
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Ring opening polymerization of macrolactones: high conversions and activities using an yttrium catalyst†
D.
Myers
a,
T.
Witt
 b,
A.
Cyriac
a,
M.
Bown
c,
S.
Mecking
*b and
C. K.
Williams
*ad
b,
A.
Cyriac
a,
M.
Bown
c,
S.
Mecking
*b and
C. K.
Williams
*ad
aDepartment of Chemistry, Imperial College London, London SW7 2AZ, UK
bDepartment of Chemistry, University of Konstanz, Universitätsstraße 10, 78457 Konstanz, Germany. E-mail: stefan.mecking@uni-konstanz.de
cCSIRO Manufacturing, Ian Wark Laboratory, Bayview Avenue, Clayton, Vic 3168, Australia
dDepartment of Chemistry, University of Oxford, Chemistry Research Laboratory, 12 Mansfield Road, Oxford, OX1 3 TA, UK
First published on 1st September 2017
Abstract
The ring-opening polymerization of macrolactones (C15–C23) enables the production of long-chain aliphatic polyesters which are crystalline polymers with melting temperatures ranging from 98–106 °C. Here, the polymerization of ω-pentadecalactone (C15), nonadecalactone (C19) and tricosalactone (C23) are investigated using an yttrium phosphasalen catalyst. The catalyst enables typical conversions to exceed >80% and the reactions occur either in neat monomer or in solution in toluene, over the temperature range 25–100 °C. The yttrium catalyst shows higher activities than previously reported aluminium–salen complexes, with TOF values in the range 200–400 h−1 in the best cases. The polymerizations occur with linear increase in molecular weight vs. conversion and enable the production of polyester with 10 < Mn< 60 kg mol−1. Using tricosalactone the polymerization thermodynamic parameters are determined and confirm the polymerization is entropically driven. The findings underscore the importance of continued catalyst development to allow higher rates of reaction which has the added benefit of accessing the highest conversions to polymer.
Introduction
Aliphatic polyesters may be sustainable alternatives to petrochemically-derived commercial polymers, especially where they are bio-derived and/or degradable.1 In this context, the search for alternatives to polyolefins, such as polyethylene (PE), are particularly important due to the enormous current scales of polyolefin production and usage.1e,2 Long-chain aliphatic polyesters show promise as more sustainable alternatives – they are able to match the properties of some classes of PE, for example showing melting temperatures, crystallinity and tensile strengths akin to PE.1c,3 Furthermore, natural triglycerides, extracted from plants or even bio-synthesized by algae, can be used as the sources of the monomers.2 The polyesters can be produced by either condensation routes or by the ring-opening polymerizations (ROP) of macrolactones.4 Although both methods are successful and could be compatible with larger scale processes, the ring-opening polymerization is a chain growth reaction and so at the same conversion higher molecular weights should be achieved. Further, using ROP it is possible to control the molar mass and chain end-groups, and to produce copolymers. There has been a growing body of work demonstrating the promising physical–chemical properties for both homo- and copolymers prepared from macrolactones, most especially using ω-pentadecalactone (PDL).3,5 Nonetheless, a difference between ROP using macrolactones, compared to smaller lactones, is that there are generally similar rates of propagation and transesterification leading to broader molecular weight dispersity values and hindering block copolymer synthesis.Lactone ring-opening polymerization is also used commercially to prepare aliphatic polyesters such as polylactide. In the case of small ring sizes (<7 membered ring), the reaction is enthalpy driven and results in a release of ring-strain upon polymer formation.1a,7 Substantial research has focussed on these smaller ring systems – there is good understanding of the thermodynamic factors controlling ring-strain and the kinetic factors enabling the production of more efficient catalysts. In contrast, larger ring-systems are less explored but are generally expected to undergo entropically driven polymerizations.5l,6b,8 The macrolactone ω-pentadecalactone (PDL) (C15) has been most widely explored and its thermodynamic parameters are consistent with entropy driven reactions:  and
and  ([PDL]eq = 0.016 M at 373 K).7 Its polymerization was pioneered more than 20 years ago using lipase enzymes5a,e,9 and has also been reported using various metal6a,10 and organic catalysts.11 It was noted that the metal based catalysts typically showed greater activity and productivity compared to enzyme systems under comparable conditions.10d Two of the best metal catalysts are lanthanide/Group 3 complexes: Nakayama et al., reported [Nd(BH4)3(THF)3] with a TOF of 343 min−1 ([PDL]0 = 2.76 M, [Nd]0 = 0.018 M, 83% conversion, THF, 60 °C)10d and Zhong et al., reported yttrium(tris(iso-propoxide)) showing a TOF of 214 min−1 ([PDL]0 = 3.8 M (neat), [Y]0 = 9.5 mM, 70% conversion, 100 °C).10b In contrast, Al–salen catalysts, which are widely available, showed TOF in the range 1–4 min−1 ([PDL]0 = 1 M, [Al]0 = 0.01 M, toluene, 100 °C).6e
([PDL]eq = 0.016 M at 373 K).7 Its polymerization was pioneered more than 20 years ago using lipase enzymes5a,e,9 and has also been reported using various metal6a,10 and organic catalysts.11 It was noted that the metal based catalysts typically showed greater activity and productivity compared to enzyme systems under comparable conditions.10d Two of the best metal catalysts are lanthanide/Group 3 complexes: Nakayama et al., reported [Nd(BH4)3(THF)3] with a TOF of 343 min−1 ([PDL]0 = 2.76 M, [Nd]0 = 0.018 M, 83% conversion, THF, 60 °C)10d and Zhong et al., reported yttrium(tris(iso-propoxide)) showing a TOF of 214 min−1 ([PDL]0 = 3.8 M (neat), [Y]0 = 9.5 mM, 70% conversion, 100 °C).10b In contrast, Al–salen catalysts, which are widely available, showed TOF in the range 1–4 min−1 ([PDL]0 = 1 M, [Al]0 = 0.01 M, toluene, 100 °C).6e
In the broader context of macrolactone ROP, the investigation of ring sizes greater than C15 (PDL) remains much less explored.5j,6a,d,e,12 We recently reported the preparation of nonadecalatone (C19) and tricosalactone (C23) from fatty acids.6e The ROP of these larger lactones was initially investigated using aluminium–salen catalysts and more recently successfully using lipase enzymes. In the case of the Al–salen catalysts, the polymerizations showed very low rates (NDL: average and unoptimized TOF = 4 h−1; TCL: average and unoptimized TOF = 0.7 h−1; [lactone]0 = 3.8 M (bulk), [Al]0 = 0.25 mol%, 100 °C) and overall conversions were very low – 23% (NDL) and 5% (TCL) (Fig. 1).6e We recently reported the use of yttrium phosphasalen catalysts which show high activity and control in the ring-opening polymerization of lactide.13 Given the poor performances of Al–salen catalysts in macrolactone ROP, it was of interest to investigate whether an yttrium–phosphasalen catalyst could be used to increase rates and overall conversions to polyester using the new macrolactones (Scheme 1).
 | ||
| Fig. 1 Ring-opening polymerizations of macrolactones catalyzed by an aluminium salen complex.6e | ||
 | ||
| Scheme 1 Yttrium phosphasalen catalysed ring-opening polymerisation of macrolactones: PDL (C15), NDL (C19) and TCL (C23). Conditions: [Lactone]0/[1] = 50–200, [Lactone]0 = 0.3–3.8 M, toluene or bulk. | ||
Results and discussion
The yttrium catalyst (1),13a NDL6e and TCL6e were synthesized according to published procedures and PDL was purchased and purified by distillation from CaH2. Firstly, the ROP of ω-pentadecalactone (PDL) was investigated in bulk monomer ([PDL]0 = 3.8 M), at 373 K, using 1 mol% loading of yttrium catalyst (vs. monomer) (Table 1, entry 1). The reaction proceeded very rapidly as shown by near complete conversion to polymer occurring within 10 s, which equates to a turn-over-frequency of TOF ∼ 126![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 h−1 or 2100 min−1 (Fig. S1–S3†). Whilst a number of catalysts are reported for the ROP of PDL, the rates observed using 1 are qualitatively fast: ca. 6 times faster than the Nd complex (TOF = 330 min−1, albeit measured at lower temperature), 16 times faster than the yttrium complex (TOF = 124 min−1), and 1200 times faster than the Al–salen complex (TOF = 1.7 min−1).5j,10b,d,f
000 h−1 or 2100 min−1 (Fig. S1–S3†). Whilst a number of catalysts are reported for the ROP of PDL, the rates observed using 1 are qualitatively fast: ca. 6 times faster than the Nd complex (TOF = 330 min−1, albeit measured at lower temperature), 16 times faster than the yttrium complex (TOF = 124 min−1), and 1200 times faster than the Al–salen complex (TOF = 1.7 min−1).5j,10b,d,f
| Entry | Lactone | T (K) | [LA]0/[1] | [Lactone]0 (M) | Conv.c (%) | Time (h) | TOF (mol mol−1 h−1) |
M
theond (kg mol−1) |
M
expne (kg mol−1) |
Đ |
|---|---|---|---|---|---|---|---|---|---|---|
| a Solvent = toluene. b No solvent. c Monomer conversion determined by 1H NMR spectroscopy. d Theoretical molecular weight determined by the following relation: Mtheon = molecular weight of monomer × conversion × no. of equivalents. e Determined by SEC analysis (trichlorobenzene, 433 K, PE standards). | ||||||||||
| 1b | PDL | 373 | 100 | 3.8 | 94 | 0.17 min | 126, 000 | 22.8 | 13.7 | 2.56 |
| 2 | PDL | 298 | 100 | 1.0 | 98 | 4 | 25 | 23.6 | 26.1 | 2.06 |
| 3 | PDL | 298 | 100 | 0.25 | 90 | 21 | 1 | 21.6 | 25.3 | 1.97 |
| 4 | PDL | 373 | 500 | 1 | 66 | 0.5 | 660 | 79.3 | 25.0 | 2.00 |
| 5b | NDL | 298 | 100 | 3.0 | 33 | 25 | 4 | 9.8 | 27.0 | 2.55 |
| 6b | NDL | 373 | 100 | 3.0 | 78 | 1 | 234 | 23.1 | 8.6 | 2.40 |
| 7 | NDL | 373 | 100 | 0.75 | 86 | 10 min | 387 | 25.5 | 18.4 | 2.17 |
| 8 | NDL | 373 | 200 | 0.3 | 37 | 5 min | 266 | 21.9 | 9.7 | 1.78 |
| 9 | NDL | 373 | 200 | 0.3 | 48 | 7 min | 247 | 28.5 | 15.2 | 1.64 |
| 10 | NDL | 373 | 200 | 0.3 | 77 | 10 min | 277 | 45.6 | 24.3 | 2.22 |
| 11 | TCL | 373 | 100 | 0.3 | 83 | 20 min | 75 | 29.3 | 21.5 | 2.54 |
| 12 | TCL | 373 | 50 | 0.3 | 97 | 5 min | 175 | 17.1 | 12.0 | 2.14 |
| 13 | TCL | 373 | 200 | 0.3 | 85 | 90 min | 34 | 59.9 | 32.1 | 2.18 |
In order to allow monitoring of the reaction, polymerizations were also investigated using a 1 M solution of monomer in toluene, at room temperature and using a 1 mol% loading of the yttrium catalyst. The conditions were selected both to decrease the reaction rate and to enable understanding of the temperature range feasible for successful catalysis. Under these conditions, catalyst 1 enabled quantitative conversion of PDL to polyester within 4 h, i.e. TOF = 25 h−1 (Table 1, entry 2) (Fig. S4 and S5†). The ability to catalyse polymerizations at room temperature was somewhat surprising but maybe useful as a means to increase conversion, it was previously reported that other fast catalysts do not operate at room temperature.10d
The kinetics of the polymerisation at room temperature were analysed by regularly withdrawing aliquots from the reaction mixture. The analysis revealed a first-order rate dependence on monomer concentration and enabled determination of kobs = 2.28 ± 0.05 × 10−4 s−1 (Fig. S6 and S7†). The polymer number average molecular weight (Mn) was determined using size-exclusion chromatography (SEC) with trichlorobenzene as the eluent, at 433 K and the instrument was calibrated with narrow molecular weight polyethylene standards. The analysis showed a linear evolution of Mnvs. monomer conversion, indicating a reasonably well-controlled polymerization, with experimental and theoretical values of molecular weight in good agreement (Fig. S8†). The dispersity values were around 2, which is as expected for macrolactone ROP and can be understood in terms of relatively similar rates of propagation and transesterification.5l
Even when the monomer concentration was further decreased, near-quantitative conversions were still achievable even using [PDL]0 = 0.25 M (298 K) (Table 1, entry 3; Fig. S11 and S12†). In the context of the thermodynamic parameters, Duda and Kowalski determined that [PDL]eq = 0.7 M (T = 370 K) a value that was calculated from experimental data reported by Lebedev and co-workers.14 On that basis, at low monomer concentrations ([PDL] < 0.7 M), polymerization would not be expected to proceed – a finding clearly at odds with the experimental results. Nonetheless, Duchateau and co-workers recently reported polymerizations at monomer concentrations as low as [PDL]0 = 0.25 M (T = 373 K).10e Duchateau and co-workers determined a lower value for the equilibrium monomer concentration: [PDL]eq = 0.016 M (T = 373 K).10e They attributed the difference between the equilibrium monomer concentrations to experimental limitations in the earlier report where extensive purification was used to isolate the polymer.10e Accordingly during purification low molecular weight polymer fractions would be removed, resulting in an under-estimation of polymer yield and an artificially high equilibrium monomer concentration.10e Here, the experimental protocol did not involve any isolation and so the equilibrium monomer concentration value is expected to be in line with that determined by Duchateau and co-workers.10e
Polymerisations were also investigated at lower catalyst loadings ([PDL]/[1] = 500, 0.2 mol%, [PDL]0 = 1 M, at 373 K) and proceeded effectively to give TOF = 660 h−1 (Table 1, entry 4; Fig. S13 and S14†). It is quite notable that the catalysts were successful at both low monomer and catalyst concentrations – there are few equivalent reports for other catalysts.5j,11d One example, reported by Duchateau et al.,12 showed approximately equivalent conversion (57%) but requiring an hour using similar loadings ([PDL]/[Al] = 433, 0.23 mol%, [PDL]0 = 1.3 M, at 373 K).
Encouraged by the activity of 1 for the ROP of PDL, the use of nonadecalactone (NDL) was subsequently investigated. As mentioned, the previous ROP of NDL applied an aluminium salen catalyst and resulted in 23% conversion after 22 h, i.e. TOF = 4 h−1 (T = 373 K).10 The first polymerizations were investigated using neat monomer at 298 K (note that NDL is liquid at this temperature) and applying 1 mol% of catalyst 1. Even under these low temperature conditions, catalyst 1 enabled 33% conversion within 25 h, i.e. TOF = 1 h−1 – with the overall conversion surpassing that previously obtained with Al–salen catalysts even at the lower reaction temperature (373 K vs. 298 K) (Fig. S15–S17†). It was also noted that the polymer precipitated, which prevented efficient agitation and so allows only approximate estimation of activity. The SEC analysis revealed that polymer molecular weights were significantly higher than expected, consistent with the heterogeneity of the reaction (Table 1, entry 5). In order to prevent polymer precipitation, the polymerisation was investigated using neat monomer, at 373 K, and under fully homogeneous conditions, the TOF increased to 234 h−1 (Table 1, entry 6; Fig. S18 and S19†). Further experiments revealed that the conversion could be increased to 94% by conducting the polymerisation in toluene (Fig. S20 and S21†). Through appropriate tuning of conditions, TOF values up to 387 h−1 were feasible (Table 1, entry 7) with molecular weights close to predicted values (Fig. S22 and S23†).
The polymerization kinetics for NDL ROP were investigated by quenching a series of separate polymerizations at specific time intervals and the crude mixtures were analysed using 1H NMR spectroscopy so as to determine the conversions of monomer and polymer (see ESI†). Analysis of the aliquots showed the high polymerization control exhibited by catalyst 1, with SEC analysis revealing a linear growth in molecular weights against conversion (Table 1, entries 8–10; Fig. S24–S29†).
Finally, the ROP of the larger tricosalactone (C23) was investigated. After 20 min, using toluene solutions at 373 K ([TCL]/[1] = 100, [TCL]0 = 0.3 M, toluene and T = 373 K), conversions of 83% were achieved, i.e. TOF = 75 h−1 (Table 1, entry 11; Fig. S30–S32†). Both the conversion and the turn over frequency of the catalyst were significantly greater than the previous report for TCL ROP using an aluminium salen catalyst (TOF = 1 h−1).6e Overall, the results using both NDL and TCL show that the low conversions obtained using Al–salen catalysts were a feature of low reaction rates and can be improved using the more active yttrium catalysts.
Next, the polymerization kinetics using TCL were obtained by quenching reactions at specific time intervals and analysing the crude mixtures by NMR spectroscopy (see ESI†). The semi-logarithmic plot of monomer concentration against time showed a linear fit indicating a first-order dependence on [TCL] and enabling determination of kobs = 3.06 ± 0.31 × 10−3 s−1 (Fig. 2). The equivalent rate constant for PDL ROP, under identical conditions, is kobs = 1.01 ± 0.11 × 10−3 s−1 – i.e. TCL is polymerized approximately three times faster than PDL using catalyst 1 (Fig. S33 and S34†).
 | ||
| Fig. 2 Polymerisation data for the ROP of TCL. Conversion vs. time plot (top left), semi-logarithmic pseudo first-order kinetic plot (top right) and molecular weight evolution with Đ vs. conversion (bottom). Conditions: [TCL]0/[1] = 100, [TCL]0 = 0.3 M, toluene, 373 K. | ||
The ROP of TCL catalyzed by 1 also showed a linear evolution of molecular weights with conversion, indicating good polymerization control. In line with these findings, the PTCL molecular weights were controlled by the catalyst loading and values in the range 17–60 kg mol−1 were obtained (Table 1, entries 11–13; Fig. S35–S38†). In order to further examine the ROP of TCL, a Van 't Hoff analysis was performed. The determination of [TCL]eq was conducted by carrying out polymerizations at various temperatures. For each reaction, exponential fits to monomer conversion vs. time allowed determination of the kobs values, the fits were in accordance with the first order dependence in monomer concentration and were carried out over >5.5 half-lives (Fig. S39–S42†). It is important to emphasise that the results are best described as semi-quantitative since monomer and polymer resonances were observed to overlap in the NMR spectra and conversions required the use of peak deconvolution techniques (see ESI†). The thermodynamic parameters were determined as:  and
and  (Fig. S43†). Despite some uncertainty in the absolute values, particularly for the entropy, the equilibrium monomer concentration was determined to be 0.03 M (373 K) and was clearly independent of the reaction temperature, as would be expected for an entropically driven ROP. The values obtained for TCL ROP are in line with the thermodynamic parameters for other macrolactones, e.g. PDL has been reported to show
(Fig. S43†). Despite some uncertainty in the absolute values, particularly for the entropy, the equilibrium monomer concentration was determined to be 0.03 M (373 K) and was clearly independent of the reaction temperature, as would be expected for an entropically driven ROP. The values obtained for TCL ROP are in line with the thermodynamic parameters for other macrolactones, e.g. PDL has been reported to show  and
and  ,7 and the 17-membered lactone ambrettolide,
,7 and the 17-membered lactone ambrettolide,  and
and 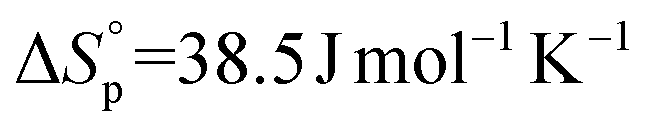 .5l
.5l
In terms of the polymers produced from the macrolactones, DSC analyses revealed crystalline polyesters with melting temperatures (Tm) at 102 °C (PNDL) and 106 °C (PTCL) (Table 1, entries 10 and 13; Fig. 3; Fig. S44 and S45†). The small increase in Tm for PTCL was attributed to the higher concentration of methylene groups in the main chain compared to PNDL (ESI†). In line with the findings, for PPDL Tm = 97 °C.5c
 | ||
| Fig. 3 DSC thermograms of PPDL, PNDL and PTCL samples. The PPDL sample is shown for comparative purposes and was prepared using an aluminium salen initiator. Second heating cycles shown. | ||
Conclusions
A highly active yttrium phosphasalen catalyst showed efficient and controlled ROP of three macrolactones: pentadecalactone (PDL), nonadecalactone (NDL) and tricosalactone (TCL). The catalyst showed rapid rates, with TOF values in the range 200–400 h−1 for NDL (100 °C, [NDL] = 0.3–0.75 M, toluene) and 34–175 h−1 for TCL (100 °C, [TCL] = 0.3 M, toluene). The polymerizations were effective under a range of conditions, including either in neat monomer or as solutions diluted in toluene, and the catalyst operated effectively over a broad temperature range: 25–100 °C. The rapid rates allowed high conversions to polyester (NDL: 86%, TCL: 97%) which is a significant improvement compared to previous investigations using Al–salen catalysts. In addition to high rates and overall conversions, the polymerizations were well controlled yielding polymers with molecular weights in the range 10–60 kg mol−1 together with broad dispersity values (Đ ∼ 2). The polymerization thermodynamic parameters were determined for tricosalactone. The polymerization is entropically driven and the equilibrium monomer conversion is 0.03 M (373 K). Overall, the findings demonstrate the relevance and potential for catalyst development in macrolactone ROP. It is likely that further optimization of catalyst and conditions will enable the more efficient production of long-chain aliphatic polyesters, as class of materials with thermal properties akin to those of polyethylene but which are distinctive by being degradable.Acknowledgements
The EPSRC (EP/L017393/1) and CSIRO-Imperial College London PhD Studentship are acknowledged for research funding. Financial support by the Stiftung Baden-Württemberg to T. W. and S. M. is acknowledged.References
- (a) M. A. Hillmyer and W. B. Tolman, Acc. Chem. Res., 2014, 47, 2390–2396 CrossRef CAS PubMed; (b) S. Slomkowski, S. Penczek and A. Duda, Polym. Adv. Technol., 2014, 25, 436–447 CrossRef CAS; (c) F. Stempfle, P. Ortmann and S. Mecking, Chem. Rev., 2016, 116, 4597–4641 CrossRef CAS PubMed; (d) J. M. Longo, M. J. Sanford and G. W. Coates, Chem. Rev., 2016, 116, 15167–15197 CrossRef CAS PubMed; (e) Y. Zhu, C. Romain and C. K. Williams, Nature, 2016, 540, 354–362 CrossRef CAS PubMed; (f) R. P. Brannigan and A. P. Dove, Biomater. Sci., 2017, 5, 9–21 RSC.
- L. Maisonneuve, T. Lebarbé, E. Grau and H. Cramail, Polym. Chem., 2013, 4, 5472–5517 RSC.
- (a) K. S. Bisht, L. A. Henderson, R. A. Gross, D. L. Kaplan and G. Swift, Macromolecules, 1997, 30, 2705–2711 CrossRef CAS; (b) I. van der Meulen, M. de Geus, H. Antheunis, R. Deumens, E. A. J. Joosten, C. E. Koning and A. Heise, Biomacromolecules, 2008, 9, 3404–3410 CrossRef CAS PubMed.
- C. Liu, F. Liu, J. Cai, W. Xie, T. E. Long, S. R. Turner, A. Lyons and R. A. Gross, Biomacromolecules, 2011, 12, 3291–3298 CrossRef CAS PubMed.
- (a) A. Kumar, B. Kalra, A. Dekhterman and R. A. Gross, Macromolecules, 2000, 33, 6303–6309 CrossRef CAS; (b) A. Kumar, K. Garg and R. A. Gross, Macromolecules, 2001, 34, 3527–3533 CrossRef CAS; (c) M. L. Focarete, M. Scandola, A. Kumar and R. A. Gross, J. Polym. Sci., Part B: Polym. Phys., 2001, 39, 1721–1729 CrossRef CAS; (d) Z. Z. Jiang, H. Azim, R. A. Gross, M. L. Focarete and M. Scandola, Biomacromolecules, 2007, 8, 2262–2269 CrossRef CAS PubMed; (e) J. L. Cai, B. S. Hsiao and R. A. Gross, Polym. Int., 2009, 58, 944–953 CrossRef CAS; (f) M. Eriksson, L. Fogelstrom, K. Hult, E. Malmstrom, M. Johansson, S. Trey and M. Martinelle, Biomacromolecules, 2009, 10, 3108–3113 CrossRef CAS PubMed; (g) J. L. Cai, C. Liu, M. M. Cai, J. Zhu, F. Zuo, B. S. Hsiao and R. A. Gross, Polymer, 2010, 51, 1088–1099 CrossRef CAS; (h) M. de Geus, I. van der Meulen, B. Goderis, K. van Hecke, M. Dorschu, H. van der Werff, C. E. Koning and A. Heise, Polym. Chem., 2010, 1, 525–533 RSC; (i) J. L. Cai, B. S. Hsiao and R. A. Gross, Macromolecules, 2011, 44, 3874–3883 CrossRef CAS; (j) I. van der Meulen, E. Gubbels, S. Huijser, R. Sablong, C. E. Koning, A. Heise and R. Duchateau, Macromolecules, 2011, 44, 4301–4305 CrossRef CAS; (k) M. P. F. Pepels, M. R. Hansen, H. Goossens and R. Duchateau, Macromolecules, 2013, 46, 7668–7677 CrossRef CAS; (l) M. P. F. Pepels, P. Soulje, R. Peters and R. Duchateau, Macromolecules, 2014, 47, 5542–5550 CrossRef CAS; (m) L. Jasinska-Walc, M. R. Hansen, D. Dudenko, A. Rozanski, M. Bouyahyi, M. Wagner, R. Graf and R. Duchateau, Polym. Chem., 2014, 5, 3306–3310 RSC; (n) L. Jasinska-Walc, M. Bouyahyi, A. Rozanski, R. Graf, M. R. Hansen and R. Duchateau, Macromolecules, 2015, 48, 502–510 CrossRef CAS; (o) M. P. F. Pepels, L. E. Govaert and R. Duchateau, Macromolecules, 2015, 48, 5845–5854 CrossRef CAS; (p) M. P. F. Pepels, W. P. Hofman, R. Kleijnen, A. B. Spoelstra, C. E. Koning, H. Goossens and R. Duchateau, Macromolecules, 2015, 48, 6909–6921 CrossRef CAS; (q) M. P. F. Pepels, R. A. C. Koeken, S. J. J. van der Linden, A. Heise and R. Duchateau, Macromolecules, 2015, 48, 4779–4792 CrossRef CAS; (r) R. Todd, S. Tempelaar, G. Lo Re, S. Spinella, S. A. McCallum, R. A. Gross, J. M. Raquez and P. Dubois, ACS Macro Lett., 2015, 4, 408–411 CrossRef CAS; (s) S. Spinella, M. Ganesh, G. Lo Re, S. Zhang, J. M. Raquez, P. Dubois and R. A. Gross, Green Chem., 2015, 17, 4146–4150 RSC; (t) J. A. Wilson, S. A. Hopkins, P. M. Wright and A. P. Dove, Biomacromolecules, 2015, 16, 3191–3200 CrossRef CAS PubMed; (u) J. A. Wilson, S. A. Hopkins, P. M. Wright and A. P. Dove, Macromolecules, 2015, 48, 950–958 CrossRef CAS; (v) M. P. F. Pepels, F. van der Sanden, E. Gubbels and R. Duchateau, Macromolecules, 2016, 49, 4441–4451 CrossRef CAS; (w) J. A. Wilson, S. A. Hopkins, P. M. Wright and A. P. Dove, ACS Macro Lett., 2016, 5, 346–350 CrossRef CAS; (x) S. Rutkowski, A. Zych, M. Przybysz, M. Bouyahyi, P. Sowinski, R. Koevoets, J. Haponiuk, R. Graf, M. R. Hansen, L. Jasinska-Walc and R. Duchateau, Macromolecules, 2017, 50, 107–122 CrossRef CAS; (y) J. Fernández, H. Amestoy, H. Sardon, M. Aguirre, A. L. Varga and J.-R. Sarasua, J. Mech. Behav. Biomed. Mater., 2016, 64, 209–219 CrossRef PubMed.
- (a) R. Nomura, A. Ueno and T. Endo, Macromolecules, 1994, 27, 620–621 CrossRef CAS; (b) A. Duda, A. Kowalski, S. Penczek, H. Uyama and S. Kobayashi, Macromolecules, 2002, 35, 4266–4270 CrossRef CAS; (c) T. Fuoco, A. Meduri, M. Lamberti, V. Venditto, C. Pellecchia and D. Pappalardo, Polym. Chem., 2015, 6, 1727–1740 RSC; (d) T. Witt, M. Häußler and S. Mecking, Macromol. Rapid Commun., 2017, 38, 1600638 CrossRef PubMed; (e) T. Witt and S. Mecking, Green Chem., 2013, 15, 2361–2364 RSC.
- D. K. Schneiderman and M. A. Hillmyer, Macromolecules, 2016, 49, 2419–2428 CrossRef CAS.
- (a) L. van der Mee, F. Helmich, R. de Bruijn, J. Vekemans, A. R. A. Palmans and E. W. Meijer, Macromolecules, 2006, 39, 5021–5027 CrossRef CAS; (b) B. Manzini, P. Hodge and A. Ben-Haida, Polym. Chem., 2010, 1, 339–346 RSC; (c) S. Strandman, J. E. Gautrot and X. X. Zhu, Polym. Chem., 2011, 2, 791–799 RSC; (d) M. P. F. Pepels, I. Hermsen, G. J. Noordzij and R. Duchateau, Macromolecules, 2016, 49, 796–806 CrossRef CAS.
- (a) H. Kikuchi, H. Uyama and S. Kobayashi, Polym. J., 2002, 34, 835–840 CrossRef CAS; (b) M. A. J. Veld, A. R. A. Palmans and E. W. Meijer, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 5968–5978 CrossRef CAS.
- (a) Z. Jedliński, M. Juzwa, G. Adamus, M. Kowalczuk and M. Montaudo, Macromol. Chem. Phys., 1996, 197, 2923–2929 CrossRef; (b) Z. Zhong, P. J. Dijkstra and J. Feijen, Macromol. Chem. Phys., 2000, 201, 1329–1333 CrossRef CAS; (c) Y. Wang and M. Kunioka, Macromol. Symp., 2005, 224, 193–205 CrossRef CAS; (d) Y. Nakayama, N. Watanabe, K. Kusaba, K. Sasaki, Z. Cai, T. Shiono and C. Tsutsumi, J. Appl. Polym. Sci., 2011, 121, 2098–2103 CrossRef CAS; (e) M. P. F. Pepels, M. Bouyahyi, A. Heise and R. Duchateau, Macromolecules, 2013, 46, 4324–4334 CrossRef CAS; (f) M. Bouyahyi and R. Duchateau, Macromolecules, 2014, 47, 517–524 CrossRef CAS; (g) J. A. Wilson, S. A. Hopkins, P. M. Wright and A. P. Dove, Polym. Chem., 2014, 5, 2691–2694 RSC; (h) T. Fuoco, A. Meduri, M. Lamberti, V. Venditto, C. Pellecchia and D. Pappalardo, Polym. Chem., 2015, 6, 1727–1740 RSC; (i) S. Naumann, P. B. V. Scholten, J. A. Wilson and A. P. Dove, J. Am. Chem. Soc., 2015, 137, 14439–14445 CrossRef CAS PubMed.
- (a) A. Pascual, J. R. Leiza and D. Mecerreyes, Eur. Polym. J., 2013, 49, 1601–1609 CrossRef CAS; (b) A. Pascual, H. Sardon, A. Veloso, F. Ruiperez and D. Mecerreyes, ACS Macro Lett., 2014, 3, 849–853 CrossRef CAS; (c) S. Naumann, A. W. Thomas and A. P. Dove, ACS Macro Lett., 2016, 5, 134–138 CrossRef CAS; (d) V. Ladelta, P. Bilalis, Y. Gnanou and N. Hadjichristidis, Polym. Chem., 2017, 8(3), 511–515 RSC.
- M. P. F. Pepels, I. Hermsen, G. J. Noordzij and R. Duchateau, Macromolecules, 2016, 49, 796–806 CrossRef CAS.
- (a) C. Bakewell, T.-P.-A. Cao, N. Long, X. F. Le Goff, A. Auffrant and C. K. Williams, J. Am. Chem. Soc., 2012, 134, 20577–20580 CrossRef CAS PubMed; (b) C. Bakewell, T.-P.-A. Cao, X. F. Le Goff, N. J. Long, A. Auffrant and C. K. Williams, Organometallics, 2013, 32, 1475–1483 CrossRef CAS; (c) C. Bakewell, A. J. P. White, N. J. Long and C. K. Williams, Angew. Chem., Int. Ed., 2014, 53, 9226–9230 CrossRef CAS PubMed; (d) C. Bakewell, A. J. P. White, N. J. Long and C. K. Williams, Inorg. Chem., 2015, 54, 2204–2212 CrossRef CAS PubMed; (e) D. Myers, A. J. P. White, C. M. Forsyth, M. Bown and C. K. Williams, Angew. Chem., Int. Ed., 2017, 56, 5277–5282 CrossRef CAS PubMed.
- (a) B. V. Lebedev, A. A. Yevstropov and Y. G. Kiparisova, Vysokomol. Soedin., Ser. A, 1983, 25, 1679 Search PubMed; (b) A. Duda and A. Kowalski, Thermodynamics and Kinetics of Ring-Opening Polymerization, in Handbook of Ring-Opening Polymerization, ed. P. Dubois, O. Coulembier and J.-M. Raque, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2009, pp. 1–51 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental section, NMR spectra and polymer characterization data. See DOI: 10.1039/c7py00985b |
| This journal is © The Royal Society of Chemistry 2017 |