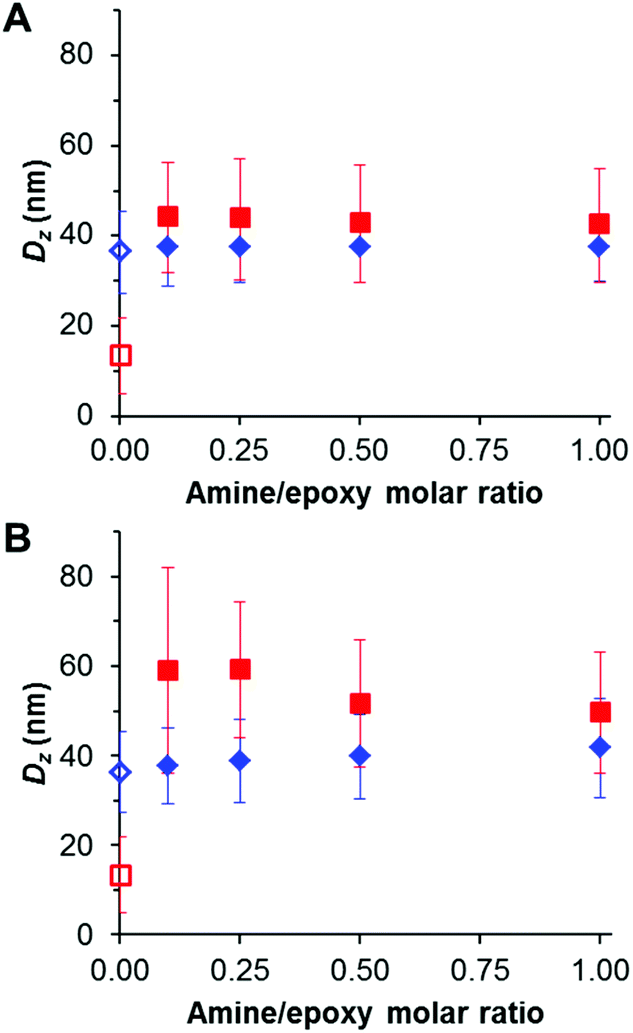 | ||
| Fig. 7 DLS analysis of PGMA45-PGlyMA100 nanoparticles crosslinked at 10% w/w solids using either (A) EDA or (B) PEG31-DA at 20 °C, followed by dilution to 0.1% w/w solids using either deionized water (blue diamonds) or DMF (red squares). The corresponding empty symbols represent the original PGMA45-PGlyMA100 nanoparticles prior to diamine crosslinking. The error bars indicate the standard deviation in the DLS particle size distributions, rather than the experimental error. | ||
Freeze-dried nanogels were also analyzed by nitrogen microanalysis, see Table 3. Increasing the amine/epoxy molar ratio used to prepare the EDA-crosslinked nanogels led to higher nitrogen contents, as expected. It is worth noting that the theoretical values shown in Table 3 were calculated assuming that each amine group reacts just once with an epoxy group (i.e. every diamine reacts with just two epoxy groups to form two secondary amines). However, in principle each primary amine can react with two epoxy groups; thus the first epoxy group reacts to produce a secondary amine, which then reacts with the second epoxy group to form a tertiary amine. Furthermore, the hydroxyl group produced via epoxy ring-opening can also react, albeit more slowly, with other epoxy groups. Both these ‘side reactions’ should lower the nitrogen content of the resulting nanogels and may well be more prevalent when using the PEG31DA crosslinker. This is because this polymeric diamine diffuses more slowly into the nanoparticle cores than EDA, which should allow more time for the initially-formed secondary amines to react with neighboring epoxy groups. FT-IR studies (Fig. S7 and S8†) did not corroborate the formation of secondary amine groups. However, the C–O stretch at around 1100 cm−1 became broader when using higher amine/epoxy molar ratios for the synthesis of the PEG31DA-crosslinked nanogels, suggesting successful incorporation of the PEG chains, see Fig. S8.†
| Diamine type | Amine/epoxy molar ratio | N content (%) | |
|---|---|---|---|
| Theory | Exp | ||
| a Below the instrument detection limit of 0.3%. | |||
| Original sample | 0.00 | 0.06 | 0.10a |
| EDA | 0.10 | 0.70 | 0.76 |
| EDA | 0.25 | 1.63 | 1.05 |
| EDA | 0.50 | 3.09 | 1.25 |
| EDA | 1.00 | 5.74 | 1.49 |
| PEG31DA | 0.10 | 0.53 | 0.02a |
| PEG31DA | 0.25 | 0.90 | 0.24a |
| PEG31DA | 0.50 | 1.21 | 0.36 |
| PEG31DA | 1.00 | 1.46 | 0.66 |
Aqueous electrophoretic behavior of crosslinked nanogels
Diamine crosslinking also conferred cationic character on the nanogels owing to protonation of pendent unreacted primary amine groups (and also the newly-formed secondary amine groups). Zeta potential vs. pH curves were constructed for two types of crosslinked nanogels prepared using an amine/epoxy molar ratio of 1.0, see Fig. 8. These dispersions possessed the highest nitrogen contents as judged by elemental microanalysis and hence were expected to be the most cationic in nature.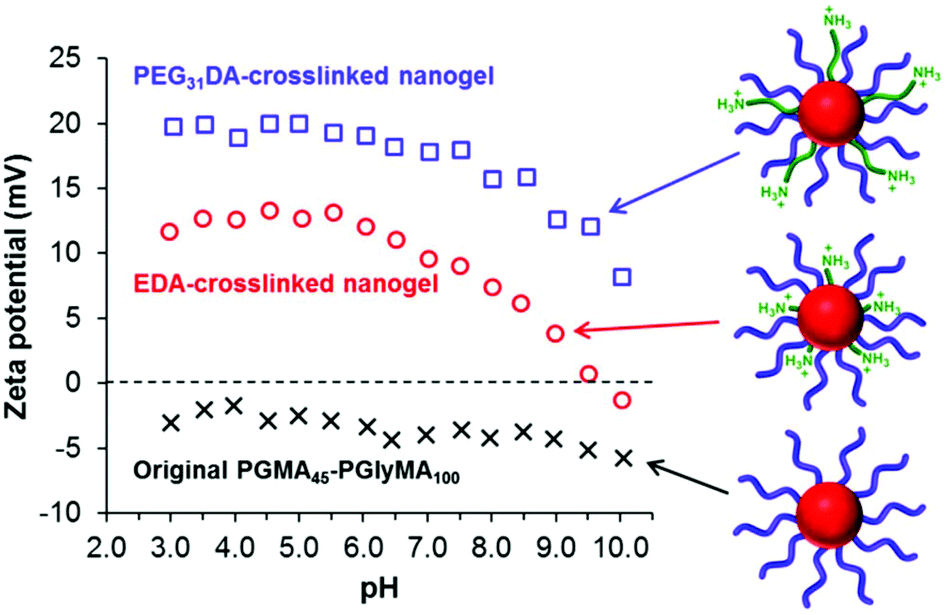 | ||
| Fig. 8 Zeta potential vs. pH curves recorded for PGMA45-PGlyMA100 nanogels crosslinked using either PEG31DA (blue squares) or EDA (red circles) at an amine/epoxy molar of 1.0. The data set obtained for the original linear PGMA45-PGlyMA100 diblock copolymer nanoparticles is also shown for comparison (black crosses). | ||
The original linear PGMA45-PGlyMA100 particles exhibit negative zeta potentials of between −2 and −6 mV across the whole pH range. This weakly anionic character is attributed to a minor fraction of carboxylic acid groups located on some of the PGMA stabilizer chain-ends, because this macro-CTA was prepared using an anionic azo initiator (ACVA). In contrast, the EDA-crosslinked nanogel has an isoelectric point of approximately pH 9.7 and becomes moderately cationic at lower pH, with a maximum zeta potential of +13 mV at around pH 4–6. Interestingly, the PEG31DA-crosslinked nanogel exhibited a positive zeta potential of at least +10 mV across the whole pH range, with a maximum zeta potential of +20 mV obtained at a pH of 3–6. The enhanced cationic nature of this latter nanogel is attributed to the long-chain nature of the PEG31DA crosslinker, which enables pendent primary amine groups not involved in epoxy ring-opening reactions to protrude further into the PGMA45 stabilizer layer and hence exert a stronger influence over the electrophoretic behavior of the nanogel. These cationic nanogels can be compared to sterically-stabilized spherical nanoparticles prepared via PISA by Semsarilar et al.,78 who statistically copolymerized a small amount of a quaternized methacrylic monomer with GMA to prepare a weakly cationic stabilizer block. In a different approach, Williams et al.79 utilized a binary mixture of cationic and non-ionic stabilizer blocks to prepare primary amine-functionalized diblock copolymer nano-objects with similar electrophoretic behavior via RAFT aqueous dispersion polymerization.
Clearly, the PISA syntheses reported herein provide multiple opportunities for the design and convenient synthesis of well-defined cationic nanoparticles. Moreover, primary amine functionality can be introduced without utilizing relatively expensive primary amine-based vinyl monomers such as 2-aminoethyl methacrylate.79
Conclusions
Well-defined epoxy-functional diblock copolymer nanoparticles can be conveniently prepared via RAFT aqueous emulsion polymerization of glycidyl methacrylate using a water-soluble poly(glycerol monomethacrylate) precursor as the stabilizer block. Optimization of the reaction conditions ensured that essentially all of the epoxy groups were retained throughout the polymerization. More specifically, polymerizations were performed at pH 4–7 and more than 99% conversion was achieved within 1 h at 50 °C, with DMF GPC analyses indicating relatively low dispersities (Mw/Mn < 1.30). However, longer reaction times led to significantly broader molecular weight distributions because the hydrolytic instability of the epoxy groups leads to latent branching. Colloidally stable diblock copolymer nanoparticles could be obtained at up to 35% w/w solids, but only when targeting relatively short core-forming blocks (DP ≤ 100). Targeting longer PGlyMA chains led to nanoparticle flocculation. Well-defined triblock copolymers could also be prepared via seeded RAFT aqueous emulsion polymerization, which extends the scope of this new PISA formulation. Gradual loss of epoxy functionality was observed during long-term storage at 20 °C, but fresh aqueous dispersions of PGMA45-PGlyMA100 nanoparticles could be derivatized via various nucleophilic ring-opening reactions. For example, addition of various diamines led to the formation of cationic core-crosslinked nanogels that remained stable when subjected to a solvent challenge. In contrast, the original linear diblock copolymer nanoparticles dissolved fully in a good solvent for both blocks (DMF). This study demonstrates that glycidyl methacrylate can be utilized for RAFT aqueous emulsion polymerization provided that appropriate care is taken to optimize the reaction conditions in order to minimize the well-known side-reactions associated with this highly versatile monomer.Acknowledgements
EPSRC is acknowledged for a Platform grant (EP/J007846/1). The European Research Council is thanked for a five-year Advanced Investigator grant for S. P. A. (PISA 320372). GEO Specialty Chemicals (Hythe, UK) is thanked for donation of the GMA monomer.References
- P. F. Cañamero, J. L. de la Fuente, E. L. Madruga and M. Fernández-García, Macromol. Chem. Phys., 2004, 205, 2221–2228 CrossRef.
- R. B. Grubbs, J. M. Dean, M. E. Broz and F. S. Bates, Macromolecules, 2000, 33, 9522–9534 CrossRef CAS.
- J. Zhu, D. Zhou, X. Zhu and G. Chen, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 2558–2565 CrossRef CAS.
- H. Yin, H. Zheng, L. Lu, P. Liu and Y. Cai, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 5091–5102 CrossRef CAS.
- M. Benaglia, A. Alberti, L. Giorgini, F. Magnoni and S. Tozzi, Polym. Chem., 2013, 4, 124–132 RSC.
- O. I. Strube, L. Nothdurft, Z. Abisheva and G. Schmidt-Naake, Macromol. Chem. Phys., 2012, 213, 1274–1284 CrossRef CAS.
- Q.-L. Li, W.-X. Gu, H. Gao and Y.-W. Yang, Chem. Commun., 2014, 50, 13201–13215 RSC.
- S. D. Kimmins, P. Wyman and N. R. Cameron, Polymer, 2014, 55, 416–425 CrossRef CAS.
- H. Li, X. Zhang, X. Zhang, K. Wang, Q. Zhang and Y. Wei, J. Mater. Chem. B, 2015, 3, 1193–1197 RSC.
- N. Ezaki, Y. Watanabe and H. Mori, Langmuir, 2015, 31, 11399–11408 CrossRef CAS PubMed.
- M. J. Bene
![[s with combining breve]](https://www.rsc.org/images/entities/char_0073_0306.gif) , D. Horák and F. Svec, J. Sep. Sci., 2005, 28, 1855–1875 CrossRef.
, D. Horák and F. Svec, J. Sep. Sci., 2005, 28, 1855–1875 CrossRef. - P. Misra, J. M. Chitanda, A. K. Dalai and J. Adjaye, Chem. Eng. J., 2016, 295, 109–118 CrossRef CAS.
- A. B. Nastasović, B. M. Ekmeščić, Z. P. Sandić, D. V. Ranđelović, M. Mozetič, A. Vesel and A. E. Onjia, Appl. Surf. Sci., 2016, 385, 605–615 CrossRef.
- S. Huš, M. Kolar and P. Krajnc, J. Chromatogr., A, 2016, 1437, 168–175 CrossRef PubMed.
- S. Ghosh and N. Krishnamurti, Eur. Polym. J., 2000, 36, 2125–2131 CrossRef CAS.
- J. A. Kling and H. J. Ploehn, J. Polym. Sci., Part A: Polym. Chem., 1995, 33, 1107–1118 CrossRef CAS.
- E.
![[Z with combining breve]](https://www.rsc.org/images/entities/char_005a_0306.gif) ůrková, K. Bouchal, D. Zde
ůrková, K. Bouchal, D. Zde![[n with combining breve]](https://www.rsc.org/images/entities/char_006e_0306.gif) ková, Z. Pelzbauer, F.
ková, Z. Pelzbauer, F. ![[S with combining breve]](https://www.rsc.org/images/entities/char_0053_0306.gif) vec, J. Kálal and H. G. Batz, J. Polym. Sci., Part A: Polym. Chem., 1983, 21, 2949–2960 CrossRef.
vec, J. Kálal and H. G. Batz, J. Polym. Sci., Part A: Polym. Chem., 1983, 21, 2949–2960 CrossRef. - W. C. Zhang, Y. Sun and L. Zhang, Ind. Eng. Chem. Res., 2015, 54, 6480–6488 CrossRef CAS.
- J. B. Tan, L. L. Fu, X. C. Zhang, Y. H. Bai and L. Zhang, J. Mater. Sci., 2016, 51, 9455–9471 CrossRef CAS.
- H. Hlídková, I. Kotelnikov, O. Pop-Georgievski, V. Proks and D. Horák, Macromolecules, 2017, 50, 1302–1311 CrossRef.
- Y. A. Liu, K. Jiang, Y. H. Ma, L. Y. Liu and W. T. Yang, Polym. Chem., 2016, 7, 2728–2739 RSC.
- R. X. Liu, A. H. Milani, T. J. Freemont and B. R. Saunders, Soft Matter, 2011, 7, 4696–4704 RSC.
- H. Zhou, X. Wang, J. Tang and Y. W. Yang, Polym. Chem., 2016, 7, 2171–2179 RSC.
- X. L. Fan, X. J. Jia, H. P. Zhang, B. L. Zhang, C. M. Li and Q. Y. Zhang, Langmuir, 2013, 29, 11730–11741 CrossRef CAS PubMed.
- S. Edmondson and W. T. S. Huck, J. Mater. Chem., 2004, 14, 730–734 RSC.
- R. Barbey and H.-A. Klok, Langmuir, 2010, 26, 18219–18230 CrossRef CAS PubMed.
- Y. Mai and A. Eisenberg, Chem. Soc. Rev., 2012, 41, 5969–5985 RSC.
- B. Charleux, G. Delaittre, J. Rieger and F. D'Agosto, Macromolecules, 2012, 45, 6753–6765 CrossRef CAS.
- P. B. Zetterlund, S. C. Thickett, S. Perrier, E. Bourgeat-Lami and M. Lansalot, Chem. Rev., 2015, 115, 9745–9800 CrossRef CAS PubMed.
- S. L. Canning, G. N. Smith and S. P. Armes, Macromolecules, 2016, 49, 1985–2001 CrossRef CAS PubMed.
- J. Jennings, G. He, S. M. Howdle and P. B. Zetterlund, Chem. Soc. Rev., 2016, 45, 5055–5084 RSC.
- M. J. Derry, L. A. Fielding and S. P. Armes, Prog. Polym. Sci., 2016, 52, 1–18 CrossRef CAS.
- C. J. Ferguson, R. J. Hughes, B. T. T. Pham, B. S. Hawkett, R. G. Gilbert, A. K. Serelis and C. H. Such, Macromolecules, 2002, 35, 9243–9245 CrossRef CAS.
- C. J. Ferguson, R. J. Hughes, D. Nguyen, B. T. T. Pham, R. G. Gilbert, A. K. Serelis, C. H. Such and B. S. Hawkett, Macromolecules, 2005, 38, 2191–2204 CrossRef CAS.
- D. E. Ganeva, E. Sprong, H. de Bruyn, G. G. Warr, C. H. Such and B. S. Hawkett, Macromolecules, 2007, 40, 6181–6189 CrossRef CAS.
- N. J. Warren and S. P. Armes, J. Am. Chem. Soc., 2014, 136, 10174–10185 CrossRef CAS PubMed.
- C. K. Poon, O. Tang, X.-M. Chen, B. T. T. Pham, G. Gody, C. A. Pollock, B. S. Hawkett and S. Perrier, Biomacromolecules, 2016, 17, 965–973 CrossRef CAS PubMed.
- A. R. Shirin-Abadi, P. G. Jessop and M. F. Cunningham, Macromol. React. Eng., 2017, 11, 1600035 CrossRef.
- C. N. Urbani and M. J. Monteiro, Macromolecules, 2009, 42, 3884–3886 CrossRef CAS.
- I. Chaduc, W. J. Zhang, J. Rieger, M. Lansalot, F. D'Agosto and B. Charleux, Macromol. Rapid Commun., 2011, 32, 1270–1276 CrossRef CAS PubMed.
- J. Rieger, Macromol. Rapid Commun., 2015, 36, 1458–1471 CrossRef CAS PubMed.
- W. J. Zhang, F. D'Agosto, P. Y. Dugas, J. Rieger and B. Charleux, Polymer, 2013, 54, 2011–2019 CrossRef CAS.
- L. Carlsson, A. Fall, I. Chaduc, L. Wagberg, B. Charleux, E. Malmstrorn, F. D'Agosto, M. Lansalot and A. Carlmark, Polym. Chem., 2014, 5, 6076–6086 RSC.
- F. L. Hatton, M. Ruda, M. Lansalot, F. D'Agosto, E. Malmstrom and A. Carlmark, Biomacromolecules, 2016, 17, 1414–1424 CrossRef CAS PubMed.
- I. Chaduc, M. Girod, R. Antoine, B. Charleux, F. D'Agosto and M. Lansalot, Macromolecules, 2012, 45, 5881–5893 CrossRef CAS.
- J. Rieger, F. Stoffelbach, C. Bui, D. Alaimo, C. Jerome and B. Charleux, Macromolecules, 2008, 41, 4065–4068 CrossRef CAS.
- J. Engstrom, F. L. Hatton, L. Wagberg, F. D'Agosto, M. Lansalot, E. Malmstrom and A. Carlmark, Polym. Chem., 2017, 8, 1061–1073 RSC.
- V. J. Cunningham, A. M. Alswieleh, K. L. Thompson, M. Williams, G. J. Leggett, S. P. Armes and O. M. Musa, Macromolecules, 2014, 47, 5613–5623 CrossRef CAS.
- B. Akpinar, L. A. Fielding, V. J. Cunningham, Y. Ning, O. O. Mykhaylyk, P. W. Fowler and S. P. Armes, Macromolecules, 2016, 49, 5160–5171 CrossRef CAS PubMed.
- L. H. Guo, Y. J. Pang, T. Qiu, Y. Meng and X. Y. Li, Polymer, 2014, 55, 4601–4610 CrossRef CAS.
- L. P. D. Ratcliffe, A. J. Ryan and S. P. Armes, Macromolecules, 2013, 46, 769–777 CrossRef CAS.
- P. Chambon, A. Blanazs, G. Battaglia and S. P. Armes, Langmuir, 2012, 28, 1196–1205 CrossRef CAS PubMed.
- N. J. W. Penfold, Y. Ning, P. Verstraete, J. Smets and S. P. Armes, Chem. Sci., 2016, 7, 6894–6904 RSC.
- J. R. Lovett, L. P. D. Ratcliffe, N. J. Warren, S. P. Armes, M. J. Smallridge, R. B. Cracknell and B. R. Saunders, Macromolecules, 2016, 49, 2928–2941 CrossRef CAS PubMed.
- J. Tan, D. Liu, C. Huang, X. Li, J. He, Q. Xu and L. Zhang, Macromol. Rapid Commun., 2017 DOI:10.1002/marc.201700195.
- L. Esser, N. P. Truong, B. Karagoz, B. A. Moffat, C. Boyer, J. F. Quinn, M. R. Whittaker and T. P. Davis, Polym. Chem., 2016, 7, 7325–7337 RSC.
- A. Blanazs, A. J. Ryan and S. P. Armes, Macromolecules, 2012, 45, 5099–5107 CrossRef CAS.
- V. J. Cunningham, M. J. Derry, L. A. Fielding, O. M. Musa and S. P. Armes, Macromolecules, 2016, 49, 4520–4533 CrossRef CAS PubMed.
- N. J. Warren, O. O. Mykhaylyk, A. J. Ryan, M. Williams, T. Doussineau, P. Dugourd, R. Antoine, G. Portale and S. P. Armes, J. Am. Chem. Soc., 2015, 137, 1929–1937 CrossRef CAS PubMed.
- I. Bannister, N. C. Billingham, S. P. Armes, S. P. Rannard and P. Findlay, Macromolecules, 2006, 39, 7483–7492 CrossRef CAS.
- Y. Li and S. P. Armes, Macromolecules, 2005, 38, 8155–8162 CrossRef CAS.
- S. Boisse, J. Rieger, K. Belal, A. Di-Cicco, P. Beaunier, M. H. Li and B. Charleux, Chem. Commun., 2010, 46, 1950–1952 RSC.
- S. Boisse, J. Rieger, G. Pembouong, P. Beaunier and B. Charleux, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 3346–3354 CrossRef CAS.
- X. W. Zhang, S. Boisse, W. J. Zhang, P. Beaunier, F. D'Agosto, J. Rieger and B. Charleux, Macromolecules, 2011, 44, 4149–4158 CrossRef CAS.
- W. J. Zhang, F. D'Agosto, O. Boyron, J. Rieger and B. Charleux, Macromolecules, 2011, 44, 7584–7593 CrossRef CAS.
- J. L. de la Haye, X. W. Zhang, I. Chaduc, F. Brunel, M. Lansalot and F. D'Agosto, Angew. Chem., Int. Ed., 2016, 55, 3739–3743 CrossRef PubMed.
- A. A. Cockram, T. J. Neal, M. J. Derry, O. O. Mykhaylyk, N. S. J. Williams, M. W. Murray, S. N. Emmett and S. P. Armes, Macromolecules, 2017, 50, 796–802 CrossRef CAS PubMed.
- W. J. Zhang, F. D'Agosto, O. Boyron, J. Rieger and B. Charleux, Macromolecules, 2012, 45, 4075–4084 CrossRef CAS.
- I. Chaduc, A. Crepet, O. Boyron, B. Charleux, F. D'Agosto and M. Lansalot, Macromolecules, 2013, 46, 6013–6023 CrossRef CAS.
- I. Chaduc, E. Reynaud, L. Dumas, L. Albertin, F. D'Agosto and M. Lansalot, Polymer, 2016, 106, 218–228 CrossRef CAS.
- S. Binauld, L. Delafresnaye, B. Charleux, F. D'Agosto and M. Lansalot, Macromolecules, 2014, 47, 3461–3472 CrossRef CAS.
- P. Chambon, A. Blanazs, G. Battaglia and S. P. Armes, Macromolecules, 2012, 45, 5081–5090 CrossRef CAS.
- C. J. Mable, N. J. Warren, K. L. Thompson, O. O. Mykhaylyk and S. P. Armes, Chem. Sci., 2015, 6, 6179–6188 RSC.
- C. J. Mable, K. L. Thompson, M. J. Derry, O. O. Mykhaylyk, B. P. Binks and S. P. Armes, Macromolecules, 2016, 49, 7897–7907 CrossRef CAS PubMed.
- Y. Kitayama, M. Yorizane, Y. Kagawa, H. Minami, P. B. Zetterlund and M. Okubo, Polymer, 2009, 50, 3182–3187 CrossRef CAS.
- M. W. Jones, L. Otten, S. J. Richards, R. Lowery, D. J. Phillips, D. M. Haddleton and M. I. Gibson, Chem. Sci., 2014, 5, 1611–1616 RSC.
- J.-F. Lutz, Angew. Chem., Int. Ed., 2007, 46, 1018–1025 CrossRef CAS PubMed.
- M. Semsarilar, V. Ladmiral, A. Blanazs and S. P. Armes, Langmuir, 2013, 29, 7416–7424 CrossRef CAS PubMed.
- M. Williams, N. J. W. Penfold, J. R. Lovett, N. J. Warren, C. W. I. Douglas, N. Doroshenko, P. Verstraete, J. Smets and S. P. Armes, Polym. Chem., 2016, 7, 3864–3873 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7py01107e |
| This journal is © The Royal Society of Chemistry 2017 |