
Regulating sequence distribution of polyethers via ab initio kinetics control in anionic copolymerization†
 *ad
*ad
Abstract
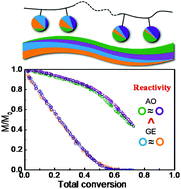
A study of the reaction kinetics of the copolymerization of two major categories of epoxy monomers, glycidyl ether (GE) and alkylene oxide (AO) derivatives, via the in situ NMR technique has been performed in a combinatorial strategy. GEs or AOs show similar reactivities when copolymerizing within either category, leading to a special azeotropic copolymerization, while they exhibit distinct reactivities between them. The underlying kinetics along with the living anionic copolymerization mechanism allow the generation of gradient and azeotropic copolymers, azeotropic-gradient terpolymers and double azeotropic-gradient tetrapolymers. Through the quantitative determination of rate constants and reactivity ratios with the aid of a numerical calculation in a simulated annealing scenario, we provide a widely applicable design principle for the sophisticated construction of sequence distribution-regulated, functional polyethers via a simple and efficient one-pot approach.
 Please wait while we load your content...
Please wait while we load your content...

