
The energy dissipation and Mullins effect of tough polymer/graphene oxide hybrid nanocomposite hydrogels†
 *a
Jie
Zheng
*b
*a
Jie
Zheng
*b
Abstract
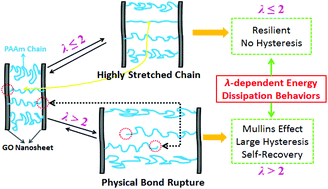
Nanocomposite hydrogels (NC gels) are considered to belong to the class of high strength hydrogels. Graphene oxide (GO), owing to its amphiphilic, mechanical, and optical properties, is widely used as a filler incorporated into different hosting materials (elastomers, plastics, and hydrogels) to improve their mechanical properties. In this work, we used in situ free radical polymerization to synthesize polyacrylamide (PAAm)/GO hybrid NC gels in the presence of GO nanosheets and a very small amount of chemical cross-linkers (N,N′-methylenebisacrylamide, MBA < 0.1 mol%). By optimizing GO and MBA concentrations, the resulting PAAm/GO gels can achieve an elastic modulus of 66 kPa, a fracture stress of 0.27 MPa, a fracture strain of 13.76 mm mm−1, deformed energy of 2.52 MJ m−3, and tearing energy of 964 J m−2. Due to the presence of physical interactions between PAAm and GO nanosheets, PAAm/GO gels demonstrate λ-dependent energy dissipation and Mullins self-recovery behaviors. The gels can rapidly recover their stiffness and toughness by 76% and 60%, respectively, after 30 min of resting at room temperature. The possible toughening mechanisms and Mullins effects of PAAm/GO gels were proposed and compared with those of filler rubbers and other high strength hydrogels. This work provides new viewpoints to develop tough hydrogels by the introduction of GO into other hydrogels with a good mechanical balance between strong chemical bonding and reversible physical bonding.
 Please wait while we load your content...
Please wait while we load your content...

