
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Facile thiolation of hydroxyl functional polymers†
O. C. J.
Andrén
 and
M.
Malkoch
*
and
M.
Malkoch
*
KTH Royal Institute of Technology, Department of Fiber and Polymer Technology, Teknikringen 56-68, SE-100 44 Stockholm, Sweden. E-mail: malkoch@kth.se
First published on 18th July 2017
Abstract
Sulfur is an important component in many biological systems. In the hands of an organic chemist it can provide an ample handle for a myriad of robust reactions including thiol–ene click chemistry. However, in polymer chemistry the thiol functionality is rarely attributed to the macromolecule due to unatainable synthetic protocols. Herein, we provide a simple and robust strategy to produce thiol-functional polymers. The chemistry capitalizes on an unsymmetrical disulfide that straightforwardly converts hydroxyl functional polymers to their thiolated counterpart. Finally, PEG hydrogels, using both thiol–ene and Michael addition, is used to showcase the possibilities presented by thiol functional polymers.
Sulfur is an element present in many of nature's building blocks, e.g. peptides, proteins and DNA. Sulfur-containing functional groups play a critical role in many important biological functions, including the thioredoxin or glutaredoxin redox systems responsible for scission of disulfides to thiols in living organisms.1 As nucleophiles, thiols can participate in a variety of selective reactions;2 mainly Michael Addition,3 Thiol–Ene Chemistry (TEC),4 Thiol–Yne Chemistry (TYC),5 thiol-epoxy chemistry6,7 and the Michael-type reaction with malemides.3,8,9 Other interesting aspects of thiols include their affinity to gold and silver, as well as a variety of stable oxidation stages ranging from −2 up to +6.10 Recent literature that exploits the versatility of thiols reports on stable hydrogels,11,12 a simplified synthetic route to dendrimers,13 and post-functionalization of macromolecules.14
There has been an increased research interest in thiol-functional building blocks, from simple monomers or polymers to materials. One direct or indirect reason for this interest is due to the acceptance of TEC as part of the “click-chemistry” family.15–17 The concept of click reactions has captivated researchers for the last 15 years and its success can be trailed to most corners of chemistry, biology and materials science, in which advanced chemistries are accomplished in a simple and reliable fashion. These reactions capitalize on benign experimental conditions, extraordinary functional-group and solvent tolerance, and high reaction efficiency and chemoselectivity. The copper(I)-catalyzed azide–alkyne cycloaddition (CuAAC) has been the dominant click reaction; however, its use is limited in biological applications. This is mainly due to the use of copper as a catalyst, more specifically the removal of copper as well as the questionable biocompatibility of the triazole adduct.18–21 Similar to strain-promoted azide–alkyne cycloaddition (SPAAC), the recent interest in photo-initiated TEC stems from the copper-free reaction conditions. The absence of copper makes TEC an attractive alternative to CuAAC by offering a synthetic pathway to generate polymers suited for biological applications22 and material science.23–25 However, as for most chemical transformations, TEC is bound to its intrinsic limitations. Hence, multiple representations of thiols are rarely found in polymeric structures as there are no viable functionalization protocols. This is largely due to the fact that conventional thiolation strategies require harsh conditions, e.g. bases,26,27 halides28 or polycondensation,29 making them incompatible with more delicate structures such as esters. These protocols also result in inherent dispersity, making them unsuitable for the preparation of precision polymers such as dendrimers.
In the literature, thiol functionality is mainly limited to low-molecular weight compounds or chain ends of poly(ethylene glycol). To alter the representation of the thiol moiety from simple mono-functional building blocks to a wide range of polymers with multiple representations of thiols would expand their use to a broad range of application-driven research.30 For instance, there are a plethora of commercial vinylic moieties that can be efficiently exploited, as well as the new hybridization avenues with inorganic substrates including gold and silver. An additional benefit for such reversed representation is the elimination of odour typically accompanying low molecular weight thiols, as macromolecular thiols are more or less odourless.
Linear pendent group polymers with thiol functionality have been accomplished utilizing protective chemistry in a one pot deprotection and functionalization protocol to access the thiol functionality.31–33 In joint efforts with Rannard and co-workers, dendrimers with peripherally masked thiols via xanthane configurations have been successfully synthesized and subsequently utilized via Michael addition with a diverse spread of acrylic monomers.34,35 While the naked thiol functional dendrimers were not isolated, the strategy did deliver a versatile synthetic route to reactive polymeric scaffolds able to utilize Michael addition chemistry for facile post functionalization protocols. Consequently, we herein report a benign and viable synthetic pathway to truly thiol functional polymers and surfaces where the thiol-functional scaffold is isolated and utilized for the formation of hydrogels.
The prerequisite of a simple thiolation strategy is to utilize easily accessible thiols that can be applied to a wide range of substrates. In this case, hydroxyl functional substrates such as monomers, linear polymers, monodisperse dendrimers and surfaces were sought as a scaffolds. A dual functional molecule comprising both a dormant thiol and a reactive carboxylic group was recognized as structurally interesting, because it would allow straightforward esterification of hydroxyl functional substrates by the introduction of dormant thiols without the presence of any competitive side reactions. As a result, a thiol in a disulfide configuration was targeted due to its stability during common coupling chemistries and efficient deprotection protocols using reducing agents, such as dithiothreitol (DTT). Commercially available 3,3′-dithiodipropionic acid (DTPA) was identified as the key building block that could potentially be altered to an asymmetric disulfide with one dormant end and one reactive carboxylic moiety. A THF solution of DTPA, benzyl alcohol and DMAP with a molar ratio of [1.2], [1], [0.2] respectively were cooled to 0 °C and reacted via N,N′-dicyclohexylcarbodiimide (DCC) mediated esterification. Having an excess of DTPA resulted in 3-((3-(benzyloxy)-3-oxopropyl)disulfanyl)propanoic acid (1) that was isolated in a 20 gram scale and a yield above 80%. Prior to any esterification attempts of hydroxyl functional substrates, the carboxylic acid was anhydride activated via DCC chemistry yielding 3-((3-(benzyloxy)-3-oxopropyl)disulfanyl)propanoic anhydride (2) (yield 73%) (see Fig. 1).
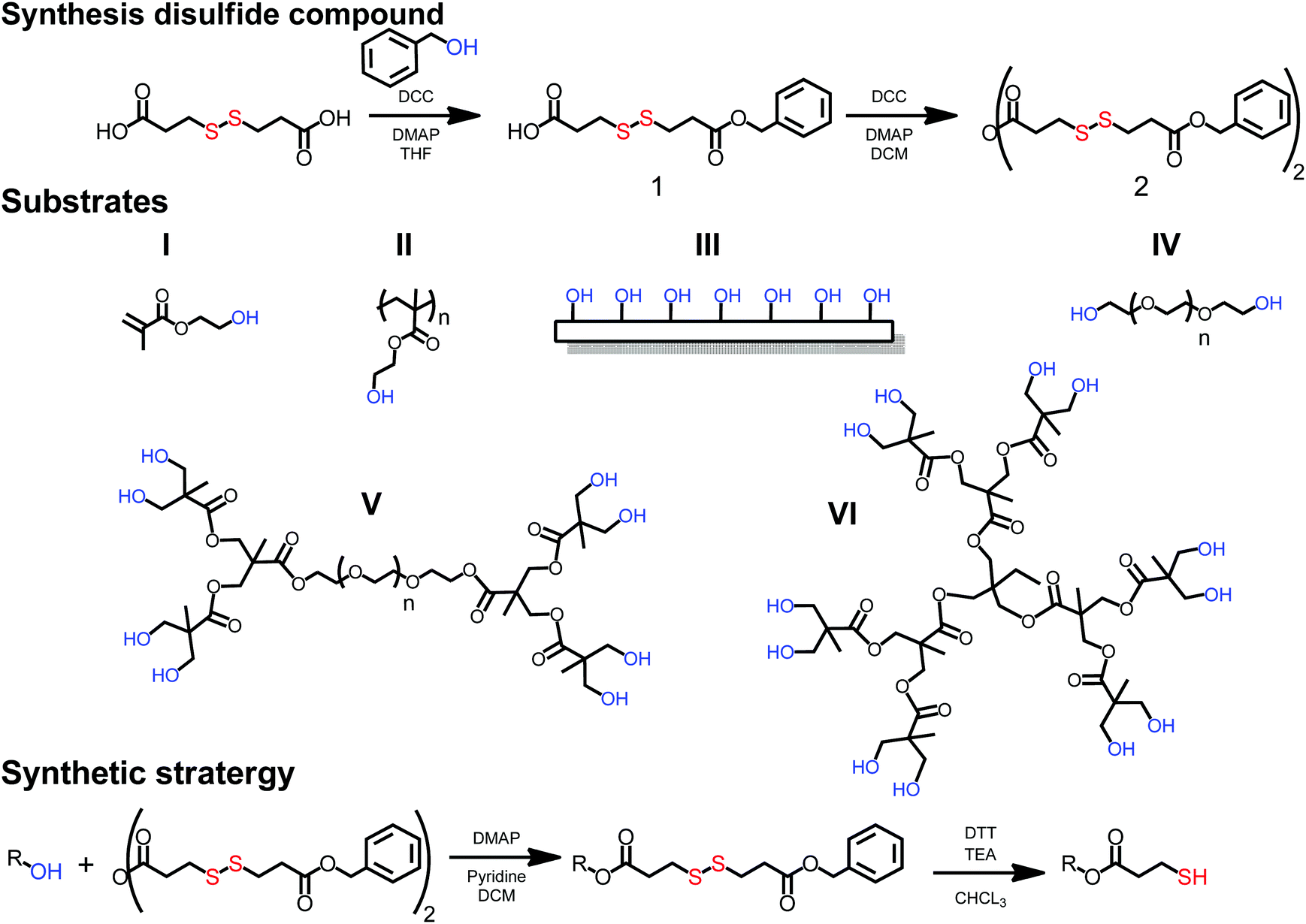 | ||
| Fig. 1 Synthesis of unsymmetrical disulfide, selected substrates: I HEMA monomer, II pendant group polymer Poly HEMA, III surface (filter paper), VI linear telechelic PEG, V 2,2-bismethylol propionic acid (bis-MPA) based linear dendritic hybrids, VI bis-MPA-based dendrimers and general esterification and deprotection strategy used to convert hydroxide functional substrates to thiol functionality. | ||
To provide solid synthetic grounds towards thiol-functional scaffolds, six substrates (I to VI) of different complexity were selected. (I) hydroxyethylmethacrylate (HEMA) as a simple monomer for ATRP polymerization; (II) poly hydroxyethylmethacrylate with a molecular weight of 6 kDa (PolyHEMA6k-OH), a linear polymer with hydroxyls as pendant groups; (III) a solid cellulose surface with an abundance of hydroxide functionalities (filter paper-OH); (IV) linear telechelic poly ethylene glycol with a molecular weight of 6 kDa (PEG6k-OH) with hydroxyl end-groups; (V) a set of linear-dendritic hybrids consisting of hydrophilic PEG core (10 kDa) and 1st to 3rd generation flawless polyester 2,2-bismethylol propionic acid (bis-MPA) dendrons displaying 4 to 16 hydroxyl groups (PEG10k-Gx-OH); and (VI) 1st to 3rd generation flawless polyester dendrimers based on bis-MPA (TMP-Gx-OH) comprising up to from 6 up to 24 peripheral hydroxyl groups.
All substrates were successfully subjected to anhydride mediated esterification using 2, generating a library of disulfide protected counterparts (Fig. 2 and ESI†). As a proof-of-concept to generate thiol-functional polymers, the optimization was initially determined on dendrimers as monodisperse polymeric substrate with undoubtable chemistry-to-structural evidence.
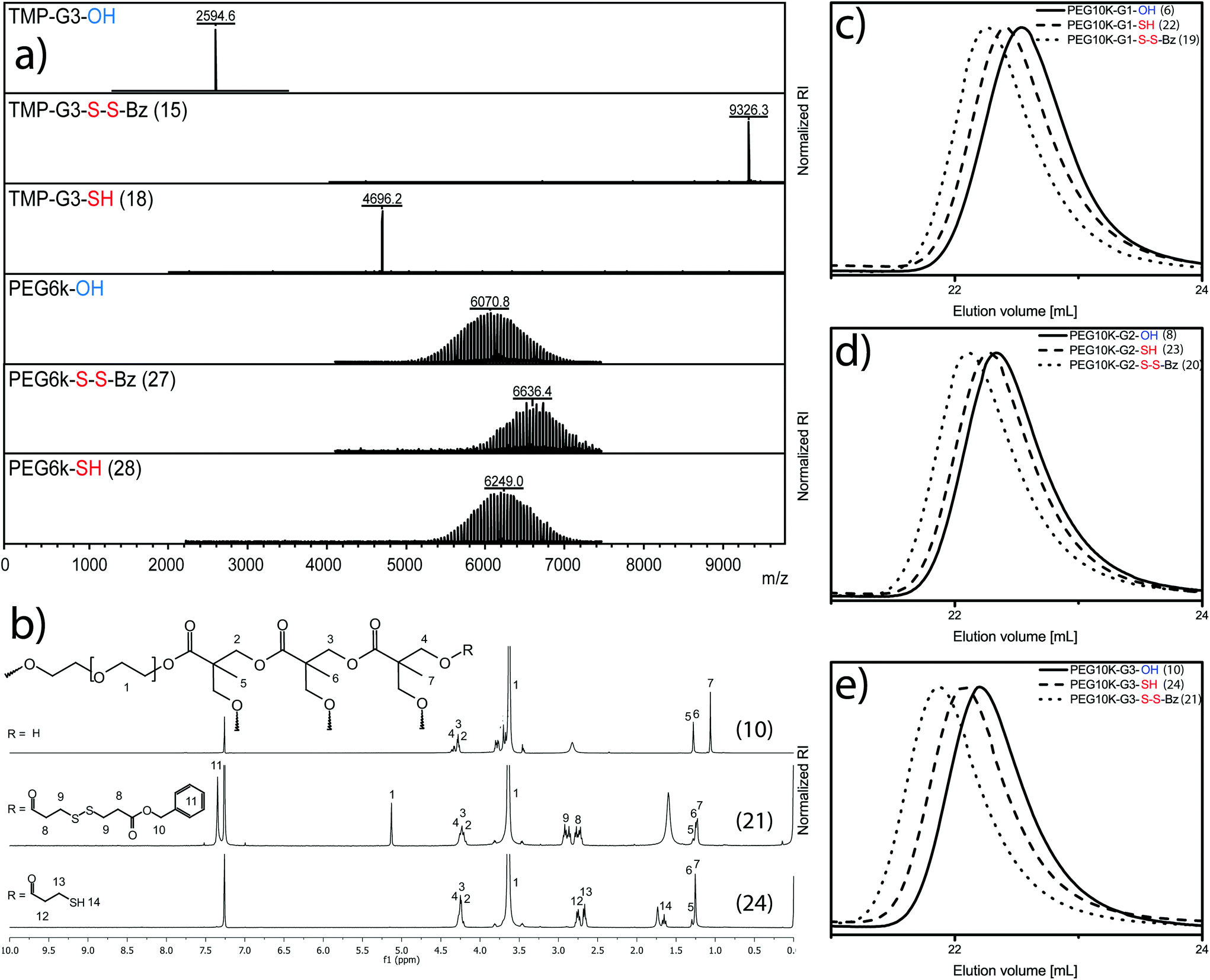 | ||
| Fig. 2 Selection of analytical data from various substrates. (a) MALDI-TOF-MS of TMP-G3-OH, TMP-G3-S-S-Bz, TMP-G3-SH, PEG6k-OH, PEG6k-S-S-Bz and PEG6k-SH. (b) 1H-NMR data for PEG10k-G3-OH, PEG10k-G3-S-S-Bz and PEG10k-G3-SH. SEC data collected in DMF for: (c) PEG10k-G1-OH, PEG10k-G1-S-S-Bz and PEG10k-G1-SH. (d) PEG10k-G2-OH, PEG10k-G2-S-S-Bz and PEG10k-G2-SH. (e) PEG10k-G3-OH, PEG10k-G3-S-S-Bz and PEG10k-G3-SH. | ||
MALDI-TOF MS in Fig. 2a exemplifies the perfection of a 3rd generation dendrimer TMP-G3-OH with 24 hydroxyl groups and a molecular weight of 2594.6 Da.
A typical reaction procedure includes the dissolution of the substrate in dichloromethane together with pyridine and DMAP. To the TMP-G3-OH solution the unsymmetrical disulfide anhydride 3 was added under stirring at 0 °C. The reaction was allowed to proceed until completion, monitored with MALDI-TOF-MS as well as 13C-NMR to ensure presence of excess of 2 with anhydride peak δ(ppm) 167.7. The satisfactory molar ratio between hydroxyls, pyridine and DMAP was found at [1]![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :[3]:[0.2] resulting in a fully substituted dendrimer, TMP-G3-S-S-Bz (15), with 24 protected disulfides and an almost four-fold molecular weight increase to 9326.3 Da (see Fig. 2a). The molar conditions were corroborated for the synthesis of the other dendrimer precursors i.e. TMP-G1-S-S-Bz (13) and TMP-G2-S-S-Bz (14). Traditional column chromatography in heptane/EtOAC was required to isolate the dendrimers.
:[3]:[0.2] resulting in a fully substituted dendrimer, TMP-G3-S-S-Bz (15), with 24 protected disulfides and an almost four-fold molecular weight increase to 9326.3 Da (see Fig. 2a). The molar conditions were corroborated for the synthesis of the other dendrimer precursors i.e. TMP-G1-S-S-Bz (13) and TMP-G2-S-S-Bz (14). Traditional column chromatography in heptane/EtOAC was required to isolate the dendrimers.
Satisfied with the purity, the dendrimers underwent benign thiol-activation in CHCl3 using 1.5 molar excess per disulfide of DTT as a reducing agent. The dendrimer, TMP-G3-SH (18), with a molecular weight of 4696.2 Da and 24 peripheral thiols was fully activated within 15 min and isolated in 94% yield. Full experimental details and analytical data that support the presented strategy of monodisperse scaffolds can be found in ESI and Fig. S1.† Consecutively, the telechelic linear polymer (III) PEG6k-OH with molecular weight by MALDI-TOF MS of 6071 Da was successfully postfunctionalized under similar conditions as for the dendrimers. In this case, a molecular weight increase was detected to 6636 Da, which corresponds to PEG6k-S-S-Bz (27). The polymer, in this case representing conventional polydisperse polymeric scaffolds, could be isolated as a pure solid and in 85% yield after precipitation in diethyl ether. The chain-end thiol-activated PEG6k-SH (28) with a molecular weight of 6249 Da was obtained following identical conditions as for the dendrimers.
Similarly, the monomer hydroxyethylmethacrylate (I) and its corresponding Poly(HEMA) (II) as well as the set of linear-dendritic hybrids (V) were postfunctionalized requiring simple precipitation protocols in diethyl ether. In the case V, a typical 1H-NMR progression is highlighted in Fig. 2b. The figure depicts the conversion from the PEG10k-G3-OH (10) to the protected PEG10k-G3-S-S-Bz (21) and finally fully activated PEG10k-G3-SH (24). A clear transition from hydroxyls to protected thiols can also be seen in Fig. 2b by the appearance of aromatic benzyl peaks at δ(ppm) 7.35. Upon deprotection and thiol activation, the spectrum alters its appearance by the absence of aromatic peaks as well as the retention of thioacetic acids characteristic CH2 peak at δ(ppm) 2.77–2.65. SEC traces for all three generation of linear-dendritic hybrids (V) show monomodal peaks with low dispersities, Fig. 2c–e.
Additionally, no residual low molecular weight compounds or uncontrolled intramolecular disulfide reactions were detected, confirming the robustness of the provided synthetic strategy. In contrast to the solution-based substrates, the solid surfaces (VI) required thorough washing with DCM upon completion including traditional soxlet-extraction. The filter papers (VI) were analyzed using contact angle (CA) measurements (see Fig. S2A†). A clear increase of contact angle (CA) was noted between the untreated filter paper-OH and filter paper subjected to 3. The CA of the hydrophilic filter paper-OH was unmeasurable, while filter-paper-S-S-Bz displayed increased hydrophobicity with a CA of 105.3° ± 5.6. Deprotection and thiol-activation yielded an expected increase in hydrophilicity to the solid filter paper-SH with CA value of 65.7° ± 11.5. In comparison, a reference filter paper was treated in the same fashion but without the main reactive compound 3. The reference paper maintained the hydrophilic nature of filter paper-OH with unmeasurable CA. All samples were run in triplicate and reported CA is represented as an average.
Taking leverage of the aqueous solubility of linear dendritic hybrids (V), which emanates from the PEG core of PEG10k-Gx-SH, combined with the multiple representation of peripheral thiol-functionalities, two sets of covalently cross-linked hydrogels were pursued utilizing the versatility of the thiol moiety. Here, hydrogels were targeted using either UV-initiated thiol–ene chemistry or base-catalysed Michael addition The cross-linkers were telechelic 10 kDa PEGs synthesized to display matching functionalities for the two chemistries i.e. PEG10k-acryl (12) and PEG10k-allyl (11). All gels were straightforwardly cross-linked at a 30 wt% dry content in EtOH:water mixture and with a molar ratio between thiols and allyl/acryl set to 1:1. The hydrogels were accomplished in situ in a rheometer, enabling real-time monitoring of the gelation. It should be noted that no spontaneous reaction occur for the thiol–ene systems prior to exposure to 365 nm UV light whereupon rapid curing can be observed.
All six mixtures formed gels using both thiol–ene chemistry and Michael addition. The curves collected during a typical cross-linking of each generation for both chemistries can be seen in Fig. 3a and b. In the case of hydrogels produced via Michael addition, the thiol-functional hybrid components and their cross-linker, PEG10k-acryl, were deposited separately on the Peltier plate and then pre-sheered to achieve mixing. This pre-emptive step was included to avoid spontaneous cross-linking that accompanies Michael addition reactions upon mixing under basic conditions. A strain (γ) = 1% and frequency (ω) = 1 Hz were used for all experiments. Both amplitude and frequency sweeps were performed to ensure that the cross-linking was conducted in the linear viscoelastic region (LVR). Amplitude and frequency sweeps for thiol–ene and Michael addition gels can be found in Fig. S3–S6.†
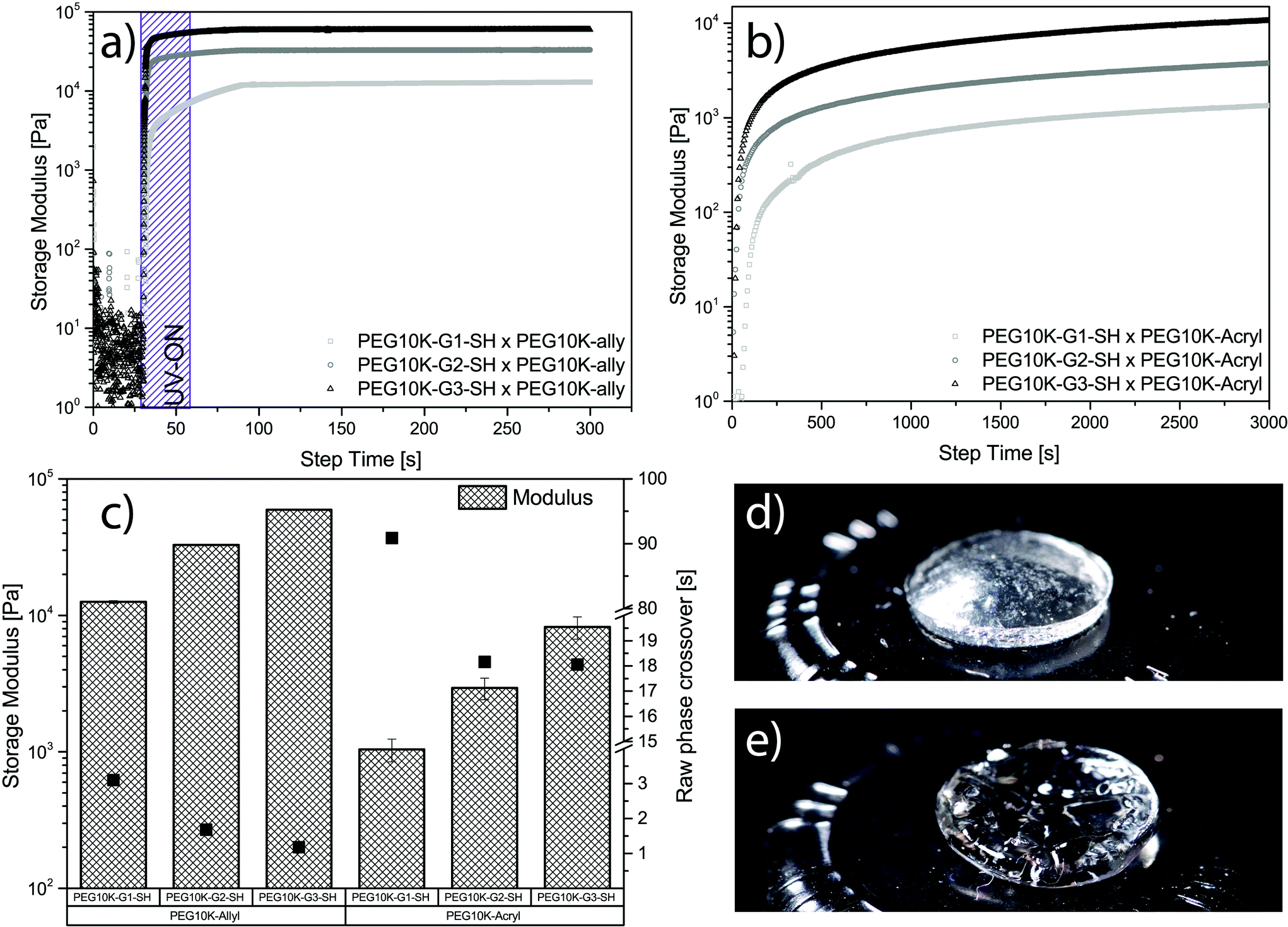 | ||
| Fig. 3 Rheological evaluation of hydrogels from scaffold V using both thiol–ene and Michael addition. (a) Gel formation using thiol–ene chemistry for PEG10k-G1-SH, PEG10k-G2-SH and PEG10k-G3-SH with PEG10k-allyl. purple area marks UV exposure. (b) Gel formation using thiol–ene chemistry for PEG10k-G1-SH, PEG10k-G2-SH and PEG10k-G3-SH with PEG10k-acryl, NaHCO3 was used as base catalysis. (c) Collection of storage modulus and raw phase crossover (gelation time) for all tested systems in triplicates. (d) Thiol–ene gel consisting of PEG10k-G3-SH and PEG10k-allyl. (e) Michael addition gel consisting of PEG10k-G3-SH and PEG10k-acryl. | ||
The thiol–ene systems were found to cross-link at a considerably faster rate when compared to the Michael addition system. A complete raw-phase crossover was observed as quickly as 1 second for PEG10k-G3-SH when cross-linked with PEG10k-allyl, whereas PEG10k-G1-SH required 3 seconds to reach the crossover (see Fig. 3a and c). Minimal standard deviation between samples, rapid curing kinetics, and the convenient on/off mechanics make TEC highly desirable for hydrogel systems. In contrast, the Michael-addition system base-activated gelation was considerably slower requiring 18 seconds until full raw-phase crossover was observed for cross-linking between PEG10k-G3-SH and PEG10k-acryl (see Fig. 3b and c). The tetra-thiolated PEG10k-G1-SH was notably slower with noted full raw-phase crossover after 91 seconds. The modulus for both hydrogel systems increase with dendritic generation, however, higher values are observed for the thiol–ene system (see Fig. 3c). The storage modulus was noted from 59.4 kPa for PEG10k-G3-SH down to 12.5 kPa for PEG10k-G1-SH using UV initiated thiol–ene chemistry. These can be compared to the values obtained for hydrogels via Michael addition, ranging from 8.2 kPa for PEG10k-G3-SH down to 1.0 kPa for PEG10k-G1-SH. A representative free-standing gel for each system can be seen in Fig. 3d and e. It is apparent that the nature of the cross-linking chemistry yields hydrogels with distinctively different physical properties.36 In this case, the gels produced by Michael addition were considerably softer. This may be due to less efficient cross-linking kinetics, noted by the longer gelation time. Overall, the multiple representations of active thiols in a polymeric configuration enabled the construction of free-standing dendritic PEG hydrogels using two different chemistries.
Conclusions
A facile synthetic pathway is herein provided that enables the introduction of reactive thiol to an array of hydroxyl functional substrates. The strategy capitalizes on designing a simple yet versatile synthetic strategy centred around an unsymmetrical disulfide, with one benzyl terminated and one anhydride-activated acid. By exploiting conventional anhydride coupling chemistry followed by disulfide cleavage using the reducing agent DTT, thiols could be introduced in a reliable and facile manner to a number of relevant substrates, ranging from simple monomers and polymers with pendant side groups to monodisperse dendrimers and solid cellulose surfaces. The multiple representation of thiols on linear dendritic hybrids were also used to engineer PEG hydrogels, both using thiol–ene “click-chemistry” as well as thiol-acrylate Michael addition.Acknowledgements
The authors acknowledge the Knut och Alice Wallenberg Foundation (KAW) (Grant number: 2012-0196) and the Seventh Framework Programme (Grant number: 60418) for financial support.References
- P. Di Mascio, M. E. Murphy and H. Sies, Am. J. Clin. Nutr., 1991, 53, S194–S200 Search PubMed.
- C. E. Hoyle, A. B. Lowe and C. N. Bowman, Chem. Soc. Rev., 2010, 39, 1355–1387 RSC.
- D. P. Nair, M. Podgorski, S. Chatani, T. Gong, W. X. Xi, C. R. Fenoli and C. N. Bowman, Chem. Mater., 2014, 26, 724–744 CrossRef CAS.
- C. E. Hoyle and C. N. Bowman, Angew. Chem., Int. Ed., 2010, 49, 1540–1573 CrossRef CAS PubMed.
- A. B. Lowe, Polymer, 2014, 55, 5517–5549 CrossRef CAS.
- M. C. Stuparu and A. Khan, J. Polym. Sci., Part A: Polym. Chem., 2016, 54, 3057–3070 CrossRef CAS.
- S. De and A. Khan, Chem. Commun., 2012, 48, 3130–3132 RSC.
- R. J. Pounder, M. J. Stanford, P. Brooks, S. P. Richards and A. P. Dove, Chem. Commun., 2008, 5158–5160, 10.1039/B809167F.
- B. H. Northrop, S. H. Frayne and U. Choudhary, Polym. Chem., 2015, 6, 3415–3430 RSC.
- C. E. M. Yanez Prieto, 3343735 Ph.D., The Pennsylvania State University, 2006.
- S. Mongkhontreerat, K. Oberg, L. Erixon, P. Lowenhielm, A. Hult and M. Malkoch, J. Mater. Chem. A, 2013, 1, 13732–13737 CAS.
- K. Oberg, Y. Hed, I. J. Rahmn, J. Kelly, P. Lowenhielm and M. Malkoch, Chem. Commun., 2013, 49, 6938–6940 RSC.
- P. Antoni, M. J. Robb, L. Campos, M. Montanez, A. Hult, E. Malmstrom, M. Malkoch and C. J. Hawker, Macromolecules, 2010, 43, 6625–6631 CrossRef CAS.
- J. N. Hunt, K. E. Feldman, N. A. Lynd, J. Deek, L. M. Campos, J. M. Spruell, B. M. Hernandez, E. J. Kramer and C. J. Hawker, Adv. Mater., 2011, 23, 2327–2331 CrossRef CAS PubMed.
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef CAS PubMed.
- C. E. Hoyle and C. N. Bowman, Angew. Chem., Int. Ed., 2010, 49, 1540–1573 CrossRef CAS PubMed.
- M. J. Kade, D. J. Burke and C. J. Hawker, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 743–750 CrossRef CAS.
- P. V. Chang, J. A. Prescher, E. M. Sletten, J. M. Baskin, I. A. Miller, N. J. Agard, A. Lo and C. R. Bertozzi, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 1821–1826 CrossRef CAS PubMed.
- C. D. Hein, X. M. Liu and D. Wang, Pharm. Res., 2008, 25, 2216–2230 CrossRef CAS PubMed.
- E. D. Oldham, S. Seelam, C. Lema, R. J. Aguilera, J. Fiegel, S. E. Rankin, B. L. Knutson and H. J. Lehmler, Carbohydr. Res., 2013, 379, 68–77 CrossRef CAS PubMed.
- N. Ma, Y. Wang, B. X. Zhao, W. C. Ye and S. Jiang, Drug Des., Dev. Ther., 2015, 9, 1585–1599 Search PubMed.
- C. E. Hoyle, T. Y. Lee and T. Roper, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 5301–5338 CrossRef CAS.
- G. Franc and A. K. Kakkar, Chem. Soc. Rev., 2010, 39, 1536–1544 RSC.
- K. M. Partridge, I. A. Guzei and T. P. Yoon, Angew. Chem., Int. Ed., 2010, 49, 930–934 CrossRef CAS PubMed.
- S. Rana and J. W. Cho, Nanoscale, 2010, 2, 2550–2556 RSC.
- J. R. Brindle and J. L. Liard, Can. J. Chem., 1975, 53, 1480–1483 CrossRef CAS.
- K. Chauhan, V. Priya, P. Singh, G. S. Chauhan, S. Kumari and R. K. Singhal, RSC Adv., 2015, 5, 51762–51772 RSC.
- C. C. Han and R. Balakumar, Tetrahedron Lett., 2006, 47, 8255–8258 CrossRef CAS.
- V. X. Truong, I. A. Barker, M. Tan, L. Mespouille, P. Dubois and A. P. Dove, J. Mater. Chem. B, 2013, 1, 221–229 RSC.
- M. Le Neindre and R. Nicolay, Polym. Chem., 2014, 5, 4601–4611 RSC.
- M. Le Neindre and R. Nicolay, Polym. Int., 2014, 63, 887–893 CrossRef CAS.
- R. Nicolay, Macromolecules, 2012, 45, 821–827 CrossRef CAS.
- T. Rudolph, P. Espeel, F. E. Du Prez and F. H. Schacher, Polym. Chem., 2015, 6, 4240–4251 RSC.
- S. E. R. Auty, O. Andren, M. Malkoch and S. P. Rannard, Chem. Commun., 2014, 50, 6574–6577 RSC.
- S. E. R. Auty, O. C. J. Andren, F. Y. Hern, M. Malkoch and S. P. Rannard, Polym. Chem., 2015, 6, 573–582 RSC.
- T. Yang, H. Long, M. Malkoch, E. K. Gamstedt, L. Berglund and A. Hult, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 4044–4054 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Full synthetic and analytical data as well as additional characterization data. See DOI: 10.1039/c7py01097d |
| This journal is © The Royal Society of Chemistry 2017 |