
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Chemoenzymatic synthesis of a peptide containing nylon monomer units for thermally processable peptide material application†
Kenjiro
Yazawa
a,
Joan
Gimenez-Dejoz
a,
Hiroyasu
Masunaga
bc,
Takaaki
Hikima
c and
Keiji
Numata
 *a
*a
aEnzyme Research Team, RIKEN Center for Sustainable Resource Science, 2-1 Hirosawa, Wako-shi, Saitama 351-0198, Japan. E-mail: keiji.numata@riken.jp
bJapan Synchrotron Radiation Research Institute, 1-1-1, Kouto, Sayo-cho, Sayo-gun, Hyogo 679-5198, Japan
cRIKEN SPring-8 Center, RIKEN 1-1-1 Kouto, Sayo-cho, Sayo-gun, Hyogo 679-5198, Japan
First published on 23rd May 2017
Abstract
Peptides exhibit no melting behavior during heating due to their strong intermolecular hydrogen bonds, which limits the use of peptides as a bulk material. Here, we added a melting point to peptides by introducing nylon monomer units via papain-catalyzed chemoenzymatic synthesis. The peptides containing 14 mol% nylon units had a melting point of approximately 200 °C.
Peptides, which are composed of amino acids with various side chain groups, have gathered attention as functional materials for use in tissue engineering and drug/gene delivery systems due to their self-assembly ability and biological activities.1–5 However, peptides have not yet been used as a structural material on a bulk scale, even though structural proteins, such as silk and collagen, are used as structural materials in nature.6–8 One of the challenges for peptidic structural material is that a peptide cannot be processed using thermal treatment.9 Peptides exhibit thermal degradation prior to melting during the heating process due to strong intermolecular interactions (i.e., hydrogen bonds).9
To induce a melting point to a peptide, the number of intermolecular hydrogen bonds must be reduced to a level similar to those in nylon and polyamide. One solution is copolymerization of amino acids and non-natural units to disturb internal hydrogen bonds. To synthesize peptides containing non-natural amino acids, solid-phase peptide synthesis (SPPS) and ring-opening polymerization of amino acid N-carboxyanhydrides (NCAs) have been widely employed.10 However, in SPPS, the peptide yield is relatively low, and laborious protection–deprotection steps along with harsh reaction conditions are neccesary.11 In the polymerization of NCAs, nylon units, which have different carbon numbers from the peptide, cannot be efficiently polymerized.12
Chemoenzymatic peptide synthesis, which is stereoselective and atom economical, is catalyzed by protease and has the potential to introduce unnatural amino acids.13–15 Papain possesses broad substrate specificity and exhibits endopeptidase, amidase, and esterase activities.15,16 Papain was previously used to introduce a nylon 4-ethyl ester (nylon4Et) at the C-terminus of oligo(L-glutamic acid ethyl ester).17 Therefore, papain is expected to copolymerize amino acids and nylon units. In this study, we copolymerize an ethyl ester of L-leucine (LeuEt), which is used as a model amino acid, and nylon monomer alkyl esters with four carbon chain lengths (i.e., methyl- and ethyl-esters of nylon 1, nylon 3, nylon 4 and nylon 6) (Fig. 1a and S1†). The copolymers of amino acids and nylons are expected to exhibit melting behavior due to reduced intermolecular hydrogen bonding.
 | ||
| Fig. 1 Reaction scheme for the papain-catalyzed chemoenzymatic synthesis of oligo(LeuEt-co-nylon4Me) (a). MALDI-TOF MS (b) and 1H NMR (c) spectra of oligo(LeuEt-co-nylon4Me). Peaks with blue circles denote oligo(LeuEt), and peaks with orange squares denote oligo(LeuEt-co-nylon4Me). | ||
The papain-catalyzed chemoenzymatic synthesis using LeuEt and methyl ester of nylon 4 monomer (nylon4Me) resulted in a white precipitate with a yield of 14 ± 2 wt% based on the initial monomer quantities. The low yield is inherent to the copolymerization of nylon monomers. Based on the monomeric composition of nylon units and monomer feeding ratios, the recognition rate of the nylon4 unit by papain was estimated to be significantly lower than that of L-leucine. Therefore, most of the nylon monomers were not polymerized by papain and remained in the buffer solution, which resulted in the low reaction yield. The chemical structure of the product was confirmed by matrix-assisted laser desorption/ionization time-of flight mass spectrometry (MALDI-TOF MS) and 1H nuclear magnetic resonance (1H NMR) (Fig. 1b and c). In the MALDI-TOF MS result, peaks with intervals of 113 g mol−1 were detected, indicating that oligo(LeuEt) was synthesized with a degree of polymerization (DP) that ranged from 5 to 9. Furthermore, the other peaks with an interval of 85 g mol−1 apart from the peaks of oligo(LeuEt) indicated that LeuEt and nylon4Me were copolymerized. The molecular weight of oligo(LeuEt-co-nylon4Me) ranged from 719 to 1058 g mol−1. Based on the MALDI-TOF MS results, we detected both methyl and ethyl groups at the C-terminus, indicating that the nylon 4 unit was successfully copolymerized in the middle of the sequence rather than at the C-terminus. The product was obtained as a precipitate, which prevents elongation of the peptide chain according to a previous study on the papain-catalyzed polymerization of L-alanine ethyl ester.18 The oligo(LeuEt-co-nylon4Me) was characterized by 1H NMR and gradient correlation spectroscopy (g-COSY) NMR (Fig. 1c and S2†), and peaks that originated from nylon4Me were detected. We assigned the peaks above 7.5 ppm to amine-related peaks derived from L-leucine, which are shown as G (Fig. S2†). This is because the local magnetic environment of the three amines is slightly different by the position, namely, at the N-terminus, at the C-terminus, or at the middle sequence. Oligo(LeuEt-co-nylon4Me) was composed of 13 ± 1 mol% nylon 4 units based on the integral values of the 1H NMR peaks corresponding to the α-proton of LeuEt and γ-proton of nylon 4 (i.e., F and H, respectively) (Fig. 1c).
The formation of an acyl-enzyme intermediate in the chemoenzymatic synthesis depends largely on the ester group19 and the carbon chain length of the substrate monomers.20 To investigate the effect of the ester group and carbon chain length of the substrates on the catalytic efficiency of papain with nylon monomers, the papain-catalyzed syntheses using various nylon monomers (e.g., nylon 1 ethyl ester (nylon1Et), nylon 3 methyl ester (nylon3Me), nylon 3 ethyl ester (nylon3Et), nylon 4 ethyl ester (nylon4Et) and nylon 6 methyl ester (nylon6Me)) were also performed (each chemical structure is shown in Fig. S1†). Nylon1Et was not polymerized by papain even with LeuEt. Based on a comparison of nylon3Et and nylon3Me as well as nylon4Et and nylon4Me (Table 1, Fig. S2–S7†), the resulting yields and compositions were not significantly different, suggesting that the methyl ester and ethyl ester groups of the nylons did not change the affinity with respect to papain. Furthermore, nylon6Me was also evaluated, and a yield of 11 ± 1 wt% was obtained. Based on the MALDI-TOF MS spectrum (Fig. S8a†), we could not confirm the existence of nylon6Me because the molecular weights of the L-leucine unit and nylon6Me are identical (i.e., 113 g mol−1). Therefore, the monomeric composition of nylon6Me was confirmed to be 13 ± 2 mol% based on the NMR spectra (Fig. S8b and S9†). An oligo(LeuEt) homooligomer was also synthesized from LeuEt as a control and confirmed by NMR and MALDI-TOF MS (Fig. S10 and 11†). The water solubility of oligo(LeuEt-co-nylon) was not significantly different from that of oligo(LeuEt). We could not synthesize the oligo(nylon) homooligomer by the papain-catalyzed chemoenzymatic polymerization. The results of the chemoenzymatic synthesis are listed in Table 1. The carbon chain length and even–odd effects of the nylon monomers did not have a significant effect on the peptide synthesis using papain.
| Product | Yield/wt% | Composition of nylon units/mol% | DPMALDI (DPNMR) | Crystallinity/% | Crystallite size/nm |
|---|---|---|---|---|---|
| Oligo(LeuEt) | 15 ± 1 | Not detected | 5–8 (7) | 71.6 ± 0.7 | 10.4 ± 0.2 |
| Oligo(LeuEt-co-nylon3Et) | 13 ± 2 | 14 ± 1 | 6–9 (7) | 67.5 ± 0.7 | 7.0 ± 1.0 |
| Oligo(LeuEt-co-nylon3Me) | 10 ± 1 | 14 ± 1 | 6–9 (7) | 66.6 ± 1.0 | 6.6 ± 0.2 |
| Oligo(LeuEt-co-nylon4Et) | 13 ± 1 | 14 ± 2 | 5–9 (7) | 67.4 ± 1.0 | 6.9 ± 0.1 |
| Oligo(LeuEt-co-nylon4Me) | 14 ± 2 | 13 ± 1 | 5–9 (7) | 67.5 ± 0.7 | 7.0 ± 0.1 |
| Oligo(LeuEt-co-nylon6Me) | 11 ± 1 | 13 ± 2 | 6–9 (7) | 67.5 ± 0.6 | 7.1 ± 0.2 |
To explain these results, we performed molecular docking simulations using nylon1Et, nylon4Et and nylon6Me as the substrates (Fig. 2 and Table S1†). The estimated free energy of binding of the best conformation for nylon1Et, nylon4Et and nylon6Me to the catalytic site of papain was −3.3, −3.9 and −4.0 kcal mol−1, respectively. This result indicates that the binding of nylon1Et was significantly weaker than that of the other two nylon units. The molecular docking simulations indicated a similar binding site for nylon1Et and nylon6Me. However, the binding direction of nylon1Et was different from that of nylon6Me. The amine group of nylon1Et was directed to Cys25, whereas the amine group of nylon6Me was located near Val157. We created two-dimensional diagrams and evaluated the docking interactions between the nylon molecules and the catalytic site of papain using the PoseView program.21 The diagrams indicated that nylon1Et did not interact with the catalytic site of papain. On the other hand, nylon4Et interacted with Trp177 and Cys22 by hydrogen bonding (Fig. S12a†). Nylon6Me interacted with Gly66 and Val157 by hydrogen bonding and interacted with Val133 by hydrophobic interactions (Fig. S12b†). These extra interactions stabilized the binding of the nylon4Et and nylon6Me in the catalytic site of papain. These different binding affinities and states of nylon monomers to the catalytic site of papain reasonably explain why papain cannot polymerize nylon1Et but does nylon4Et and nylon6Me.
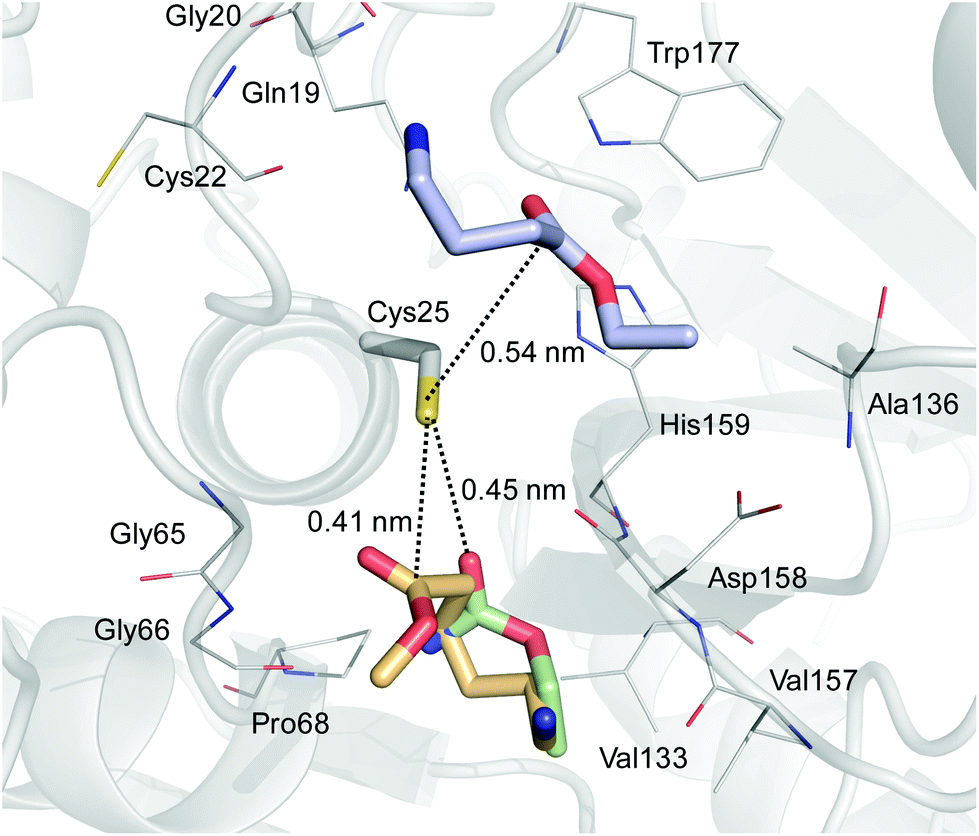 | ||
| Fig. 2 Best binding conformation of nylon units to the catalytic site of papain. Papain is displayed as a gray cartoon. The Cys25 catalytic residue is shown as a gray stick, and the residues implicated in the binding of nylon units are presented as gray lines. Nylon1Et, nylon4Et and nylon6Me are shown as green, blue and orange sticks, respectively. The distance between the carbon atom of the carbonyl group and the thiol group of Cys25 is shown as dashed lines. | ||
To evaluate the effects of the presence of nylon units on the thermal properties of oligo(LeuEt-co-nylon), we conducted differential scanning calorimetry (DSC) and thermogravimetric analysis (TGA) (Fig. 3). The DSC profiles in the first heat scan indicated that the endothermic peak ranged from 50 °C to 120 °C (Fig. 3a), and this peak corresponded to water removal from the peptide based on the TGA results.22 The water content was calculated based on the initial weight decrease in the TGA profiles (Fig. S13a and Table S2†). Oligo(LeuEt) contained less water than oligo(LeuEt-co-nylon), suggesting that the water content was not related to the amide bond density but to more solid-state structures. The endothermic peak of oligo(LeuEt-co-nylon) was observed from 170 °C to 250 °C. Oligo(LeuEt) did not exhibit that kind of endothermic peak, indicating that oligo(LeuEt-co-nylon) had a melting point from 170 °C to 250 °C due to the presence of the nylon units. The peak-top temperature of the endothermic transition decreased in the following order: oligo(LeuEt-co-nylon6Me) > oligo(LeuEt-co-nylon3Me) = oligo(LeuEt-co-nylon3Et) > oligo(LeuEt-co-nylon4Me) = oligo(LeuEt-co-nylon4Et). According to a previous study using ab initio quantum calculations of the crystal structures of nylon, the methylene chains of nylon exist in a parallel or antiparallel orientation depending on the interplay between the hydrogen bonds and methylene packing.23 The oligo(LeuEt-co-nylon) synthesized in this study contained approximately 14 mol% of nylon units. The nylon composition could contribute to the orientation of the hydrogen-bond networks, resulting in different peak-top temperatures in the melting transition. In addition, an endothermic peak was observed above 270 °C due to the thermal degradation of the peptide according to the TGA results (Fig. 3b). The thermal degradation temperatures at 5% and 10% weight loss for oligo(LeuEt) and oligo(LeuEt-co-nylon) are summarized in Fig. S13b and Table S2.† The effect of the carbon chain length of the nylon monomers on the thermal properties was not significant. Based on the derivative plot of the TGA data (Fig. 3d), the rapid weight decrease arising from thermal degradation began at 270 °C. Remarkably, a decrease in weight was not detected from 170 °C to 250 °C in the TGA profiles, which confirmed that the endothermic peak detected in the DSC measurement was derived from a melting transition rather than thermal degradation (Fig. 3a).
 | ||
| Fig. 3 (a) DSC, (b) TGA, (c) magnified TGA plot, and (d) first derivative of the TGA plot of (i) oligo(LeuEt-co-nylon3Et), (ii) oligo(LeuEt-co-nylon3Me), (iii) oligo(LeuEt-co-nylon4Et), (iv) oligo(LeuEt-co-nylon4Me), (v) oligo(LeuEt-co-nylon6Me), and (vi) oligo(LeuEt) at 10 °C min−1. | ||
To investigate the effect of the nylon monomer units on the crystal structure and crystalline state of the (LeuEt-co-nylon), wide-angle X-ray scattering (WAXS) measurements were performed. The one-dimensional profile is plotted in Fig. 4a as the circular average of the scattering intensity of the corresponding two-dimensional profile (Fig. S14†).24 The WAXS data indicated three sharp peaks due to the (020), (210), and (211) planes with d-spacing values of 1.07 nm, 0.45 nm, and 0.36 nm, respectively, which confirm the formation of beta-sheet crystals according to the WAXS data of polyleucine.25 The peak originated from a crystal region of nylon was not detected. This result indicates that oligo(LeuEt-co-nylon) could not form a crystalline region of nylon in this study. The degree of polymerization of oligo(LeuEt-co-nylon) was approximately 7, and then the monomeric composition of the nylon unit in oligo(LeuEt-co-nylon) was approximately 13 mol%. Thus, the length of nylon sequences was not enough to form nylon crystals, even though nylon polymers are crystalline. The crystallinities of oligo(LeuEt) and oligo(LeuEt-co-nylon) are summarized in Fig. S15a† and Table 1. The crystallinity of oligo(LeuEt) was 71.6 ± 0.7%. However, the crystallinity of oligo(LeuEt-co-nylon) was slightly lower. Based on the water content (Fig. S13a†), the decrease in crystallinity after the introduction of the nylon monomer units appeared to contribute to the water content because water molecules preferentially access the amorphous regions compared to the crystal regions.26 We calculated the crystallite size using the full width at half-maximum of the peaks corresponding to the beta-sheet crystal structure with a d-spacing value of 0.45 nm according to Scherrer's theory.27 The crystallite size of oligo(LeuEt) was 10.4 ± 0.2 nm. However, the crystallite size of oligo(LeuEt-co-nylon) was significantly smaller (i.e., approximately 7.0 nm) (Fig. S15b† and Table 1). This result indicates that the presence of the nylon monomer units prevented the growth of crystallites.
 | ||
| Fig. 4 (a) One-dimensional WAXS data of (i) oligo(LeuEt), (ii) oligo(LeuEt-co-nylon3Et), (iii) oligo(LeuEt-co-nylon3Me), (iv) oligo(LeuEt-co-nylon4Et), (v) oligo(LeuEt-co-nylon4Me), and (vi) oligo(LeuEt-co-nylon6Me). (b) Temperature-dependent WAXS profiles of oligo(LeuEt-co-nylon4Et). | ||
The IR spectra of oligo(LeuEt) and oligo(LeuEt-co-nylon) were measured to evaluate the effect of nylon monomers on hydrogen bond formation (Fig. S16†). The IR spectra indicated the N–H stretching at around 3300 cm−1, which was assigned to the hydrogen bonding between amide groups in the peptide backbone (Fig. S16†).28 The IR spectra also showed the N–H stretching at around 3700 cm−1,28 which was assigned to the hydrogen bonding between amide groups in the peptide backbone and water molecules in the case of oligo(LeuEt-co-nylon). The hydrogen bonding was converted from amide–amide to water–amide bonding by the presence of nylon units. Therefore, the introduction of nylon units into oligo(LeuEt) disturbed the intermolecular hydrogen bonding of the peptide.
Furthermore, we performed temperature-dependent WAXS to investigate the effect of the nylon monomer units on the relationship between the structural and thermal properties of oligo(LeuEt-co-nylon) (Fig. 4b and S17–S22†). As a representative result, one-dimensional temperature-dependent WAXS profiles of oligo(LeuEt-co-nylon4Et) are shown in Fig. 4b. The change in the crystallinity as a function of temperature (Fig. S23†) indicates that a gradual decrease in the crystallinity occurs from 170 °C to 250 °C followed by a sharp decrease at temperatures higher than 250 °C. The same trend was observed for the other oligo(LeuEt-co-nylon) samples. Because the DSC results indicated a melting point that ranged from 170 °C to 250 °C in Fig. 3a, the first gradual decrease in crystallinity originated from the melting transition of oligo(LeuEt-co-nylon). The presence of nylon monomer units disturbed the molecular packing and efficient crystallization of the peptide main chains, inducing a melting transition. After the melting transition, small peaks derived from beta-sheet crystals remained above 200 °C, which is most likely due to the crystalline region consisting of a homosequence of oligo(LeuEt) being highly packed and stable. Therefore, a melting point was not observed. For the oligo(LeuEt) homooligomer without nylon units, a slight decrease in crystallinity was observed from 150 °C to 250 °C followed by a sharp decrease in crystallinity above 250 °C. The decreases in crystallinity were due to the partial degradation of the oligo(LeuEt) crystal and the thermal degradation of the main chain of the peptide, respectively. These results are due to the endothermic peak (melting behavior) not being detected from 150 °C to 250 °C by DSC and the weight loss from the thermal degradation being detected above 250 °C by TGA.
In this study, we utilized a papain-catalyzed reaction under mild aqueous conditions to synthesize oligo(LeuEt-co-nylon) containing approximately 14 mol% nylon units. The oligo(LeuEt-co-nylon) exhibited a melting point, and the nylon unit prevented the growth of peptide crystals consisting of Leu units. The presence of the nylon monomer units most likely changed the density of the intermolecular hydrogen bonds, inducing the melting transition. Although we could induce a melting point in the peptide by adding nylon monomer units, the monomeric compositions of the nylon unit were still low (i.e., approximately 10 mol%). Despite the low reaction efficiency, our reaction system can be easily conducted under mild conditions and can prevent racemization of amino acids. The reaction conditions including the choice of enzyme catalyst for the chemoenzymatic synthesis should be optimized to increase both the reaction yield and monomeric compositions of the nylon units from the copolymerization. Our next step is to develop thermally processable structural peptide materials. Tsuchiya and Numata have recently reported a multi-block copolymer that has a crystal structure similar to that of spider silk.29 In combination with the methodology mentioned in this study, we will be able to synthesize a nylon-containing peptide that is thermally processable as well as a silk-like amino acid sequence. These peptide designs may provide new insight into peptide/protein-based thermally processable materials for use as structural materials in our daily life.
This work was supported by the Impulsing Paradigm Change through the Disruptive Technologies Program (ImPACT).
Notes and references
- J. A. Chuah, A. Matsugami, F. Hayashi and K. Numata, Biomacromolecules, 2016, 17, 3547–3557 CrossRef CAS PubMed.
- H. Cui, M. J. Webber and S. I. Stupp, Biopolymers, 2010, 94, 1–18 CrossRef CAS PubMed.
- E. Gazit, Chem. Soc. Rev., 2007, 36, 1263–1269 RSC.
- K. Numata and D. L. Kaplan, Adv. Drug Delivery Rev., 2010, 62, 1497–1508 CrossRef CAS PubMed.
- Y. M. Wong, H. Masunaga, J. A. Chuah, K. Sudesh and K. Numata, Biomacromolecules, 2016, 17, 3375–3385 CrossRef CAS PubMed.
- K. Numata, Polym. J., 2015, 47, 537–545 CrossRef CAS.
- J. D. van Beek, S. Hess, F. Vollrath and B. H. Meier, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 10266–10271 CrossRef CAS PubMed.
- C. Vepari and D. L. Kaplan, Prog. Polym. Sci., 2007, 32, 991–1007 CrossRef CAS PubMed.
- P. Cebe, X. Hu, D. L. Kaplan, E. Zhuravlev, A. Wurm, D. Arbeiter and C. Schick, Sci. Rep., 2013, 3, 1130 CrossRef PubMed.
- R. B. Merrifield, J. Am. Chem. Soc., 1963, 85, 2149–2154 CrossRef CAS.
- I. Coin, M. Beyermann and M. Bienert, Nat. Protocols, 2007, 2, 3247–3256 CAS.
- F. Guzman, S. Barberis and A. Illanes, Electron. J. Biotechnol., 2007, 10, 279–314 CAS.
- R. A. Gross, A. Kumar and B. Kalra, Chem. Rev., 2001, 101, 2097–2124 CrossRef CAS PubMed.
- J.-i. Kadokawa and S. Kobayashi, Curr. Opin. Chem. Biol., 2010, 14, 145–153 CrossRef CAS PubMed.
- K. Yazawa and K. Numata, Molecules, 2014, 19, 13755–13774 CrossRef PubMed.
- G. Li, V. K. Raman, W. Xie and R. A. Gross, Macromolecules, 2008, 41, 7003–7012 CrossRef CAS.
- K. Yazawa and K. Numata, Polymers, 2016, 8, 194 CrossRef.
- P. J. Baker and K. Numata, Biomacromolecules, 2012, 13, 947–951 CrossRef CAS PubMed.
- J. M. Ageitos, K. Yazawa, A. Tateishi, K. Tsuchiya and K. Numata, Biomacromolecules, 2016, 17, 314–323 CrossRef CAS PubMed.
- R. J. de Beer, B. Zarzycka, H. I. Amatdjais-Groenen, S. C. Jans, T. Nuijens, P. J. Quaedflieg, F. L. van Delft, S. B. Nabuurs and F. P. Rutjes, ChemBioChem, 2011, 12, 2201–2207 CrossRef CAS PubMed.
- K. Stierand, P. C. Maass and M. Rarey, Bioinformatics, 2006, 22, 1710–1716 CrossRef CAS PubMed.
- K. Yazawa, K. Ishida, H. Masunaga, T. Hikima and K. Numata, Biomacromolecules, 2016, 17, 1057–1066 CrossRef CAS PubMed.
- S. Dasgupta, W. B. Hammond and W. A. Goddard, J. Am. Chem. Soc., 1996, 118, 12291–12301 CrossRef CAS.
- A. P. Hammersley, S. O. Svensson, M. Hanfland, A. N. Fitch and D. Hausermann, High Pressure Research, 1996, 14, 235–248 CrossRef.
- R. A. Miranda, E. Finocchio, J. Llorca, F. Medina, G. Ramis, J. E. Sueiras and A. M. Segarra, Phys. Chem. Chem. Phys., 2013, 15, 15645–15659 RSC.
- R. M. Hodge, G. H. Edward and G. P. Simon, Polymer, 1996, 37, 1371–1376 CrossRef CAS.
- A. Monshi, M. R. Foroughi and M. R. Monshi, World J. Nano Sci. Eng., 2012, 02, 154–160 CrossRef.
- M. W. Urban and C. D. Craver, Structure-Property Relations in Polymers, American Chemical Society, 1993 Search PubMed.
- K. Tsuchiya and K. Numata, ACS Macro Lett., 2017, 6, 103–106 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, nylon monomers used in this study (Fig. S1); MALDI-TOF MS and 1H NMR spectra (Fig. S3, S4, S8, S10); g-COSY NMR spectra (Fig. S2, S5–S7, S9, S11); best docking interaction (Fig. S12); water content and thermal degradation temperature (Fig. S13); WAXS profiles (Fig. S14); crystallinity and crystallite size (Fig. S15); IR spectra (Fig. S16); temperature-dependent WAXS profiles (Fig. S17–S22); temperature-dependent crystallinity change (Fig. S23); estimated free energy of binding (Table S1); water content and thermal degradation temperature (Table S2). See DOI: 10.1039/c7py00770a |
| This journal is © The Royal Society of Chemistry 2017 |