
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The living anionic polymerization of activated aziridines: a systematic study of reaction conditions and kinetics†
Elisabeth
Rieger
,
Tassilo
Gleede
,
Katja
Weber
,
Angelika
Manhart
,
Manfred
Wagner
and
Frederik R.
Wurm
 *
*
Max-Planck-Institut für Polymerforschung (MPI-P), Ackermannweg 10, D-55128 Mainz, Germany. E-mail: wurm@mpip-mainz.mpg.de
First published on 6th April 2017
Abstract
“A living race” – polymerization kinetics of anionic polymerizations depends strongly on the solvent polarity and reactivity of the growing chain end. Both the carb- and oxyanionic polymerization is under control at the university lab and on the industrial level, however, no information for the aza-anionic polymerization of aziridines has been reported systematically. This work studies the polymerization of two activated aziridines (2-methyl-N-mesylaziridine (MsMAz) and 2-methyl-N-tosylaziridine (TsMAz)) by real-time 1H NMR spectroscopy. This technique allows monitoring the consumption of the monomer precisely during the polymerization under different conditions (temperature, solvent, initiator and counter-ion variation). From the experimental data, propagation rate constants (kp) were calculated and analyzed. The polymerization of MsMAz was monitored at different temperatures (20, 50, and 100 °C). The increase of temperature increases the speed of polymerization, but keeps the living behavior. Furthermore, the influence of different solvents on the polymerization speed was examined, proving solvating solvents such as DMSO and DMF as the fastest solvents. Two different initiators, the potassium salts of N,N′-(1,4-phenylenebis(methylene))dimethanesulfonamide (BnBis(NHMs)), the first bifunctional initiator for the AROP of aziridines, and of N-benzyl-sulfonamide (BnNHMs) were compared. The variation of the counter ions Li+, Na+, K+, and Cs+ (generated from the respective bis(trimethylsilyl)amide salts) proved successful polymerization of both monomers with all counter ions. Slight variations have been detected in the order: Cs+ > Li+ > Na+ > K+, which is in strong contrast for the AROP of epoxides, shows a strong gegenion-dependent kinetic profile. This allows the use of commercially available initiators, such as BuLi for the synthesis of PAz. With these results in hand, the azaanionic polymerization can be used as a valuable tool in the family of anionic polymerization for the preparation of structurally diverse polysulfonamides and polyamines under a broad variety of conditions, while maintaining the living behavior.
Introduction
The knowledge of polymerization kinetics allows us to construct complex polymeric architectures by different polymerization techniques. 60 years after the discovery of the living anionic polymerization, their solvent, counter ion and temperature dependencies are taught in introductory polymer classes. Conditions are known for the carb- and oxyanionic polymerization.1–3However, such detailed and fundamental investigations are missing for the living anionic ring-opening polymerization (AROP) of aziridines and will be presented in this work.
The azaanionic polymerization of activated aziridines was recently established.4–9 To enable anionic polymerization of aziridines, the acidic proton at the nitrogen needs to be substituted by an activating group, e.g. a sulfonamide group (Scheme 1). To date, only such activated aziridines undergo anionic polymerization, but also a few other aziridine-containing polymers have been prepared and studied as functional polymers for postmodification.10–14
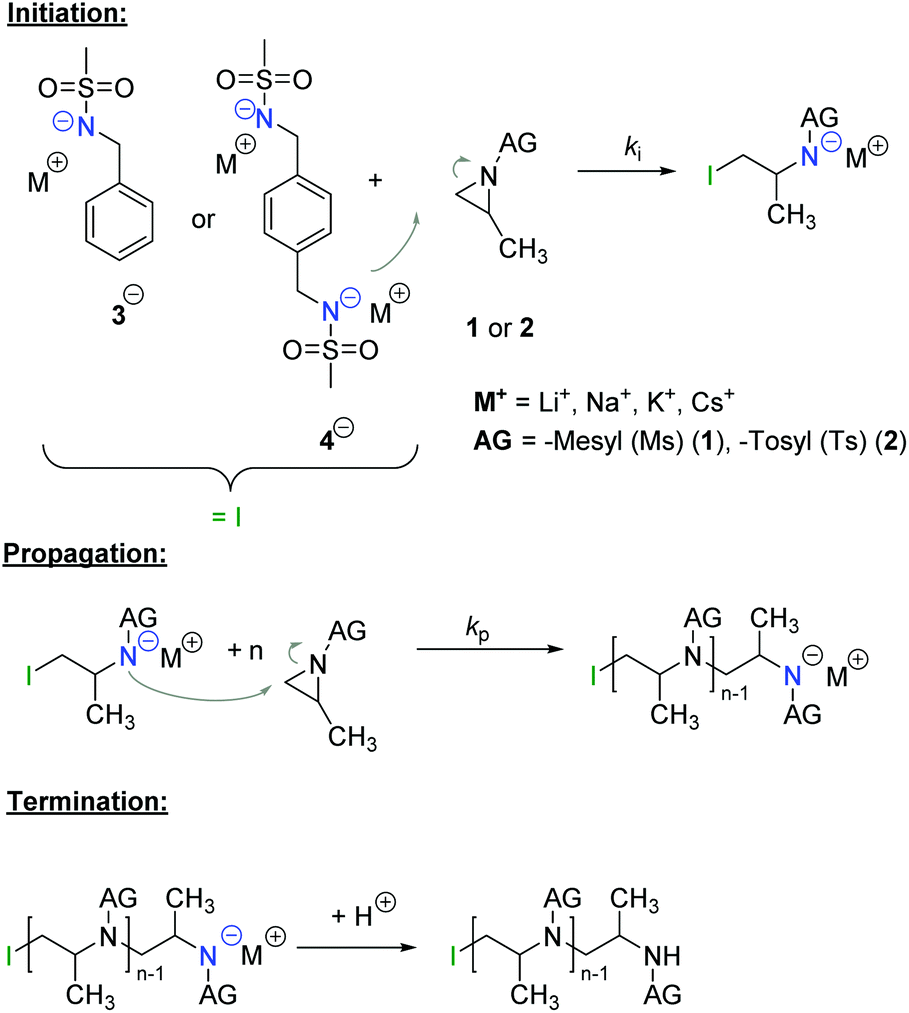 | ||
| Scheme 1 Mechanism of the living anionic ring-opening polymerization of activated aziridines (AG = activation group). | ||
The AROP of aziridines allows us to prepare well-defined poly(ethylene imine) derivatives.4,9 We have developed new monomers and initiator-systems during the last few years, expanding this still rather unexplored approach to polysulfonamides and amines.5–8,15,16
With a similar ring-strain of 111 kJ mol−1 for ethylene imine as for ethylene oxide (114 kJ mol−1), the anionic ring-opening polymerization should be feasible.17,18 In contrast to unsubstituted ethylene imine, which can only be polymerized via a cationic mechanism, leading to branched PEI (poly(ethylene imine)),19N-protected aziridines can also be polymerized anionically, due to their activating group. The sulfonamide substitutes the acidic proton at the aziridine and acts as an electron-withdrawing group. This results not only in the general possibility for anionic AROP, but further in different reactivities of the monomers, leading to sequential incorporation.7
Herein, 2-methyl-N-mesyl-aziridine (MsMAz, 1) and 2-methyl-N-tosylaziridine (TsMAz, 2) were used to elucidate the polymerization kinetics under different conditions. The results from this study will allow us to use activated aziridines for the preparation of well-defined polymer architectures by anionic polymerization in the future.
Experimental section
Chemicals
All solvents and reagents were purchased from Sigma-Aldrich, Acros Organics or Fluka and used as received unless otherwise mentioned. All deuterated solvents were purchased from Deutero GmbH and were distilled from CaH2 or sodium and stored in a glovebox prior to use. All monomers and initiators were dried extensively by azeotropic distillation with benzene prior to polymerization. Cesium bis(trimethylsilyl)amide was synthesized according to a literature protocol.4,20 2-Methyl-N-mesyl-aziridine (MsMAz, 1), 2-methyl-N-tosylaziridine (TsMAz, 2) and N-benzyl methanesulfonamide (BnNHMs, 3) were synthesized according to our previously published protocol.7 The synthesis of the bifunctional initiator (BnBis(NHMs), 4) can be found in the ESI.†Methods
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :[I]0 = 30:1, if not otherwise stated. The initiator-solution in 1 mL deuterated solvent was prepared separately (exemplarily for the polymerization of MsMAz (1) (70 mg) in 0.7 mL DMF-d7 and the initiator-system: BnNHMs (3) ((32.0 mg), potassium bis(trimethylsilyl)-amide (KHMDS) (34.4 mg) in 1 mL DMF-d7). A conventional NMR-tube was filled with the reaction mixture and sealed with a rubber-septum. Prior to initiation, the pure monomer–solvent mixture was measured at 50 °C. From the stock solution of the initiator, 100 μL were added to the monomer mixture, mixed quickly and inserted into the spectrometer. All 1H NMR kinetics were recorded using a Bruker Avance III 700. All spectra were referenced internally to residual proton signals of the deuterated solvent dimethylformamide-d7 at 8.03 ppm, dimethylsulfoxide-d6 at 2.50 ppm, tetrahydrofuran-d8 at 3.58 ppm, benzene-d6 7.16 ppm, and cyclohexane-d12 at 1.38 ppm. The π/2-pulse for the proton measurements was 13.1 μs. The spectra of the polymerizations were recorded at 700 MHz with 32 scans (equal to 404 s (acquisition time of 2.595 s and a relaxation time of 10 s after every pulse)) over a period of at least 3 h. No B-field optimizing routine was used over the kinetic measurement time. The spin–lattice relaxation rate (T1) of the ring-protons, which are used afterwards for integration, was measured before the kinetic run with the inversion recovery method.21
:[I]0 = 30:1, if not otherwise stated. The initiator-solution in 1 mL deuterated solvent was prepared separately (exemplarily for the polymerization of MsMAz (1) (70 mg) in 0.7 mL DMF-d7 and the initiator-system: BnNHMs (3) ((32.0 mg), potassium bis(trimethylsilyl)-amide (KHMDS) (34.4 mg) in 1 mL DMF-d7). A conventional NMR-tube was filled with the reaction mixture and sealed with a rubber-septum. Prior to initiation, the pure monomer–solvent mixture was measured at 50 °C. From the stock solution of the initiator, 100 μL were added to the monomer mixture, mixed quickly and inserted into the spectrometer. All 1H NMR kinetics were recorded using a Bruker Avance III 700. All spectra were referenced internally to residual proton signals of the deuterated solvent dimethylformamide-d7 at 8.03 ppm, dimethylsulfoxide-d6 at 2.50 ppm, tetrahydrofuran-d8 at 3.58 ppm, benzene-d6 7.16 ppm, and cyclohexane-d12 at 1.38 ppm. The π/2-pulse for the proton measurements was 13.1 μs. The spectra of the polymerizations were recorded at 700 MHz with 32 scans (equal to 404 s (acquisition time of 2.595 s and a relaxation time of 10 s after every pulse)) over a period of at least 3 h. No B-field optimizing routine was used over the kinetic measurement time. The spin–lattice relaxation rate (T1) of the ring-protons, which are used afterwards for integration, was measured before the kinetic run with the inversion recovery method.21
Results and discussion
The AROP of N-activated aziridines can be initiated by a deprotonated secondary sulfonamide, which can be formed in situ e.g. by the use of bis(trimethylsilyl)amides. This freshly prepared nucleophile opens the ring most likely at its less substituted side and thus forms the propagating sulfonamide anion.4 Propagation occurs via nucleophilic attack of this azaanion at the next monomer and it continues, as long as the monomer is available. As it is a living polymerization, no termination occurs, in the absence of impurities, and controlled termination by adding an electrophile is possible (Scheme 1).4,7All propagation rates are calculated from the linear first-order kinetic plots, using the equations shown below for living polymerizations (eqn (I) and (II)). Eqn (I) shows the reduction of the monomer concentration [M] over time, [P−] stands for the number of growing chains and is equivalent to the initiator concentration [I], because in the living anionic polymerization (LAP) each initiating site starts a growing polymer chain ([P−] = [I]). Integration gives the linear eqn (II), therefore plotting ln([M]0/[M]t) versus time results in a straight line, where the slope (kapp) gives the propagation rate (kp), when divided by the original initiator concentration [I]0. First-order kinetics, which are required for simplification of the equation, are evidenced if ln([M]0/[M]t) increases linearly and has already been reported for the LAP of some sulfonyl aziridines.2,4
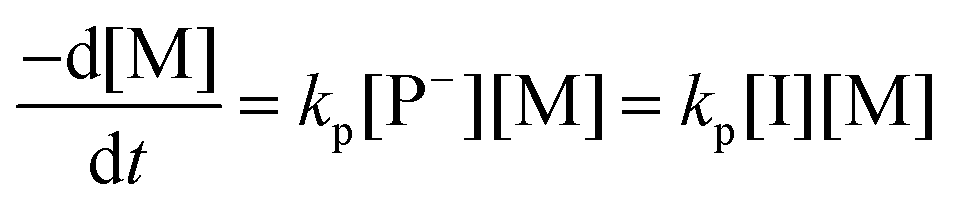 | (I) |
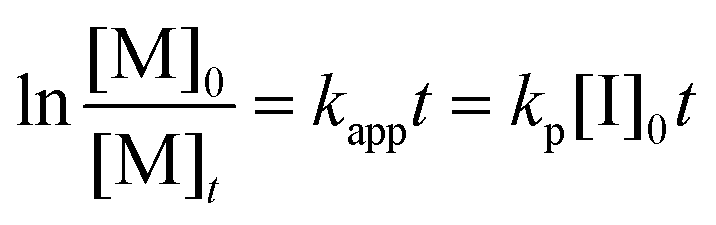 | (II) |
However, a systematic kinetic investigation of the AROP has not been reported to date. Here we chose 2-methyl-N-mesylaziridine (MsMAz, 1) and 2-methyl-N-tosyl-aziridine (TsMAz, 2) as two monomers with different activating groups that alter the monomer reactivity, to study the polymerization in different solvents, namely dimethylsulfoxide (DMSO-d6), dimethylformamide (DMF-d7), tetrahydrofuran (THF-d8), benzene-d6 and cyclohexane-d12 (CyHex) at a constant temperature of 50 °C. In DMF-d7 also different temperatures (20, 50, and 100 °C) were investigated. Two sulfonamide-based initiators were used and the influence of the counter ions on propagation rates was studied. N-Benzyl methanesulfonamide (BnNHMs, 3), deprotonated by lithium (LiMDS), sodium (NaMDS), potassium (KMDS) and cesium (CsMDS) bis(trimethylsilyl)amide, was used as a monofunctional initiator. N,N′-(1,4-Phenylenebis(methylene))dimethanesulfonamide (BnBis(NHMs), 4) was designed as a novel bifunctional initiator for the AROP and also deprotonated with KMDS, which will allow the preparation of ABA triblock copolymers and is currently under investigation in our lab.
Real-time 1H NMR spectroscopy was used to monitor the polymerization under these different conditions.22,23 Requirements for this method are reaction times in the range of minutes to hours and reagents with distinguishable resonances in their spectrum (Fig. 1). In N-sulfonyl-aziridines, the three ring-protons are detected as two doublets (CH2) and one multiplet (CH) in the region from 3 to 2 ppm of the 1H NMR spectrum (Fig. 1A). These chemical shifts are sensitive probes for the monomer reactivity: the more they are shifted downfield in the spectrum, the stronger the activation, i.e. the electron-withdrawing effect of the sulfonamide. This allowed us to use the different monomer reactivities and to prepare sequenced copolymers.7 As the monomer is consumed during the polymerization, the monomer signals vanish and simultaneously the growing polymer-backbone emerges between 3.5 and 4.5 ppm (Fig. 1B). By integration of the well-separated monomer peaks over time and normalization to the amount of unreacted monomer, plotting of the monomer conversion vs. the reaction time or the total conversion is possible (Fig. 1C and D).
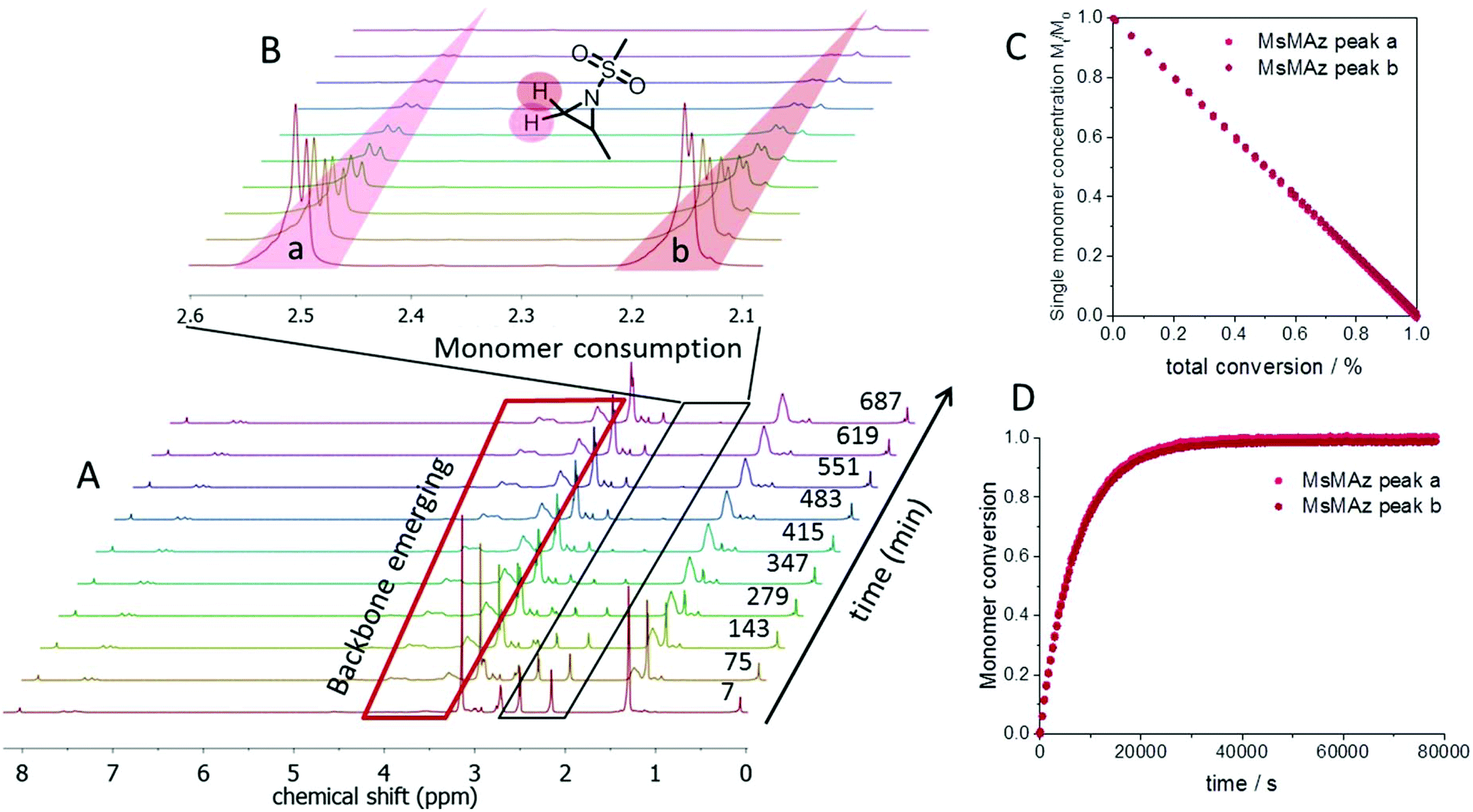 | ||
| Fig. 1 (A) Selection of the 1H NMR spectra of the azaanionic polymerization of MsMAz (1) with BnNKMs (3) as the initiator in DMF-d7, at 50 °C. (B) Relevant signals of the monomer ring-protons, showing the consumption of the monomer. (C) Normalized single monomer concentration versus total conversion. (D) Monomer conversion versus reaction time. | ||
The azaanionic polymerization proceeds in a living manner which was proven by chain extension experiments. Both the formation of diblock copolymers of 1 and 2, and the chain extension of 2 resulted in a complete shift of the molecular weight distributions in SEC experiments and thus underlines the living character of the chain ends (Fig. S16–S19†).
The propagation rate constants kp were calculated from the integrals of the monomer signals of the first 13 spectra (ca. 1.5 h reaction time). For “fast” polymerizations (100% conversion in less than 1 h) only the first 4 values were used for the determination of the slope (equivalent to the apparent propagation rate constant (kapp)) of the linear fits. Division of kapp with the initial initiator-concentration reveals the initiator-independent propagation rate coefficient kp.
From every reaction in the NMR tube a small aliquot was taken and analyzed by size exclusion chromatography (SEC). All polymers exhibited monomodal and narrow molecular weight distributions (Đ typically < 1.1, Tables 1–4 and the ESI†) and reached full monomer conversion in most cases (see below). The molecular weights of the PAz determined from SEC are underestimated on our setup compared to the absolute values calculated from NMR by end group analysis (Tables 1–4). Since for all SEC analyses PEO-standards were used for conventional calibration, the molecular weights calculated from NMR-data should be considered for comparisons.
| Initiator | BnNKMs (3-K) | BnNKMs (3-K) | BnNKMs (3-K) |
|---|---|---|---|
| a Number-average molecular weight and molecular weight dispersity determined via SEC in DMF (vs. PEO standards). b Number-average molecular weight determined by NMR analyses. | |||
| Ratio [I]:[M] |
01:30 |
01:30 |
01:30 |
| Monomer | MsMAz (1) | MsMAz (1) | MsMAz (1) |
| Additive | HMDS | HMDS | HMDS |
| Solvent | DMF-d7 | DMF-d7 | DMF-d7 |
| T/°C | 100 | 50 | 20 |
| k p /10 −3 L mol −1 s −1 | 123.85 | 10.53 | 0.98 |
|
M
na/g mol−1 |
2000 | 2200 | 2000 |
|
M
nb/g mol−1 |
4000 | 3600 | 2200 |
| Đ | 1.08 | 1.06 | 1.08 |
| Reaction time/h | 0.50 | 8.00 | 17.00 |
| Conversion/% | >99 | >99 | 60 |
| Initiator | BnNKMs (3-K) | BnNKMs (3-K) | BnNKMs (3-K) | BnNKMs (3-K) | BnNKMs (3-K) | BnNKMs (3-K) | BnNKMs (3-K) |
|---|---|---|---|---|---|---|---|
| a Two identical polymerization mixtures (I or II). b Number-average molecular weight and molecular weight dispersity determined via SEC in DMF (vs. PEO standards). c Number-average molecular weight determined by NMR. d Samples taken after 17 h reaction, no full conversion. | |||||||
| Ratio [I]:[M] |
01:30 |
01:30 |
01:30 |
01:30 |
01:30 |
01:30 |
01:30 |
| Monomer | MsMAz (1) | MsMAz (1) | MsMAz (1) | MsMAz (1) | MsMAz (1) | MsMAz (1) | MsMAz (1) |
| Additive | HMDS | HMDS | HMDS | HMDS | HMDS | HMDS | HMDS |
| Solventa | DMSO-d6-Ia | DMSO-d6-IIa | DMF-d7 | THF-d8-Ia | THF-d8-IIa | Benzene-d6 | CyHex-d12 |
| T/°C | 50 | 50 | 50 | 50 | 50 | 50 | 50 |
| k p/10−3 L mol−1 s−1 | 13.87 | 12.46 | 10.53 | 0.87 | 0.65 | 0.56 | — |
|
M
nb/g mol−1 |
2100 | 2500 | 2200 | 1600 | 1500 | 700 | — |
|
M
nc/g mol−1 |
4100 | 5100 | 3600 | 1500d | 1700d | —d | —d |
| Đ | 1.11 | 1.09 | 1.06 | 1.09 | 1.09 | 1.17 | — |
| Reaction time/h | 8.00 | 8.00 | 8.00 | >17 | >17 | >17 | >17 |
| Conversion/% | >99 | >99 | >99 | 30 | n.d. | n.d. | n.d. |
| Initiator | BnNKMs (3-K) | BnBis(NKMs) (4-K) | BuLi |
|---|---|---|---|
| a Number-average molecular weight and molecular weight dispersity determined via SEC in DMF (vs. PEO standards). b Number-average molecular weight determined by NMR analyses. | |||
| Ratio [I]:[M] |
01:30 |
01:50 |
01:30 |
| Monomer | MsMAz (1) | MsMAz (1) | MsMAz (1) |
| Additive | HMDS | HMDS | — |
| Solvent | DMF-d7 | DMF-d7 | DMF-d7 |
| T/°C | 50 | 50 | 50 |
| k p/10−3 L mol−1 s−1 | 10.53 | 9.02 | 18.08 |
|
M
na/g mol−1 |
2200 | 2700 | 2200 |
|
M
nb/g mol−1 |
3600 | 3700 | n.d. |
| Đ | 1.06 | 1.09 | 1.08 |
| Reaction time/h | 8 | 6 | 5 |
| Conversion/% | 99 | >99 | >99 |
| Initiator | BnNLiMs (3-Li) | BnNNaMs (3-Na) | BnNKMs (3-K) | BnNCsMs (3-Cs) |
|---|---|---|---|---|
| a Number-average molecular weight and molecular weight dispersity determined via SEC in DMF (vs. PEO standards). b Number-average molecular weight determined by NMR analyses. | ||||
| Ratio [I]:[M] |
01:30 |
01:30 |
01:30 |
01:30 |
| Monomer | TsMAz (2) | TsMAz (2) | TsMAz (2) | TsMAz (2) |
| Additive | HMDS | HMDS | HMDS | HMDS |
| Solvent | DMF-d7 | DMF-d7 | DMF-d7 | DMF-d7 |
| T/°C | 50 | 50 | 50 | 50 |
| k p/10−3 L mol−1 s−1 | 90.30 | 60.91 | 39.79 | 98.69 |
|
M
na/g mol−1 |
3000 | 2900 | 2300 | 2800 |
|
M
nb/g mol−1 |
n.d. | n.d. | n.d. | n.d. |
| Đ | 1.09 | 1.09 | 1.09 | 1.06 |
| Reaction time/h | 8 | 8 | 8 | 8 |
| Conversion/% | >99 | >99 | >99 | >99 |
Temperature variation
The polymerization of MsMAz (1) was initiated with BnNHMs (3)/KMDS in DMF-d7 at 20, 50 and 100 °C (Table 1; note: under these conditions 1 eq. hexamethyldisilazane (HMDS) is present as an inherent additive in the polymerization mixture, its influence will be discussed later on). At 20 °C after 17 hours the conversion reached 60% with a propagation rate of kp = 0.98 × 10−3 L mol−1 s−1. At 50 °C full conversion was achieved after ca. 8 hours, revealing a kp of 10.53 × 10−3 L mol−1 s−1, which is ca. ten times higher compared to 20 °C. When the polymerization was performed at 100 °C, full conversion was achieved after 30 min with a kp-value of ca. 123.85 × 10−3 L mol−1 s−1 (Fig. S1†). At all temperatures, the polymerization remains living, and the addition of new monomer allows the formation of block copolymers.7,8,15Solvent variation
The solvent polarity and the solvation of the living chain ends have a tremendous influence on the propagation rate in ionic polymerizations.2,3,24 For the azaanionic ROP of MsMAz (1) at 50 °C, polar solvents such as DMSO-d6 (kp = 13.17 ± 0.7 × 10−3 L mol−1 s−1 (mean value from repeated measurements I and II)) and DMF-d7 (10.53 × 10−3 L mol−1 s−1) are the suitable solvents and reach full conversion after ca. 8 hours and with narrow molecular weight distributions (Fig. 2 and Table 2).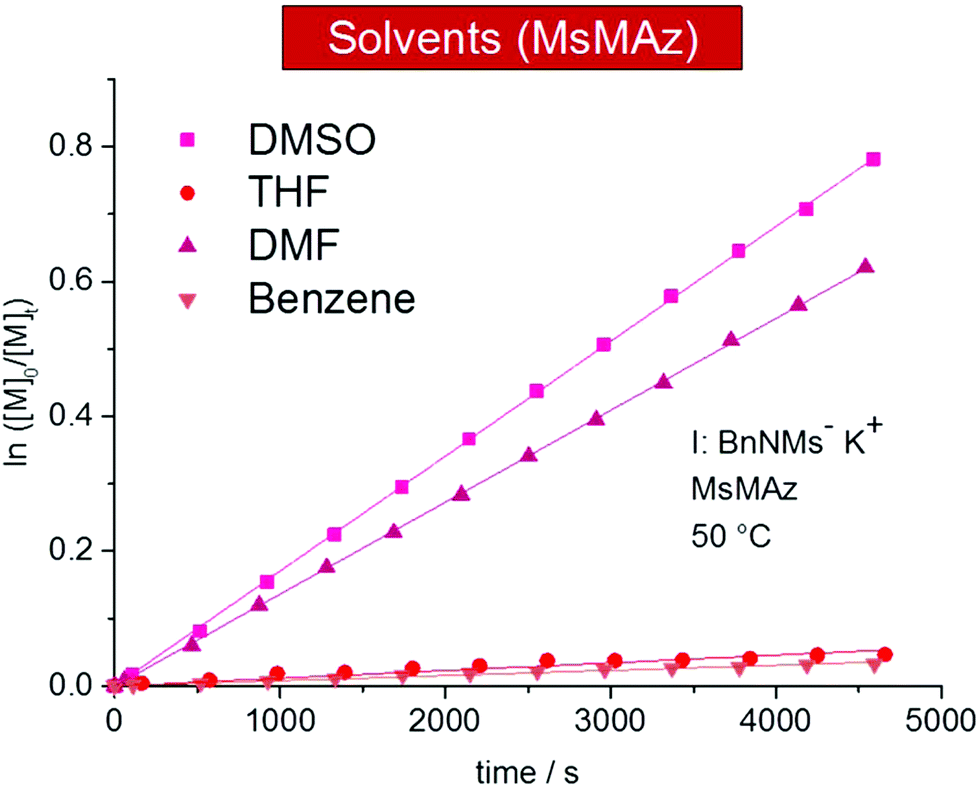 | ||
| Fig. 2 Kinetic plots of ln([M]0/[M]t) vs. time of MsMAz (1), BnNKMs (3) at 50 °C in different solvents (data listed in Table 2). | ||
The polymerization in THF-d8 was remarkably slower with kp = 0.76 ± 0.11 × 10−3 L mol−1 s−1 (mean value from repeated measurements I and II). Also in benzene-d6 only slow propagation was detected (kp = 0.56 × 10−3 L mol−1 s−1). After 17 h a conversion of 30% was reached. Cyclohexane-d12, a typical solvent for carbanionic polymerization, did not result in chain growth. This trend directly reflects the solvation of the living chains and reveals aprotic polar solvents such as DMSO and DMF as the solvents of choice for the AROP of sulfonyl aziridines. However, also in the other solvents the polymerizations remain living and might be considered for special monomers.
Initiator variation
Deprotonated sulfonamides are used as the initiators for the azaanionic ROP of activated aziridines. KMDS was previously used for this series of experiments at 50 °C in DMF-d7 (Table 3). It has to be noted, that in all cases an equimolar amount of HMDS is produced during the initiator formation (also compare sections below).Comparing different sulfonamide initiators, (Fig. 3) the potassium salt of N-benzyl methanesulfonamide (BnNKMs, 3) (kp = 10.53 × 10−3 L mol−1 s−1) and the novel bifunctional initiator (BnBis(NKMs), 4) exhibit a propagation constant of kp (BnBis(NKMs)) = 9.02 × 10−3 L mol−1 s−1. MALDI-ToF mass spectrometry of the polymers prepared with both initiators proves their successful incorporation in the polymer chain and the absence of any additional distribution (Fig. S11 and S12†). This allows the synthesis of ABA triblock-copolymers based on aziridines which is currently under investigation.
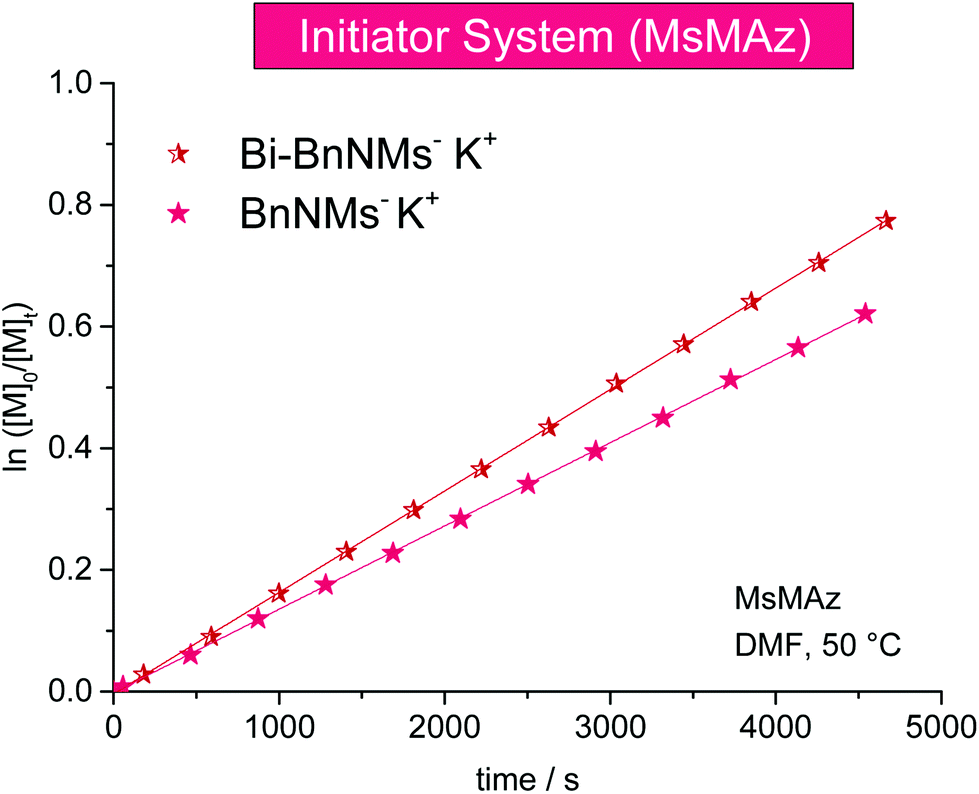 | ||
| Fig. 3 Kinetic plots of ln([M]0/[M]t) vs. time for the azaanionic polymerization of MsMAz (1) with different initiators at 50 °C (data listed in Table 3). | ||
As the studies with different counter ions revealed (see below) that also lithium cations can propagate the AROP of aziridines, n-butyllithium (n-BuLi) was tested as a commercially available initiator for the AROP of MsMAz (1) which demonstrated the fastest reaction rate under these conditions (kp = 18.08 × 10−3 L mol−1 s−1, cf. Fig. S4† and discussion for counter ions). MALDI-ToF mass spectrometry proved the incorporation of the butyl chain and shows a single mass distribution (Fig. S13†).
Influence of counter-ions
It is known from ionic polymerizations that the solvation of the growing chain and the respective counter ion plays an important role in the polymerization kinetics: the stronger the binding between the growing chain and the counter ion (and the lower the solvation efficiency of the solvent), the slower the propagation. For the anionic polymerization of styrene, for example, lithium counter ions show an increase of the polymerization kinetics compared to sodium counter ions. For epoxides, typically potassium and cesium show the highest propagation rates, while lithium alkoxides do not or only very slowly propagate, as they coordinate strongly to the Pearson-hard alkoxide.2,25–32 In the case of sulfonamide anions such studies had not been performed; in previous work, potassium salts proved to be efficient. We studied the polymerization of 1 and 2 (DMF-d7, 50 °C, BnNHMs (3) as the initiator) with different counter ions by deprotonating 3 with different bis(trimethylsilyl)amide salts (lithium, sodium, potassium salts are commercially available, CsMDS was prepared according to the literature4,20). For both monomers, propagation with all counter ions was observed (Fig. 4, S2† and Table 4), probably due to the rather soft nature of the propagating anion. Under these conditions the order of Cs+ > Li+ > Na+ > K+ was found for both monomers (Fig. 4A).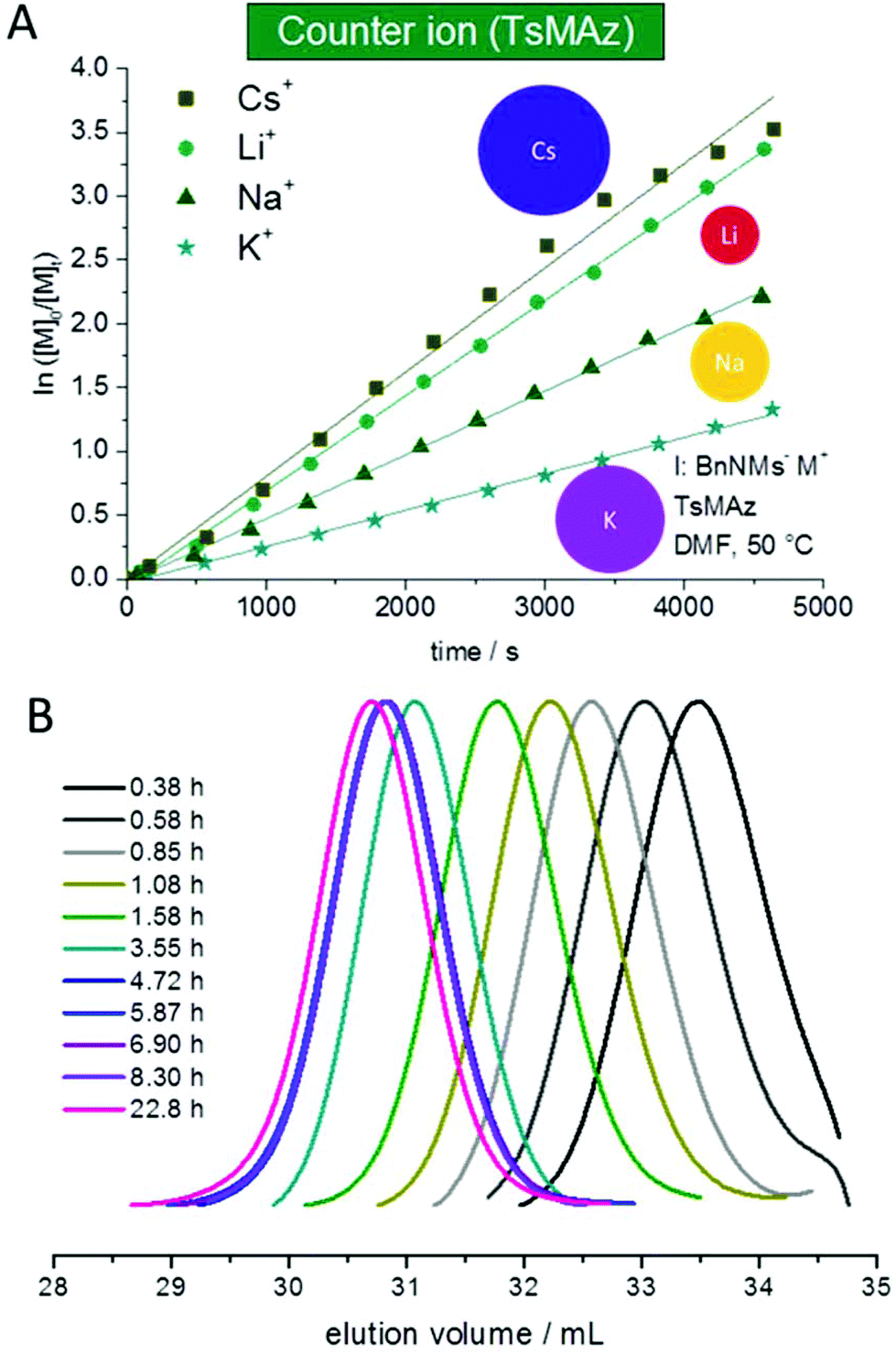 | ||
| Fig. 4 (A) Kinetic plots of ln([M]0/[M]t) vs. time for the azaanionic polymerization of TsMAz (2) with BnNHMs (3, initiator) in DMF-d7 at 50 °C with different bis(trimethylsilyl)amide-salts. (B) SEC-kinetics of MsMAz (1) and BnNKMs (3) at 50 °C in DMF (RI-signal, PEO-standard), (Table 5). | ||
For MsMAz (1) as a less reactive monomer the same trend was observed (Cs+ > Li+ > Na+ > K+), however, the differences were less pronounced (Fig. S2† and Table 5). Noteworthily, in all cases living polymerization with reasonable polymerization rates of the activated aziridines is observed (cf.Fig. 4B, S14 and 15†). This indicates a higher solvation of the propagating azaanion chains under these conditions, in contrast to the epoxide polymerization, where hardly propagation is observed with lithium as a counter ion, also in highly solvating solvents. Also the cation dependence on the polymerization kinetics, which does not follow the cation size can be explained by the Pearson acid base concept33 that the sulfonamide anion is weakly coordinated by its respective cation compared to an alkoxide, which strongly binds to lithium cations also in polar organic solvents as mentioned above. The “softer” sulfonamide anion exhibits higher binding to intermediate sized cations sodium and potassium, but less binding to the hard lithium and soft cesium cations. In addition, comparing the two monomers 1 and 2, with the smaller electron withdrawing effect of the mesyl group in MsMAz (1) compared to the tosyl-group in 2, a more nucleophilic growing chain end is produced. This leads to the less expressed trend in the propagation rate of the different counter ions, as the interaction between the azaanion at the chain end and the cationic counter ion is stronger.
| Initiator | BnNLiMs (3-Li) | BnNNaMs (3-Na) | BnNKMs (3-K) | BnNCsMs (3-Cs) | BnNKMsc (3-K) | BnNKMs (3-K) |
|---|---|---|---|---|---|---|
| a Number-average molecular weight and molecular weight dispersity determined via SEC in DMF (vs. PEO standards). b Number-average molecular weight determined by NMR analyses. c Prepared by deprotonation with KOH. | ||||||
| Ratio[I]:[M] |
01:30 |
01:30 |
01:30 |
01:30 |
01:30 |
01:30 |
| Monomer | MsMAz (1) | MsMAz (1) | MsMAz (1) | MsMAz (1) | MsMAz (1) | MsMAz (1) |
| Additive | HMDS | HMDS | HMDS | HMDS | — | 2 eq. HMDS |
| Solvent | DMF-d7 | DMF-d7 | DMF-d7 | DMF-d7 | DMF-d7 | DMF-d7 |
| T/°C | 50 | 50 | 50 | 50 | 50 | 50 |
| k p/10−3 L mol−1 s−1 | 15.39 | 11.66 | 10.53 | 15.74 | 11.19 | 8.45 |
|
M
na/g mol−1 |
2400 | 2400 | 2200 | 2100 | 2200 | 2000 |
|
M
nb/g mol−1 |
4000 | 3900 | 3600 | 3800 | 3700 | 4000 |
| Đ | 1.07 | 1.06 | 1.06 | 1.08 | 1.07 | 1.10 |
| Reaction time/h | 8.00 | 7.00 | 8.00 | 7.00 | 8.00 | 9.00 |
| Conversion/% | >99 | >99 | 97 | >99 | 98 | >99 |
To identify the influence of the inherent additive hexamethyldisilazane (HMDS), which is generated in a molar amount after the deprotonation of BnHMs (3) by the bis(trimethylsilyl)amide salt, on the chain end reactivity, polymerizations of 1 with different amounts of HMDS were performed (note: MALDI ToF mass spectrometry revealed that only polymers, initiated by 3 are produced under these conditions, cf. the ESI†).
To study the influence of HMDS on the polymerization kinetics several experiments were conducted: (i) 3 was deprotonated with KMDS (i.e. one equivalent of HMDS is produced with respect to the initiator); (ii) to exclude HMDS in the polymerization, BnNHMs (3) was deprotonated with potassium hydroxide (KOH) and dried by azeotropic removal of the emerging water with benzene before adding the monomers; (iii) another polymerization was conducted under the same conditions, however with two equivalents of HMDS with respect to the initiator. Comparing the propagation rate constants of these three polymerizations in the presence of 1 or 2 eq. or without HMDS, a decrease of the polymerization kinetics with an increasing amount of HMDS was detected (BnNKMs in DMF (no HMDS) at 50 °C kp = 11.19 × 10−3 L mol−1 s−1, BnNKMs, 1 eq. HMDS in DMF at 50 °C kp = 10.53 × 10−3 L mol−1 s−1, BnNKMs, 2 eq. HMDS in DMF at 50 °C kp = 8.45 × 10−3 L mol−1 s−1). These results prove that HMDS influences the polymerization kinetics, probably due to coordination to the anionic chain end and the formation of a complex. The same trend was observed for the polymerization of 1 with lithium as the counter ion: for MsMAz, 50 °C, DMF, BuLi (no HMDS) a kp = 18.1 × 10−3 L mol−1 s−1 was determined (Table 3), while the presence of HMDS in the system reduced the kp to 15.4 × 10−3 L mol−1 s−1 (Table 5 first entry, MsMAz, 50 °C, DMF, BnNLiMs, HMDS).
To examine the influence of DMF as a highly solvating solvent, polymerizations with lithium, potassium and cesium, with the “standard procedure”, i.e. BnNHMs (3), the respective bis(trimethylsilyl)amide salt, at 50 °C, were carried out in THF. In all cases the polymerization proceeds much slower in THF compared to DMF, irrespective of which counter ion was used (Table 6 and Fig. S3†). This indicates a lower solvation of the living chain ends in THF, reducing the polymerization kinetics (at least by a factor of 10). In contrast, the conventional oxyanionic polymerization and the recently reported organocatalytic ring-opening polymerization of sulfonyl-aziridines16 proceed smoothly in THF and reach full conversion in the course of several hours.
| Initiator | BnNLiMs (3-Li) | BnNKMs (3-K) | BnNKMs (3-K) | BnNCsMs (3-Cs) |
|---|---|---|---|---|
| a In the case of KMDS the measurements were repeated and are marked with I, respectively II. b Number-average molecular weight and molecular weight dispersity determined via SEC in DMF (vs. PEO standards). c Number-average molecular weight determined by NMR analyses. d Samples taken after minimum 17 h reaction, no full conversion. | ||||
| Ratio [I]:[M] |
01:30 |
01:30 |
01:30 |
01:30 |
| Monomer | MsMAz (1) | MsMAz (1) | MsMAz (1) | MsMAz (1) |
| Additivea | HMDS | HMDS-I | HMDS-II | HMDS |
| Solvent | THF-d8 | THF-d8 | THF-d8 | THF-d8 |
| T/°C | 50 | 50 | 50 | 50 |
| k p/10−3 L mol−1 s−1 | 0.72 | 0.87 | 0.65 | 1.36 |
|
M
nb/g mol−1 |
700 | 1600 | 1500 | 1400 |
|
M
nc/g mol−1 |
1300d | 1500d | 1700d | 2500d |
| Đ | 1.17 | 1.09 | 1.09 | 1.08 |
| Reaction time/h | >24 | >17 | >17 | >18 |
| Conversion/% | 33 | 38 | 43 | 63 |
With these results in hand, the combination of the azaanionic polymerization with other ionic polymerization techniques will be used to produce various macromolecular architectures and the use of commercially available lithium-based initiators (e.g. butyllithium).
Conclusions
We report on the systematic polymerization kinetics of the living anionic polymerization of N-activated aziridines, exemplary with MsMAz and TsMAz, two activated aziridines of different reactivities, by real-time 1H NMR spectroscopy. We found that their polymerization follows living conditions at temperatures between 20 and 100 °C. The comparison of different solvents for the polymerization proved that polar aprotic solvents exhibit the fastest polymerization kinetics with the order of DMSO ≥ DMF ≫ THF ≥ benzene, and no propagation in cyclohexane, depending on the solvation of the living chain end. The use of different initiators, namely sulfonamides BnNHMs and a novel bifunctional sulfonamide (BnBis(NHMs)) were compared with each other and we additionally identified n-butyllithium as a potent commercial alternative.However, the sulfonamide initiators are ideal to study the influence of the counter ion on the polymerization kinetics. The sulfonamide initiator was deprotonated with the respective metal bis(trimethylsilyl)amide (Li, Na, K, or Cs). In all cases fast propagation of the anionic polymerization was observed, which is in strong contrast to epoxide polymerization, where lithium alkoxides show only very slow propagation rate constants. For activated aziridines the following trend was observed: Cs+ > Li+ > Na+ > K+ with reasonable kp values in all cases in DMF, indicating a high solvation of all propagating azaanions in DMF, with a less pronounced effect of the counter ion compared to alkoxide chains. In contrast, in THF only a weak counter ion dependency and low reaction kinetics have been observed.
We believe that this fundamental work will help to further understand and foster the field of the anionic polymerization of aziridines. In particular, the less pronounced counter ion effect compared to the well-known anionic polymerization of epoxide makes the AROP of sulfonylaziridines easy and switching for example from carb- to aza-anionic polymerization or the use of simple commercially available butyllithium. N-Sulfonyl-activated aziridines undergo AROP under various conditions producing well-defined polysulfonamides and -amines after hydrolysis. This defined access to such structures was not possible to date and we believe that aziridines will become a valuable tool for combinations with other anionic polymerizations for diverse applications, for example as a well-defined alternative for branched poly(ethylene imine)s.
Acknowledgements
The authors thank the Deutsche Forschungsgemeinschaft (DFG WU750/7-1) for support. The authors thank Stefan Spang for the NMR measurements. E. R. thanks the BMBF/MPG network MaxSynBio. Open Access funding provided by the Max Planck Society.References
- M. Szwarc, M. Levy and R. Milkovich, J. Am. Chem. Soc., 1956, 78, 2656–2657 CrossRef CAS.
- G. Odian, Principles of Polymerization, John Wiley & Sons, New Jersey, USA, 2004 Search PubMed.
- N. Hadjichristidis and A. Hirao, Anionic Polymerization: Principles, Practice, Strength, Consequences and Applications, Springer Japan, 2015 Search PubMed.
- I. C. Stewart, C. C. Lee, R. G. Bergman and F. D. Toste, J. Am. Chem. Soc., 2005, 127, 17616–17617 CrossRef CAS PubMed.
- L. Thomi and F. R. Wurm, Macromol. Rapid Commun., 2014, 35, 585–589 CrossRef CAS PubMed.
- L. Thomi and F. R. Wurm, Macromol. Symp., 2015, 349, 51–56 CrossRef CAS.
- E. Rieger, A. Alkan, A. Manhart, M. Wagner and F. R. Wurm, Macromol. Rapid Commun., 2016, 37, 833–839 CrossRef CAS PubMed.
- E. Rieger, A. Manhart and F. R. Wurm, ACS Macro Lett., 2016, 5, 195–198 CrossRef CAS.
- L. Reisman, C. P. Mbarushimana, S. J. Cassidy and P. A. Rupar, ACS Macro Lett., 2016, 5, 1137–1140 CrossRef CAS.
- M. Kobayashi, K. Uchino and T. Ishizone, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 4126–4135 CrossRef CAS.
- A. P. Spork and T. J. Donohoe, Org. Biomol. Chem., 2015, 13, 8545–8549 CAS.
- H.-J. Jang, J. T. Lee and H. J. Yoon, Polym. Chem., 2015, 6, 3387–3391 RSC.
- D. C. McLeod and N. V. Tsarevsky, Macromol. Rapid Commun., 2016, 37, 1694–1700 CrossRef CAS PubMed.
- H. K. Moon, S. Kang and H. J. Yoon, Polym. Chem., 2017, 8, 2287–2291 RSC.
- T. Homann-Müller, E. Rieger, A. Alkan and F. R. Wurm, Polym. Chem., 2016, 7, 5501–5506 RSC.
- C. Bakkali-Hassani, E. Rieger, J. Vignolle, F. R. Wurm, S. Carlotti and D. Taton, Chem. Commun., 2016, 52, 9719–9722 RSC.
- J. B. Sweeney, Chem. Soc. Rev., 2002, 31, 247–258 RSC.
- M. Bednarek, P. P. Kubisa and S. Penczek, Macromolecules, 1999, 32, 5257–5263 CrossRef CAS.
- B. D. Monnery and R. Hoogenboom, in Cationic Polymers in Regenerative Medicine, The Royal Society of Chemistry, 2015, pp. 30–61 Search PubMed.
- S. Neander and U. Behrens, Z. Anorg. Allg. Chem., 1999, 625, 1429–1434 CrossRef CAS.
- R. Freeman, H. D. W. Hill and R. Kaptein, J. Magn. Reson., 1972, 7, 327–329 CAS.
- A. Alkan, A. Natalello, M. Wagner, H. Frey and F. R. Wurm, Macromolecules, 2014, 47, 2242–2249 CrossRef CAS.
- A. Natalello, A. Alkan, P. von Tiedemann, F. R. Wurm and H. Frey, ACS Macro Lett., 2014, 3, 560–564 CrossRef CAS.
- M. Van Beylen, D. N. Bhattacharyya, J. Smid and M. Szwarc, J. Phys. Chem., 1966, 70, 157–161 CrossRef CAS.
- K. S. Kazanskii, A. A. Solovyanov and S. G. Entelis, Eur. Polym. J., 1971, 7, 1421–1433 CrossRef CAS.
- M. Szwarc, in Living Polymers and Mechanisms of Anionic Polymerization, Springer, Berlin, Heidelberg, Germany, 1983, vol. 49, pp. 1–177 Search PubMed.
- K. Matyjaszewski and A. H. E. Müller, Controlled and Living Polymerizations: From Mechanisms to Applications, John Wiley & Sons, Weinheim, Germany, 2009 Search PubMed.
- J. Herzberger, K. Niederer, H. Pohlit, J. Seiwert, M. Worm, F. R. Wurm and H. Frey, Chem. Rev., 2016, 116, 2170–2243 CrossRef CAS PubMed.
- M. van Beylen, S. Bywater, G. Smets, M. Szwarc and D. J. Worsfold, Adv. Polym. Sci., 1988, 86, 87–143 CrossRef CAS.
- H. Jeuck and A. H. E. Müller, Makromol. Chem., 1982, 3, 121–125 CrossRef CAS.
- F. S. Dainton, G. C. East, G. A. Harpell, N. R. Hurworth, K. J. Ivin, R. T. Laflair, R. H. Pallen and K. M. Hui, Makromol. Chem., 1965, 89, 257–262 CrossRef CAS.
- J. E. L. Roovers and S. Bywater, Trans. Faraday Soc., 1966, 62, 701–706 RSC.
- A. F. Holleman and E. Wiberg, Lehrbuch der Anorganischen Chemie, Walter de Gruyter Co., Berlin, Germany, 34th edn, 1995 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7py00436b |
| This journal is © The Royal Society of Chemistry 2017 |