
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Olefin cross metathesis and ring-closing metathesis in polymer chemistry
Fern
Sinclair
a,
Mohammed
Alkattan
 ab,
Joëlle
Prunet
b and
Michael P.
Shaver
*a
ab,
Joëlle
Prunet
b and
Michael P.
Shaver
*a
aEastCHEM School of Chemistry, Joseph Black Building, University of Edinburgh, David Brewsster Road, Edinburgh EH9 3FJ, UK. E-mail: michael.shaver@ed.ac.uk
bWestCHEM, School of Chemistry, University of Glasgow, Joseph Black Building, Glasgow, G12 8QQ, UK
First published on 24th April 2017
Abstract
The use of olefin cross metathesis in preparing functional polymers, through either pre-functionalisation of monomers or post-polymerisation functionalisation is growing in both scope and breadth. The broad functional group tolerance of olefin metathesis offers a wealth of opportunities for introducing a broad range of functional groups into the polymer backbone, tuning polymer properties and expanding potential applications. Similarly, ring-closing metathesis offers the ability to tune the polymer macrostructure and microstructure to similar effect. In this review, we explore the importance of understanding selectivity in olefin cross metathesis in designing functional polymers, the manipulation of this reactivity to prepare (multi)functional polymers, and show how polymer systems can be constructed to favour ring closing and change backbone structure and properties.
 Fern Sinclair | Fern was awarded a first class Master of Chemistry from the University of Edinburgh in 2013, including one year industrial experience working with Cytec Industries, USA. Upon completion of her studies the University of Edinburgh awarded her the prestigious Principal's Career Development Scholarship where she is currently completing her PhD under the supervision of Dr Michael Shaver. Her research is focussed on using olefin cross metathesis for the modification of biodegradable polymers through both novel monomer synthesis and copolymerisation techniques. |
 Mohammed Alkattan | Mohammed graduated from Damascus University with a BSc in Applied Chemistry. After two years of practical experience as a chemist in pharmaceutical labs he obtained an MSc in Drug Chemistry with distinction from Newcastle University. Mohammed then gained some research experience on nanotechnology for polymer-drug formulations at the University of Nottingham and AstraZeneca. He then moved to the University of Edinburgh in 2015 to undertake a PhD and is investigating post-polymerization modification of polyethers and polyesters via olefin metathesis. |
 Joëlle Prunet | Dr Joëlle Prunet attended the Ecole Normale Supérieure (Paris) as an undergraduate and obtained her Ph.D. from Harvard University with Professor David A. Evans in 1993. Afterwards she joined the CNRS as Chargée de Recherche at the Ecole Polytechnique. In 2003 she was promoted to Directrice de Recherche and obtained a teaching position at the Ecole Polytechnique. In 2009, she was appointed to the University of Glasgow where she is currently Head of Organic Teaching. Her research interests include synthetic organic methodologies, and the use of metathesis reactions for the synthesis of bioactive natural products and more recently post-polymerisation functionalisation. |
 Michael P. Shaver | Michael Shaver leads the Green Materials Laboratory in the School of Chemistry at the University of Edinburgh. After undergraduate education at Mount Allison University and a PhD at the University of British Columbia, he was awarded an NSERC Post-doctoral Fellowship to study at Imperial College London. From there he crossed the Atlantic Ocean again as an Assistant, and then Associate Professor, at the University of Prince Edward Island. A growing research team prompted a move to the University of Edinburgh as a Chancellor's Fellow and Reader, where he now leads a diverse research group developing new catalysts and functional polymers using ring-opening and controlled radical polymerisations. |
1. Introduction
Polymers are essential to our lives, providing the structure and function that underpins advances in materials science, medicine, energy and more. Polymeric materials have expanded well beyond commodity plastics to access specialty applications enabled by precise control of their architectures, molecular weights and dispersity, including methodologies to control polymer microstructure, macrostructure and co-monomer composition.1 A less explored strategy is chemical functionalisation. Due to the size of individual macromolecules, and entanglement of polymer chains, direct chemical modification is sometimes difficult but has received renewed attention in recent years with the growth of efficient methodologies to incorporate the desired functionality. While many chemical groups could provide a platform for further modification, arguably one of the most versatile groups is the alkene functionality. In polymer science, transformations including Michael additions, epoxidations and radical thiol–ene additions have all played important roles in polymer modification.2While each of these methodologies has their strengths, they also present limitations on functional groups, polymer backbone tolerance or introduce unnatural chemical linkers which may alter stability or properties. The most important feature of olefin metathesis is preservation of the double bond post metathesis, which can be further modified using a range of techniques that is not offered in competing organic transformations. Olefin metathesis provides an important alternate functionalisation strategy, and is of growing importance to polymer science. The word metathesis means to change place; olefin metathesis is thus defined as the exchange of carbon–carbon double bonds. Chauvin's mechanistic proposal (Scheme 1)3 has since developed into a diverse and essential chemical reaction.
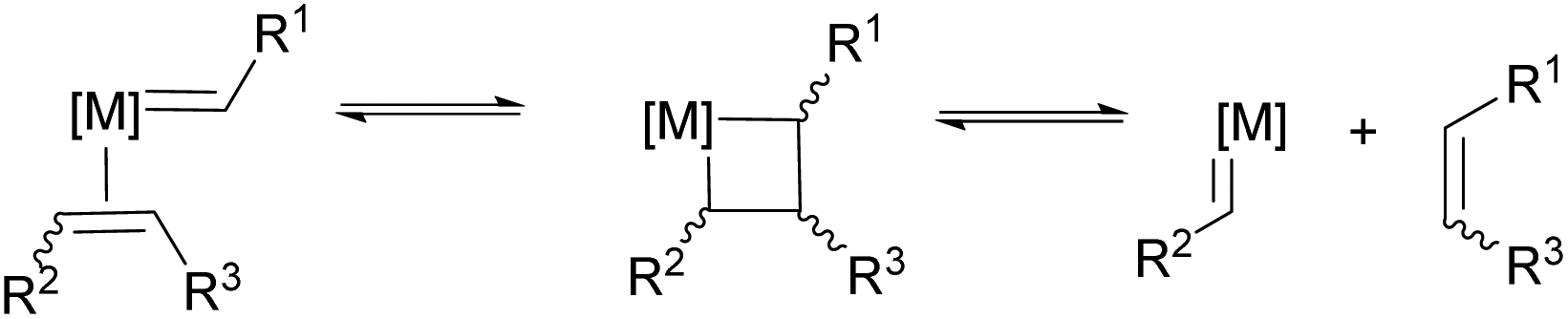 | ||
| Scheme 1 General mechanism for olefin metathesis and the formation of a new cross product. | ||
Olefin metathesis is an umbrella term for a series of reactions, the most prevalent of which are ring-opening metathesis (ROM), ring-closing metathesis (RCM), cross metathesis (CM), ring-opening metathesis polymerisation (ROMP) and acyclic diene metathesis polymerisation (ADMET). ROM, as the name implies, opens rings to afford new, derivatised small molecules featuring two olefin fragments. When no cross-partner is present, homopolymerisation is favoured, as in ROMP (from the ring form) or ADMET (from a diolefin monomer); these fields have been extensively reviewed4–10 and fall outside the scope of this review. For RCM and CM, however, the reactions have been developed with small molecule synthesis in mind and while they are well established in organic synthesis,11–13 much less work has been reported in the application of these reactions to polymer chemistry. The importance of these reactions in organic synthesis and polymer chemistry is facilitated by the development of exceptional catalysts, largely led by the groups of Schrock, Grubbs and Hoveyda. Schrock has developed highly active Mo-based metathesis catalysts (Fig. 1, 5), while Grubbs and Hoveyda have developed easily handled Ru-based catalysts (Fig. 1, 1–3), which are more commonly used today. In general, Shrock's early transition-metal catalysts have higher activities reacting preferentially with carboxylic acids, alcohols and aldehydes5 and are sensitive to oxygen and water. The Ru-based catalysts sacrifice activity for greater functional group tolerance with an inherent oxygen and moisture stability and preferentially react with alkenes.5 Various derivations on the first-generation catalyst, 1, have improved activity and selectivity. Comprehensive reviews of olefin metathesis catalysts alongside mechanistic studies complement this review.14–16
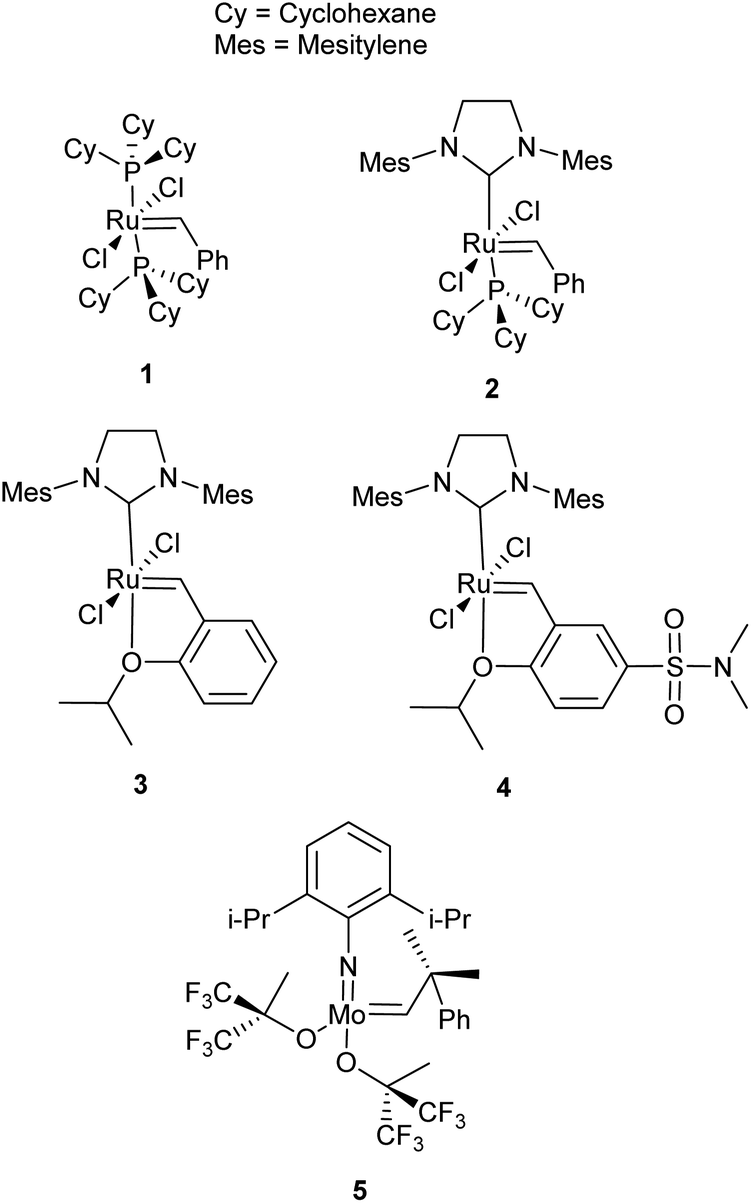 | ||
| Fig. 1 Olefin metathesis catalysts; 1 Grubbs first generation catalyst, 2 Grubbs second generation catalyst, 3 Hoveyda–Grubbs second generation catalyst, 4 a derivative of Hoveyda–Grubbs catalyst (Zhan-1B) and 5 Schrock's catalyst. | ||
Particularly in CM, where the metathesis reaction is between two chemically distinct alkenes, selectivity can be a challenge. To help overcome this problem, Grubbs devised an empirical model to aid in the design of selective CM reactions.17 This model, alongside the aforementioned development of more active metathesis catalysts, now permits the application of CM reactions to more complex systems, including polymers. The model categorises alkenes into four different types based on their ability to homodimerise (react with themselves) and the ability of those homodimers to participate in a secondary CM reaction (Scheme 2). Type I alkenes undergo fast homodimerisation, with the homodimers formed readily able to participate in a secondary metathesis reaction. Type II alkenes undergo slow homodimerisation; the homodimers formed are unlikely to react in a secondary metathesis reaction. Type III alkenes are unable to homodimerise, but can couple with alkenes of Type I or II. Finally, Type IV alkenes cannot undergo CM at all, but do not interfere with catalyst activity towards other alkenes. Both sterics and electronics influence alkene categorisation. As steric bulk surrounding the alkene increases the ease of homodimerisation decreases and thus the alkene categorisation increases. Moreover, electron-rich alkenes are more reactive compared to electron-deficient alkenes. In general terms, electron-rich, sterically unhindered alkenes can be categorised as type I alkenes, whereas electron-deficient sterically hindered alkenes can be categorised as type IV alkenes, with a gradient of reactivity existing in between these extremes.
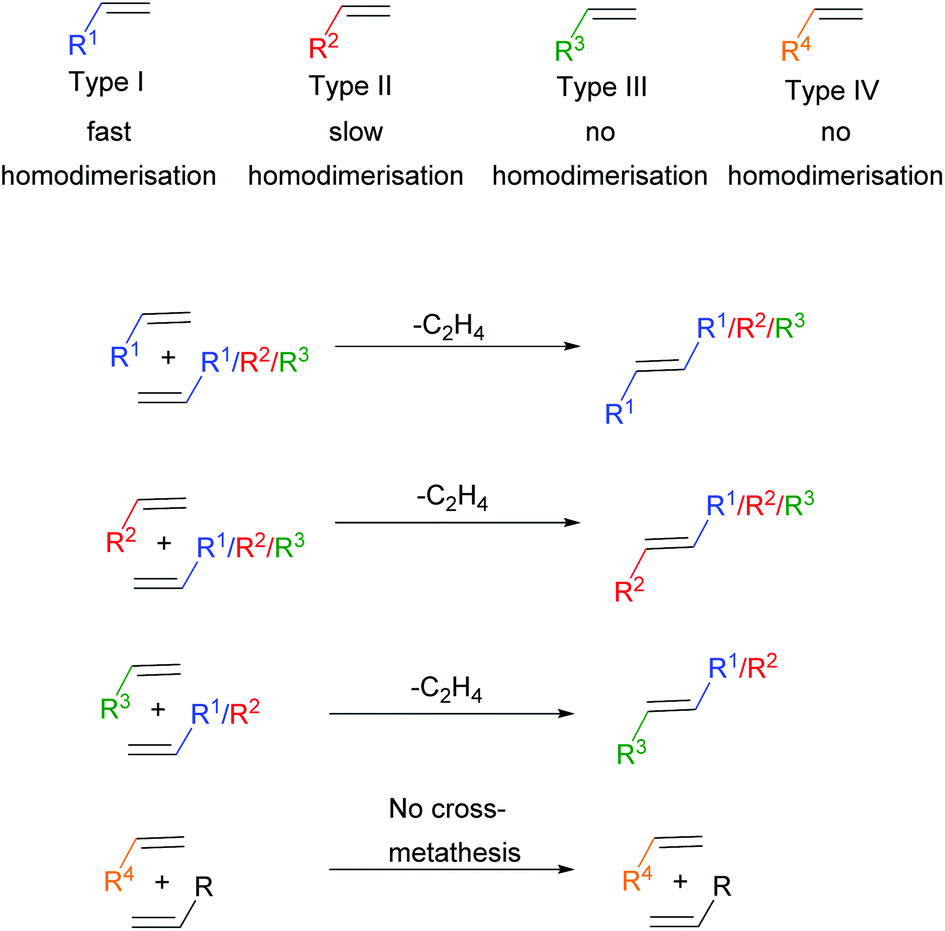 | ||
| Scheme 2 Alkene categorisation into type I, type II, type III or type IV. The colour codes shown are specific to the alkene types throughout the review. | ||
The key to selective CM is to react two alkenes of different types, where there is no competition between the rates of homodimerisation. For example, reacting a Type I alkene in the presence of a Type II alkene should lead to selective CM (Scheme 3, Scenario 1). The rate of homodimerisation of the Type II alkene is very slow (Scenario 2), while the fast homodimerisation of Type I affords a new alkene that will still undergo secondary metathesis, providing an alternative route to the desired cross-product (Scenario 3). Consequently, the Type II alkene will preferentially react with the Type I alkene in a selective CM reaction, with the reactivity of the formed cross product towards secondary metathesis remaining low. For this reason, selective CM is difficult in the presence of two different type I alkenes as the rate of homodimerisation of both alkenes are similar and the reactivity of the homodimers and cross product formed towards secondary metathesis is high. This reaction results in a statistical product mixture that is only overcome by using a large excess of one of the cross partners, normally as much as 10 times or more. Generally, reacting two alkenes of different types requires an excess of the least reactive alkene (normally 2 times or more), to promote a high yield to the cross product. The rate of homodimerisation of the less reactive alkene will be slow (or not observed in the case of a type III alkene), thus in excess and in the presence of a more reactive alkene, selective cross metathesis will be favoured.
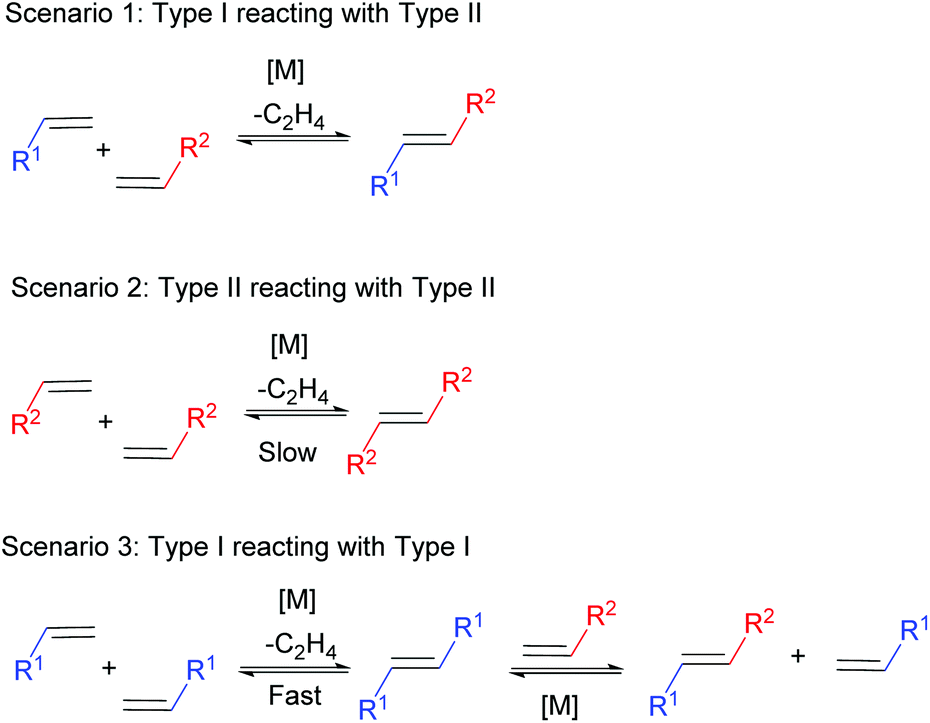 | ||
| Scheme 3 Selective cross metathesis reaction between a type I alkene and a type II alkene. | ||
It is important to note that the choice of catalyst is also key in predicting reactivity. For example, an alkene can be categorised as a Type II alkene when reacting with Grubbs first generation catalyst, 1, but may alternatively be classified as a Type I alkene in the presence of Hoveyda–Grubbs second generation catalyst, 3. Moreover, a gradient of reactivity exists within each alkene category, adding further complications to selectivity predictions.17 For this reason, it is recommended to test the reactivity of your alkene towards different cross partners to enable its categorisation prior to largescale synthesis or polymerisations.
This review will focus on the application of this model to olefin selective CM and RCM reactions in polymer synthesis. In particular, emphasis will be made on reaction scope, synthesis strategies that achieve high conversions, and how alkene type impacts reaction success in CM. The review will extend this discussion to examine RCM reactions that alter the polymer macrostructure and function, and the reaction conditions that favour this transformation. For each, sterics play a particularly important role, especially in instances where long macromolecular chains can hinder access to the catalyst active site. The review is meant to serve as a tutorial for those working in either organic synthesis and catalysis or polymer chemistry with the hopes of providing a foundation for future cross-disciplinary research.
2. Olefin cross metathesis
2.1 Pre-polymerisation cross metathesis for monomer derivatisation
Despite extensive literature describing the application of olefin CM in small molecule transformations, few reports have exploited this methodology for monomer synthesis. This pre-polymerisation derivatisation is advantageous, as the product polymers are fully functionalised, and the strategy would quickly access a family of monomers (and then polymers) from which structure–property relationships could be built. There are many reports that use olefin CM to generate potential precursors for step-growth polymerisation, in particular the functionalisation of renewable fatty acids, including the work of Dixneuf, Bruneau and Meier.18–22 Yet, reports that demonstrate successful CM for the formation of novel monomers and prove their corresponding polymerisation is more rare.23–25 In 2010, Cádiz et al. targeted modified vegetable-oils as alternative feedstocks to fossil fuel derived monomers, a topical challenge in polymer science.23 Vegetable oils are commonly copolymerised with olefinic styrenes to improve their physical properties, however the resultant thermosets have limited applications due to their high flammability. Cádiz and co-workers identified the opportunity to functionalise vegetable oils via CM (Scheme 4) to introduce inherent flame retardant properties. Thus, CM between vegetable oil derivative methyl 10-undeceneoate, 6, and a phosphonate containing styrene derivative, 7, generated a novel monomer, 8. Cationic copolymerisation of divnyl benzene, styrene, soybean oil and 8 affords vegetable oil based thermosets. The phosphonate group – well recognised for its flame retardant properties – effectively reduced product flammability.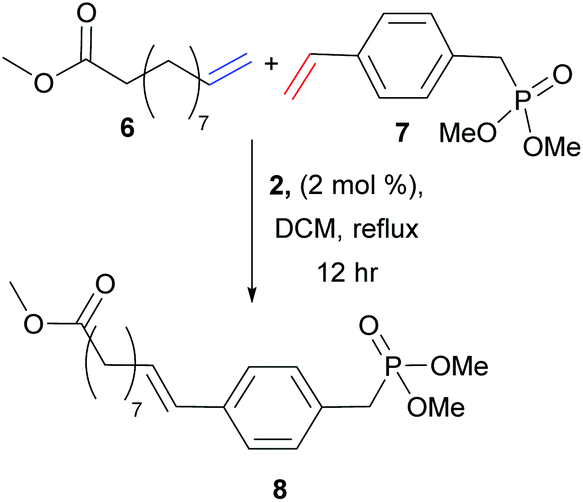 | ||
| Scheme 4 Vegetable oil-based thermosets generated from the cationic copolymerisation of divinyl benzene, styrene, soybean oil and 8, which was generated from the olefin cross metathesis of methyl 10-undeconate, 6, with styrene phosphonate, 7. | ||
In 2014, Meier and Winkler sought to generate polyamides from plant oil based monomers (Scheme 5).24 Polyamides are highly sought-after engineering plastics, but CM of amine functionalised substrates is difficult due to catalyst deactivation. Traditional strategies of protecting group chemistry or amine quaternisation can be used to overcome this challenge. The authors thus devised a novel methodology to take advantage of CM to prepare renewable polyamides. Precursor 9, a renewable substrate derived from plant oils and readily deprotected to form a primary amine, was used as a feedstock for a CM with methyl acrylate to generate 10. The Cbz protecting group was easily removed and the olefin reduced to afford monomer 11, which was finally polymerised to yield renewable polyamide 12, matching the thermal performance of commercial polyamides.
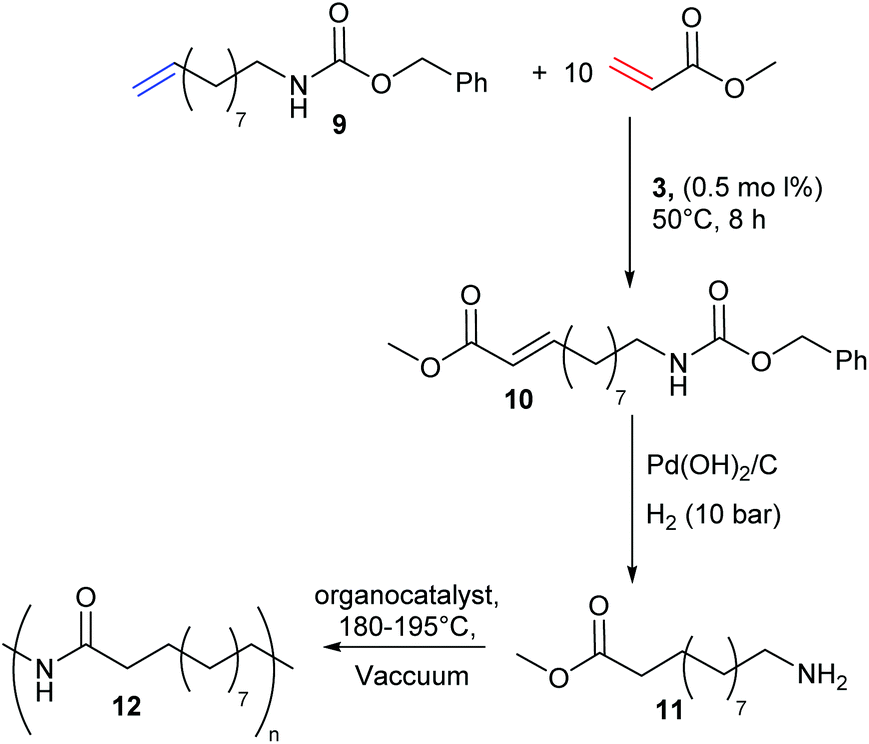 | ||
| Scheme 5 Renewable polyamide synthesis via sequential olefin cross metathesis and hydrogenation. | ||
More recently, in 2016, Shaver et al. reported the successful CM of an alkene substituted lactone, 13, with methyl acrylate to generate monomer 14.25 Ring-opening polymerisation of this bulky propiolactone afforded the biodegradable aliphatic polyester 15 (Scheme 6).
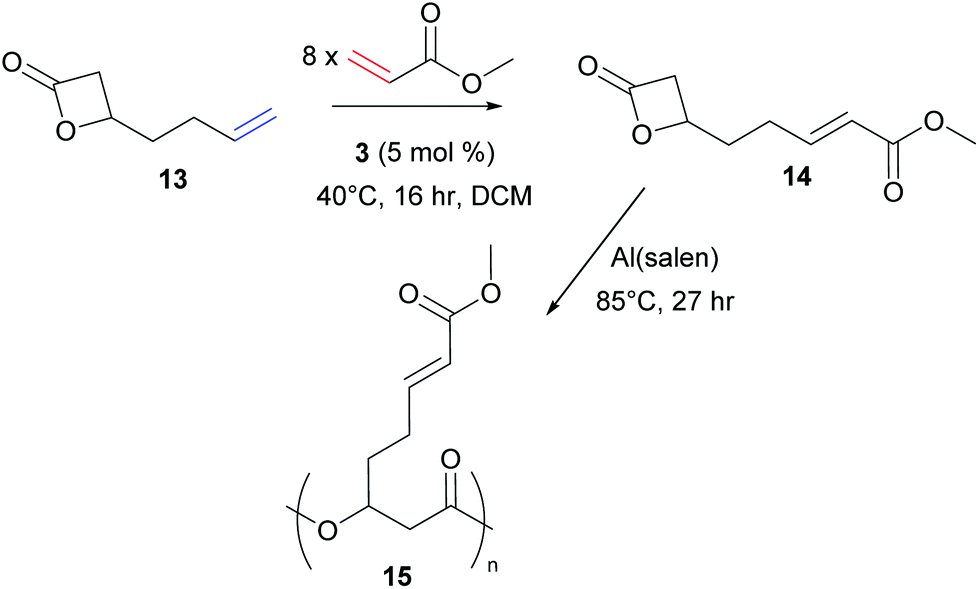 | ||
| Scheme 6 Functionalisation of a biodegradable monomer via olefin cross metathesis followed by ring-opening polymerisation. | ||
Comparing these three CM reactions, the importance of alkene reactivity is clear. All three groups used long alkyl tethers in one partner (6, 9, 13), positioning the terminal alkene far from an influencing functional group, suggesting these feedstocks would be Type I. The shorter chain (two carbon linker), 13, was experimentally confirmed to be a Type I olefin by Shaver and co-workers. Both Meier and Shaver use methyl acrylate, a known Type II alkene, as a coupling partner. By using a high loading of acrylate, CM is favoured as homodimerisation of the Type II alkene is slow. The groups achieve very high functionalisation, with conversions of 91–99% achieved under relatively mild conditions.24,25
Conversely, Cádiz and co-workers achieve much lower product yields, with optimum conversions of only 60% using a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio of alkenes.23 In this metathesis reaction, 10-undeceneoate, 6, can be classed as a type I alkene and the phosphorus styrene derivative, 7, is likely categorised as a Type II alkene, due to steric bulk in the ortho position. The lack of a substantial excess of one partner can account for the decrease in the obtained yield of the desired product. This work also highlights the importance of catalyst choice, as the team showed that catalyst 2, a more active metathesis catalyst, improved cross product formation to 60% from 44% with catalyst 1.
:1 ratio of alkenes.23 In this metathesis reaction, 10-undeceneoate, 6, can be classed as a type I alkene and the phosphorus styrene derivative, 7, is likely categorised as a Type II alkene, due to steric bulk in the ortho position. The lack of a substantial excess of one partner can account for the decrease in the obtained yield of the desired product. This work also highlights the importance of catalyst choice, as the team showed that catalyst 2, a more active metathesis catalyst, improved cross product formation to 60% from 44% with catalyst 1.
While rare, the use of CM to prepare monomers provides an important route to new performance polymers, but also provides a simple exemplification of the importance of alkene reactivity in shaping product selectivity. Importantly, this consistency and selectivity is the principal driving force for using CM prior to polymerisation – the polymers afforded are true homopolymers, with each repeat unit identically functionalised, avoiding concerns of low conversions affording incomplete functional group incorporation.
2.2 Post-polymerisation cross metathesis for polymer functionalisation
Rather than using CM to create monomers, the CM reaction can alternatively be used in post-polymerisation functionalisation. This is a more robust methodology, having been applied to a wide array of polymer backbones and cross partners. While CM has also been used for end-group functionalisation26,27 and cross-linking for self-healing polymers,28,29 the development of CM to directly functionalise polymer backbones is of growing importance.25,30–36 To the best of our knowledge Coates et al. were the first to propose CM functionalisation of alkene containing polyolefins as a general strategy for developing functional polymer architectures in 2004.30 The authors synthesised a variety of copolymers, exemplified by polymer 16 (Scheme 7). Coates et al. demonstrated the versatility of using olefin CM as a tool to functionalise polymers using seven alkenes to modify polymer properties. This included: simple aliphatic alkenes to introduce long chain branching; 4-penten-1-ol to introduce polarity and hydrogen bonders; ethyl acrylate to add non-protic polarity; and fluorinated acrylates to increase phase separation. CM reactions were catalysed by 2 with conversions of 45–91%. The simple aliphatic alkenes and fluorinated acrylates resulted in higher incorporation into the polymer chain as boiling point of the alkene increased. In general, an excess of 10 equivalents of the cross partner was used to favour high conversions.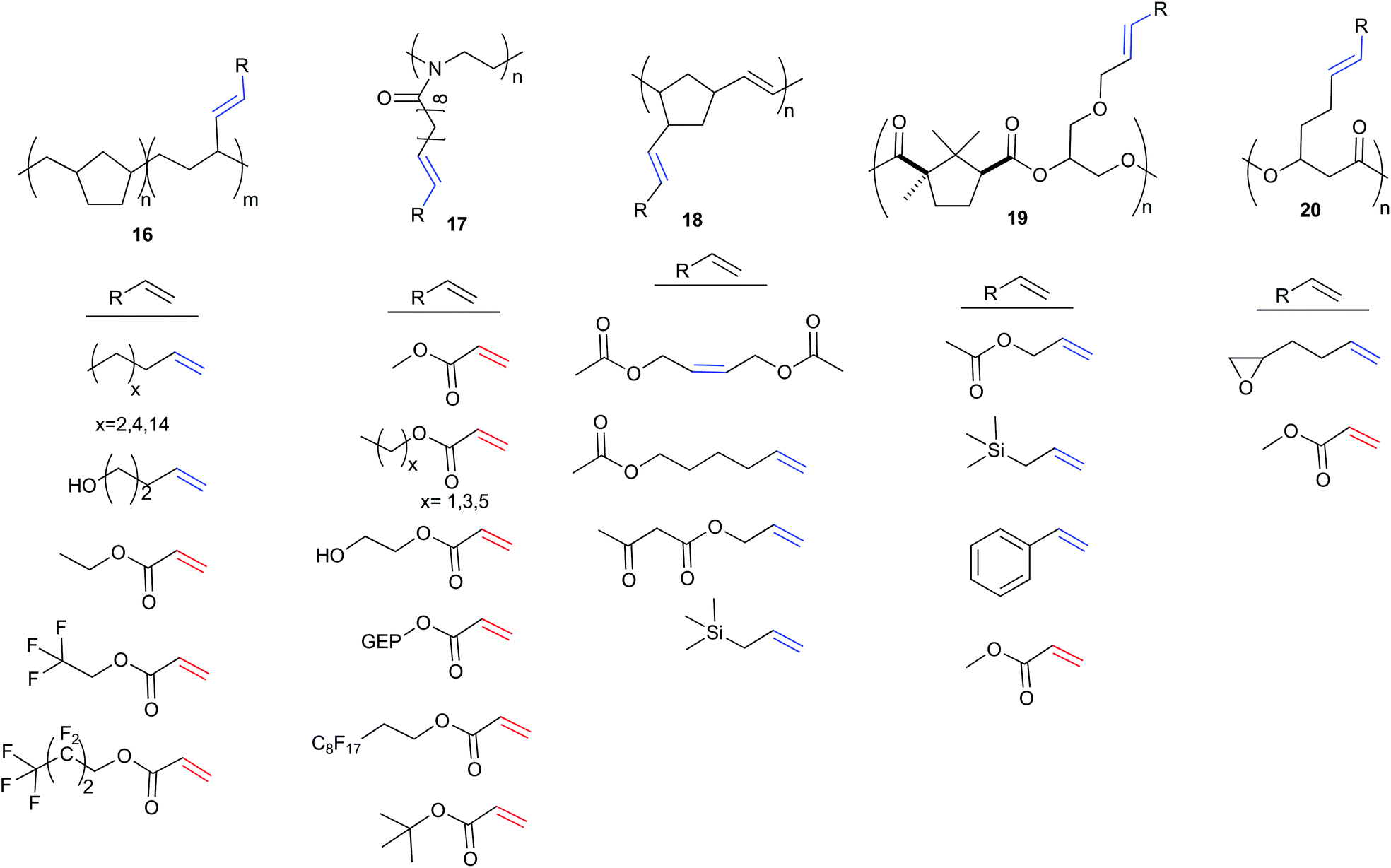 | ||
| Scheme 7 Polymers and their cross-partners for olefin cross metathesis. | ||
In 2012, Hoogenboom and Meier et al. explored the CM of a polymer derived from 10-undecenoic acid, poly(2-oxazoline) (17, Scheme 7) in a “grafting onto approach”,31 similar to earlier work by Kolbe and Meier.36 Post-polymerisation modification was achieved with 8 different acrylates using functional group tolerant Hoveyda–Grubbs second generation catalyst, 3.31 In this work, reaction optimisation were used to limit competing self-metathesis of the polymer chains. The long pendent arm of polymer 17 positions the alkene far from the polymer backbone, promoting higher reactivity and categorises this polymer as a reactive Type I alkene. The fast homodimerisation would thus lead to cross-linking in uncontrolled conditions. Solvent, quantity of cross partner, temperature and catalyst concentration were all essential in optimising this, or in fact any, CM reaction to yield the highest cross metathesis/self-metathesis ratio. Dilute conditions favour less self-metathesis, as the double bonds of the polymers are kept apart, albeit at the expense of catalyst activity. Hoogenboom and Meier et al. explored the impact of reaction conditions on the conversion to cross product using methylacrylate, a type II alkene, as the cross partner. They showed that increasing either temperature or catalyst loading increased conversion, while a decrease in acrylate equivalents increased conversion. Ideal conditions, which both promoted complete conversion and minimised self-metathesis, gave complete functionalisation of the polymer (>99%, 40 °C, 7 equivalents of methyl acrylate, 4 mol% 3). The authors hypothesised that both solvent and acrylate (when installed on the chain) hinder the interaction of two different polymer chains and effectively prevent self-metathesis. This would imply that larger acrylates should promote less self-metathesis, and give rise to a lower associated dispersity (Đ, a measure of the range of macromolecule weights in a sample). They compared the cross metathesis of 17 under optimised reaction conditions with varying acrylate sizes (C1–C6). As expected, hexyl acrylate was best at preventing self-metathesis.
In 2014, Zedník and colleagues illustrated the gradient of reactivity that exists within alkene categories. The scope of alkene cross partners was expanded in the CM of poly(5-vinyl-2-norborene) using catalyst 4 (18, Scheme 7).32 Partners were all type I alkenes; cis-1,4-diacetoxybutene, 5-hexenyl acetate, allyl acetoacetate and allyltrimethylsilane. The short pendent arm of polymer 18 makes this alkene more sterically encumbered, hindering the olefin CM reaction. Unlike previous work that achieved high conversions with acrylate cross partners, the highest degree of polymer functionalisation was achieved with a 2-fold excess of cis-1,4-diacetoxybutene (59% incorporation). The rate of homodimerisation of cis-1,4-diacetoxybutene is thus slower than the rate of selective cross metathesis with the polymer. When an equimolar amount of cross-partner is employed, functionalisation decreases (32%) and cross-linking is favoured, evidenced by a doubling of the polymer molecular weight. Changing the cross partner to 5-hexenyl acetate resulted in low incorporation (10–30%) but no associated increase in molecular weight was observed. In this scenario, the rate of homodimerisation of the cross partner, 5-hexenyl acetate, must be faster than the rate of selective cross metathesis with the polymer. Finally, allyl acetoacetate and allyltrimethylsilane gave very low functionalisation (11% and 6% respectively). The preference of the cross partners to homodimerise, coupled with their steric bulk effectively prevents post-polymerisation modification. Thus, polymer backbone design is essential when considering this synthesis strategy, as is the recognition that, although all of the cross partners can be categorised as Type I alkenes, there remains significant variation in individual reagent reactivity.
In 2016, Thomas and Prunet et al. reported the first modification of a polyester via olefin CM.33 They synthesised copolymers of camphoric anhydride with various olefin containing epoxides, to generate a set of four novel copolymers that contained, in the backbone, Type I, Type II and Type III alkenes. Based on Grubbs’ empirical model and catalyst 3, Thomas and Prunet et al. coupled the Type II and III alkene polyesters (not shown in Scheme 7) with a reactive Type I cross partner (allyltrimethylsilane). However, no selective cross metathesis was observed. The pendent olefin arms in these polyesters were short, and hence more sterically encumbered when in a polymer backbone – especially compared to the prepared Type I polyesters. CM of the Type I polymers with type II alkene methyl acrylate was facile, with high conversions (>90%) and no traces of self-metathesis. The scope of the reaction was expanded to include styrene, allyl acetate and allyltrimethylsilane (19, Scheme 7).
Most recently, Shaver et al. reported the ring-opening polymerisation of β-heptenolactone (β-HL), bearing a pendent type I alkene arm (Polymer 20, Scheme 7).25 CM with both methyl acrylate (Type II) and 1,2-epoxy-5-hexene (Type I) using Hoveyda–Grubbs second generation catalyst 3, in the presence of a large excess (8 equivalents) of the cross partner, could fully functionalise the polymer chains (>99%). Importantly, the methodology could be extended to copolymers of β-HL and lactide. Starting from 85:19 units of lactide:β-HL, 15 different alkenes ranging from Type I to Type III (21, Scheme 8) were incorporated, the largest substrate scope to date. As expected, the Type II and Type III alkenes gave high conversions (>99%) when reacted with the type I copolymer, due to the slow rates of homodimerisation of these cross partners, while the reactive Type I alkenes gave incomplete incorporation.
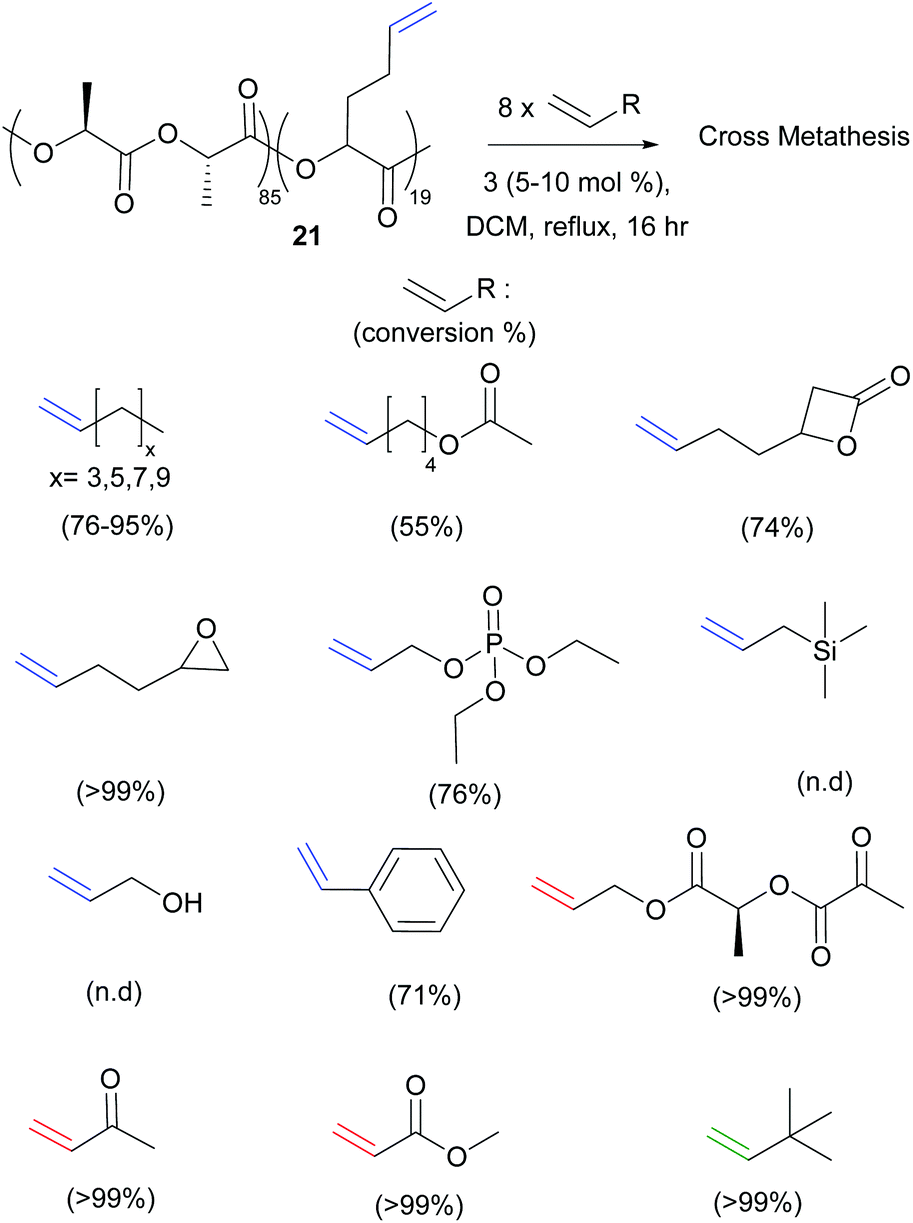 | ||
| Scheme 8 Olefin cross metathesis of biodegradable copolymers of lactide with type I through to type IV alkenes. | ||
Additionally, the group developed a methodology to introduce two distinct functionalities into the same polymer backbone via olefin CM. Type I alkene cross-partners never afforded complete functionalisation of the polymer backbone, with parent alkenes (10–40%) remaining even with more alkene cross-partner and higher catalyst loadings. This was an outcome of the competing metathesis reactions that exist in the presence of two Type I alkenes; homodimerisation of the alkene cross partner, selective cross metathesis between the cross partner and the polymer and secondary metathesis of both the homodimer of the cross partner and of the polymer functionalised with the cross partner. While these competing reactions make selective CM between two Type I alkenes problematic, it offered an interesting approach to introduce two unique functionalities into the polymer backbone. Reacting the copolymer with a Type II partner (i.e. methyl acrylate) first, followed by reaction with a reactive Type I alkene, post precipitation, (i.e. 1,2-epoxy-5-hexene) results in incorporation of both functionalities (22, scenario 1, Scheme 9). If the order of alkene addition is reversed, full incorporation of the Type II partner is observed (23, scenario 2, Scheme 9).
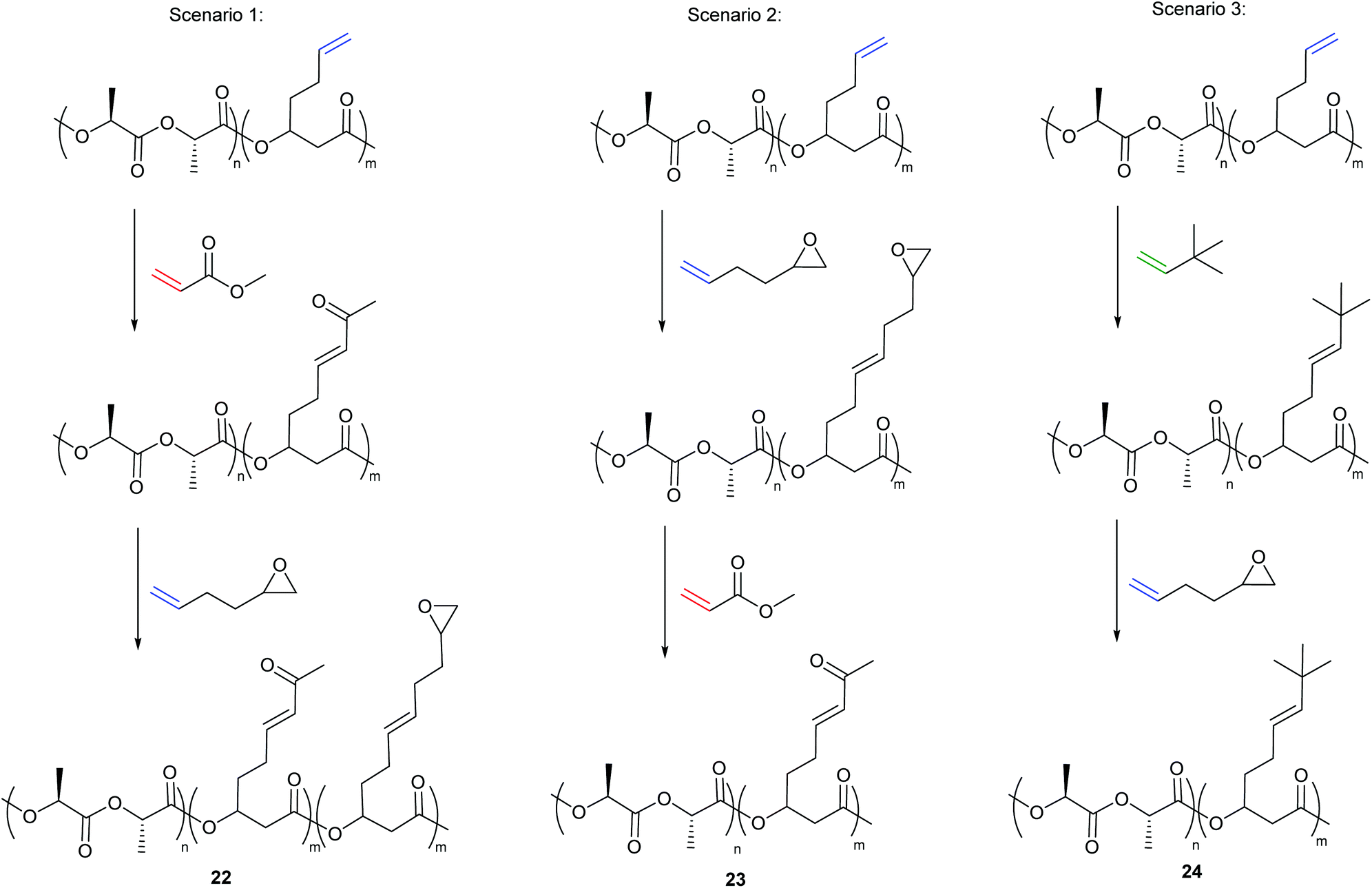 | ||
| Scheme 9 Double cross metathesis based on alkene reactivity to introduce two unique functionalities into the polymer backbone. | ||
By reacting the polymer with the Type II alkene first, an alkene-substituted polymer is formed that is less susceptible to secondary metathesis, but remains reactive towards the added Type I alkene. In the latter case, the polymer functionalised with the Type I alkene first readily undergoes secondary metathesis and completely exchanges with the added Type II alkene, as the rate of secondary metathesis of the polymer functionalised with the epoxide is faster than the rate of homodimerisation of methyl acrylate (type II alkene). Interestingly, when the system is reacted with a Type III alkene first (i.e. 3,3-dimethylbut-1-ene) followed by a reactive type I alkene (1,2-epoxy-5-hexene), no incorporation of the type I alkene is observed (24, Scenario 3, Scheme 9) as the polymer formed can be categorised as a Type IV alkene that is unable to participate in secondary metathesis.
Post-polymerisation functionalisation via olefin CM has a significant impact on the thermal properties of the resulting polymers. Functional group incorporation can significantly alter the glass transition temperature of the polymer as seen in the work of the groups of Coates, Hoogenboom and Meier, Thomas and Prunet, where the glass transition temperature was increased substantially. It can also affect the crystallinity of the polymer as seen in the work of Shaver et al. by changing the intramolecular bonding occurring between polymer chains. The application scope and expansion of new materials can be improved, adding groups such as alcohols to increase hydrophilicity, silanes for binding to solid supports, phosphonates for fire retardancy and epoxides for cross-linking and further derivatisation. Moreover as the alkene is still present post-functionalisation, unlike many other alkene transformations, further potential modification of the polymer can be carried out.
2.3. Post-polymerisation olefin cross metathesis of dendrimers
While not strictly polymers, the CM strategy can also be used to functionalise dendrimers. With multiple arms decorated with terminal alkenes, the competition is not simply between cross-partners but also from cyclisation of two adjacent arms – providing a segue to the next section of this review covering ring-closing metathesis. Dendrimers are spherical macromolecules containing a central core, which are typically synthesised via divergent (the dendrimer is assembled from the core out using a multifunctional core and a series of reactions) syntheses or convergent (the dendrimer is assembled from the periphery inwards via the combination of small molecules, ending with attachment to a central core) syntheses.CM has been used as a tool to build dendrimers, acting as part of an iterative sequence,37 though only a handful of examples exist that use CM to directly functionalise the polymer architecture.38–40 Astruc and co-workers pioneered dendrimer functionalisation via post-synthesis olefin CM.38 Using a divergent synthesis, dendrimer 25 (Scheme 10) was constructed and reacted with acrylic acid and methyl acrylate using catalyst 2, to yield functionalised dendrimers 26a and 26b respectively. As discussed previously, selectivity is problematic in olefin CM – and the proximity of chains made intramolecular self-metathesis (RCM) particularly favourable as the reaction generated favoured 5-membered rings.
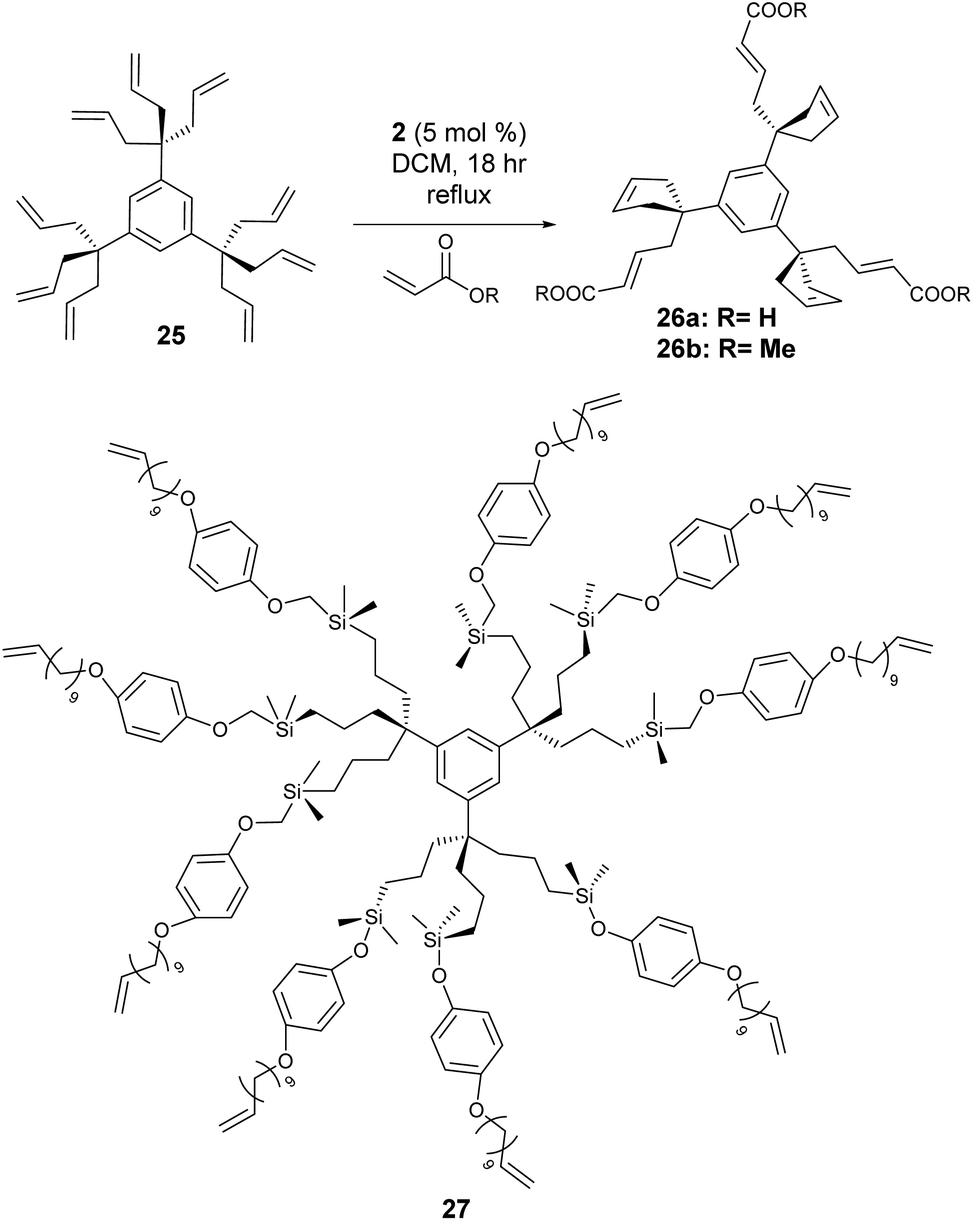 | ||
| Scheme 10 Functionalisation of dendrimers via olefin cross metathesis. | ||
To overcome this problem, the length of the dendrimer tethers was increased via hydrosilyation and a variety of dendrimers were generated consisting of 9, 27 (27, Scheme 10) and 81 atom tethers. With RCM now disfavoured due to the resultant ring size, selective CM between the host alkene and the dendrimer occurred to produce water soluble poly(carboxylic acid) dendrimers. Astruc expanded the scope of alkenes to include acrylates containing ferrocenyl groups to produce responsive dendrimers.39
Fréchet and co-workers used olefin CM to transform a generic poly(benzyl ether) dendrimer into a functionalised derivative.40 The dendrimer was synthesised using a convergent approach containing internal allyl ether groups in the final architecture. Upon CM with a UV-active allylated pyrene derivative, 60% incorporation of the cross-partner was achieved, although some competing RCM was observed. This competing metathesis reaction is more favourable than dendrimer-partner coupling due to the close packing of the allyl branches.
3. Ring-closing metathesis
As discussed in the previous section, RCM is often actively avoided, but this complementary reaction can itself be an important tool to shape the synthesis of specialty polymers and dendrimers. As RCM produces rings and capsules of varying sizes, properties and applications, it targets modification of macrostructure rather than introduction of new functionality. The reaction is driven entropically by the evolution of ethylene gas and, as with CM, reaction conditions greatly influence product formation. High dilutions are required to limit intermolecular cross-linking and promote intramolecular RCM – if the reaction mixture is too concentrated, CM will occur promoting ADMET polymerisation and through it extensive cross-linking (Scheme 11).41–44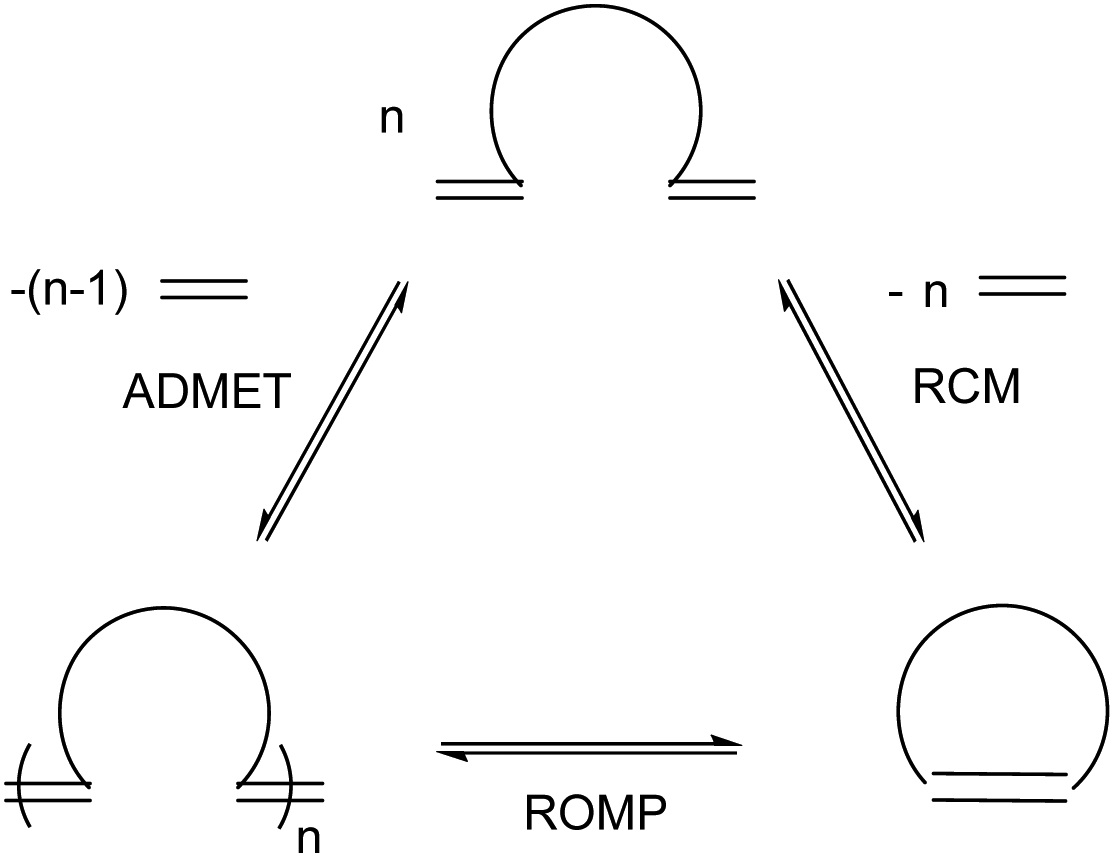 | ||
| Scheme 11 Relationship of various metathesis reactions at equilibrium. ADMET: acyclic diene metathesis, RCM: ring-closing metathesis, ROMP: ring-opening metathesis polymerisation. Reprinted (adapted) with permission from (S. Monfette and D. E. Fogg, Chem. Rev., 2009, 109, 3783–3816.). Copyright (2009) American Chemical Society. | ||
Even with this entropic advantage, ring strain also dictates ring formation. Five, six or seven membered rings are readily achieved through RCM, while the formation of highly strained four-membered rings is thermodynamically unfavourable. The formation of large rings is also challenging, as the probability of two olefins interacting to form a ring is lower for longer pendent chains.
Consequently, RCM of medium (8–11 membered rings) and large rings (≥12) is more difficult. The medium rings not only have a kinetic disadvantage, but also suffer from unfavourable transannular interactions. However, among many cyclisation reactions, RCM has gained enormous popularity in recent years as a tool to make these challenging rings.45 Construction of rings does not only represent a central theme in natural product synthesis but also produces feedstocks for ROMP. It is thus no coincidence that these cycles dominant use is as feedstocks for ring opening metathesis polymerisations (ROMP). Finally, catalyst choice is again important for achieving high conversions in RCM reactions. As with CM, Ru-based catalysts dominate thanks to their ease-of-use and high functional group tolerance.46,47
3.1 Ring-closing metathesis of dendrimers
Dendrimer formation and functionalisation via RCM has led to many important applications including drug delivery agents,48 molecular imprints,49 and covalent organic nanotubes.50 In 1999, Zimmerman demonstrated the first successful RCM of a dendrimer (28, Scheme 12), which coupled terminal homoallyl ether groups to generate a ring-closed polymer 29.48 The uniformity of the ring size and shape in RCM allows a high degree of control over the dimensions, and thus overall properties of these unique structures. Judicious control of reaction conditions was essential for achieving complete conversion and for achieving the desired product. High concentrations led to CM between dendrimers, promoting higher molecular weights and dispersities. Optimum conditions (4 mol% 1, [dendrimer] < 10−5 M) favoured RCM exclusively.51 Increasing steric bulk around the terminal alkene also favoured RCM at higher concentrations, while compact catalysts with minimal bulk (catalyst 2) gave improved yields. Catalyst 2 also benefits from a higher thermal stability, permitting a secondary rearrangement of the dendrimers to form extensively intramolecular cross-linked dynamic molding structures.52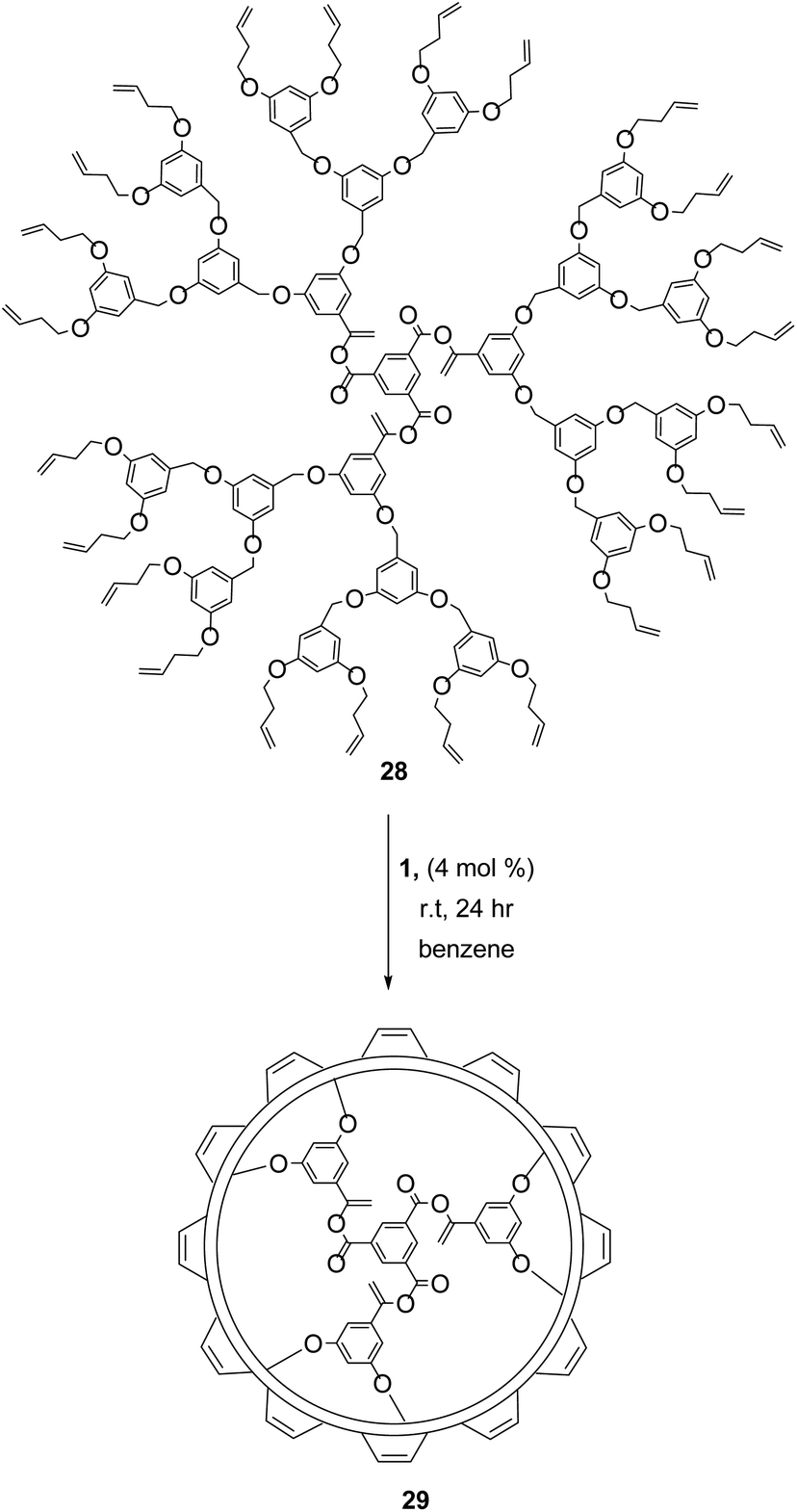 | ||
| Scheme 12 Ring-closing metathesis of dendrimers with homoallyl ether terminal group. Reprinted (adapted) with permission from (M. S. Wendland and S. C. Zimmerman, J. Am. Chem. Soc., 1999, 121, 1389–1390). Copyright (1999) American Chemical Society. | ||
The reversibility of olefin metathesis reactions, offers another tool for controlling polymer properties, changing the size and rigidity of the formed dendrimer. Theoretically, complete RCM should lead to a decrease in ethylene mass (per two terminal olefin units) of the overall mass, but experimental molecular weight determinations suggest a much greater mass loss. Moreover, increased reaction times result in a decrease in the hydrodynamic volume of the dendrimer to reach the most thermodynamically stable conformation. Thus, the size of individual macromolecules can be tuned by the degree of RCM and thus the size of the resultant nanoparticles.53
3.2 Nanoparticles from ring-closing metathesis
This use of RCM to knit-in macromolecules can be extended to the synthesis of nanoparticles from polymers as well. In particular, the use of three-armed star tethers allows competing RCM and longer intramolecular cross-linking to form so-called nanosize star imprinted polymers.54,55 The rigidity of the peripheral shell, built using RCM, and the length of tether that shapes ring sizes, can control both diffusion and guest/drug encapsulation. Larger “loops” improved complexation and encapsulation, at the expense of peripheral stability.56 An excellent example of this feature is shown in Scheme 13, transforming the cross-linkable polymer 30 into defined nanoparticle 31. The latent remaining olefin functionality can be further derivatised by post-RCM dihydroxylation to produce water-soluble nanoparticles of controlled size and shape.57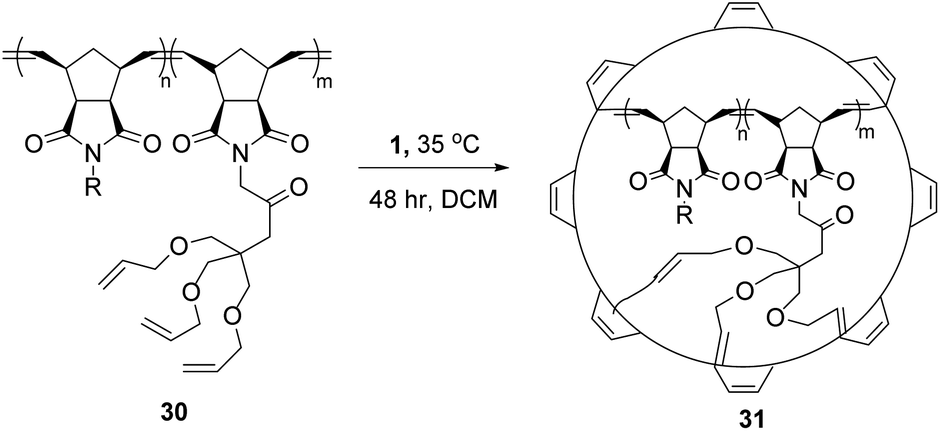 | ||
| Scheme 13 Synthesis of organic nanoparticles using ring-closing metathesis process. Reproduced from Ref. Y. Bai, H. Xing, G. a. Vincil, J. Lee, E. J. Henderson, Y. Lu, N. G. Lemcoff and S. C. Zimmerman, Chem. Sci., 2014, 5, 2862. with permission from the Royal Society of Chemistry. | ||
Coates et al. investigated different reaction conditions on a vinyl functionalised polymer, 32 (Scheme 14), to observe different olefin metathesis reactions and analyse the outcomes via different characterisation techniques. At high concentrations of polymer (>10 mg mL−1), both CM and RCM occurred. However, working at dilute polymer solutions (1 mg mL−1), the apparent molecular weight decreased steadily, and the molecular weight distribution remained narrow, indicating that only intramolecular RCM occurred.58
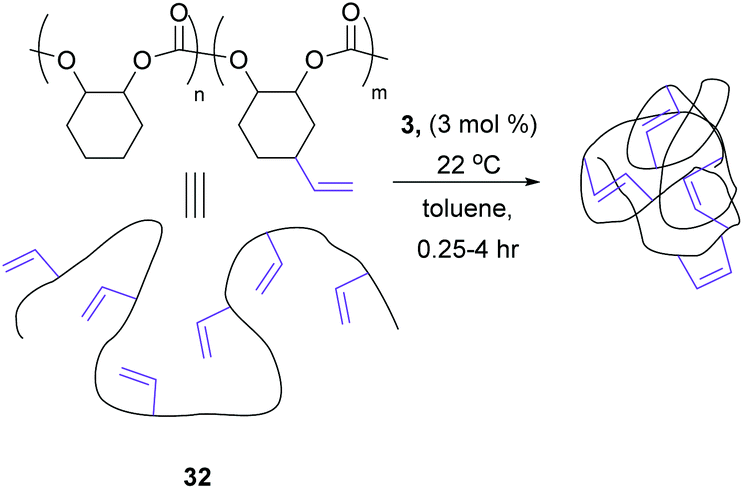 | ||
| Scheme 14 Exclusive ring-closing metathesis on a vinyl functionalised polymer to produce nanoparticles. Reprinted (adapted) with permission from (A. E. Cherian, F. C. Sun, S. S. Sheiko and G. W. Coates, J. Am. Chem. Soc., 2007, 129, 11350–11351). Copyright (2007) American Chemical Society. | ||
3.3 Cyclopolymers from ring-closing metathesis
Cyclopolymers are linear polymers consisting of in-chain cyclic structures. Several strategies have been devised to prepare cyclopolymers using olefin metathesis. This provides a powerful and easy method to prepare polymers that contain alternating double and single bonds along the polymer main chain with a cyclic recurring unit. For instance, tandem RO/RCM of unsaturated bicyclic monomer 33 produces 34 (Scheme 15).59 Modified ADMET polymerisation of diyne monomers also produces cyclopolymers (35 to 36, Scheme 15).60,61 Neither of these strategies exploit a direct RCM to produce cyclopolymers. Coates and Grubbs et al. first showed this was possible, reacting atactic 1,2-polybutadiene with catalyst 1 to afford the cyclopolymer 37 (Scheme 16). Analysing the data, they observed a lower molecular weight (Mn) and broader polydispersity (Đ) of the polycyclopentene compared to the starting material. They attributed this to metathetical degradation of the infrequent 1,4-vinyl units along the polymer backbone. Kinetic studies show the reaction proceeds quickly to 90% conversion in 30 min, followed by a much slower observed rate, reaching 98% conversion in 21 h. Importantly, this suggests the catalyst randomly, and quickly, closes adjacent olefins along the backbone until only isolated olefins remain. Slow rearrangement through secondary metathesis continues up and down the chain until all olefins are cyclised.62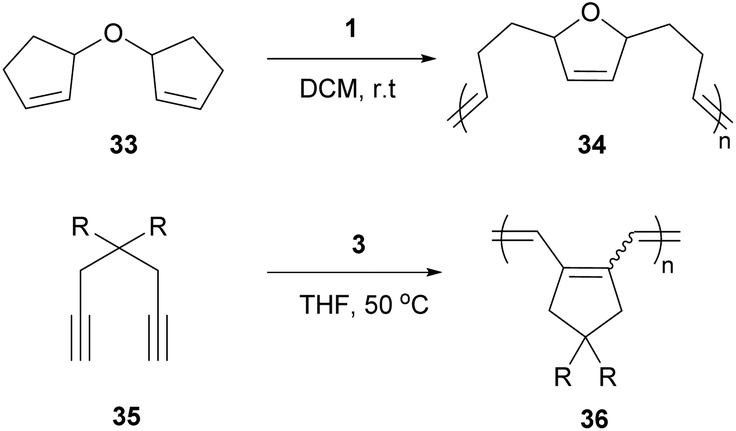 | ||
| Scheme 15 Current routes to generate cyclopolymers via metathesis polymerisation. | ||
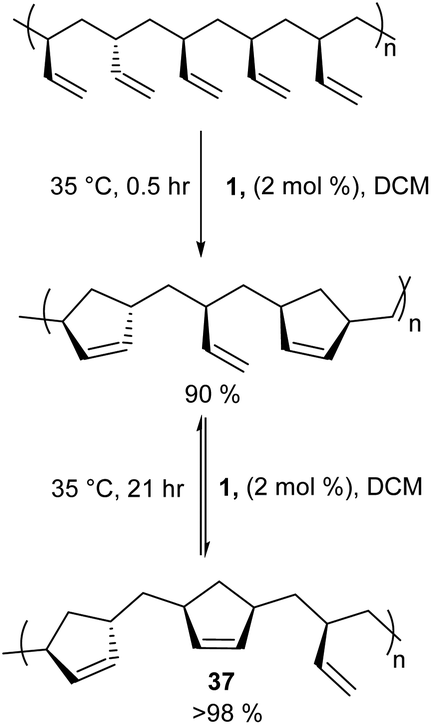 | ||
| Scheme 16 Atactic 1,2-polybutadiene cyclization via ring-closing metathesis. | ||
In 2015, Onitsuka extended this work to produce a chiral cyclopolymer (38, Scheme 17). As RCM can restrict the conformation of the main chain, it can lock asymmetric centres to create a fixed chiral polymer, rather than a rotatable chiral chain. These local conformational restrictions lead to a specific compact structure of the entire polymer which influences both physical properties and biological activity.63
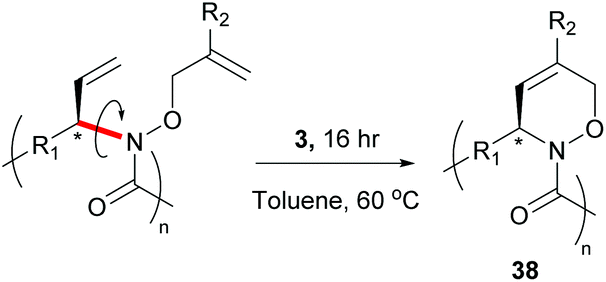 | ||
| Scheme 17 Ring-closing metathesis on a chiral linear polymer to generate a chiral cyclopolymer. Reprinted (adapted) with permission from (N. Kanbayashi, T. A. Okamura and K. Onitsuka, Macromolecules, 2015, 48, 8437–8444). Copyright (2015) American Chemical Society. | ||
3.4 Cyclic polymers from ring-closing metathesis
Differing from cyclopolymers, cyclic polymers (CPs) have gathered particular attention due to their unusual topology. These polymers lack end groups and exhibit a substantial decrease in their hydrodynamic volume compared to their linear counterparts. Additionally it has been shown that CPs have better circulation half-lives than their linear counterparts making them attractive candidates for possible drug carriers and other biological applications.64 In addition to their interesting properties, CPs also present a unique challenge to synthetic chemists, as there are few procedures that favour the formation of these “endless” polymers to high purity. Current synthetic strategies have their drawbacks. End-to-end linking reactions is a common route to generate CPs, however this method is limited due to competing chain-extension reactions that are favoured with increasing chain length. More traditionally, CPs are synthesized via high-dilution cyclisation of dianionic linear polymers in the presence of a two-site coupling agent yet, RCM offers a more attractive approach to making CPs. Binder and colleagues compared the macrocyclisation of polyisobutylenes (PIB's) via RCM and azide/alkyne-“click”-chemistry. The results indicated both methods produced a mixture of cyclic and oligomeric products. The main advantage of RCM is the reaction is reversible whereas “click”-chemistry is not, thus fixing the generated structure without chance of correction. This therefore made RCM the more attractive option.65RCM has acted as an effective tool in forming the cyclic version of linear polymers. Polystyrene-b-poly (ethylene oxide) (cyclic PS-b-PEO), a micelle formed from a cyclic amphiphile, displays a significantly enhanced thermal stability compared to its linear counterpart.66 Other examples include cyclic poly(phosphoester) and cyclic stereoblock polylactides (39, Scheme 18), which were both formed from RCM and could display better biological activity than their linear forms.67,68
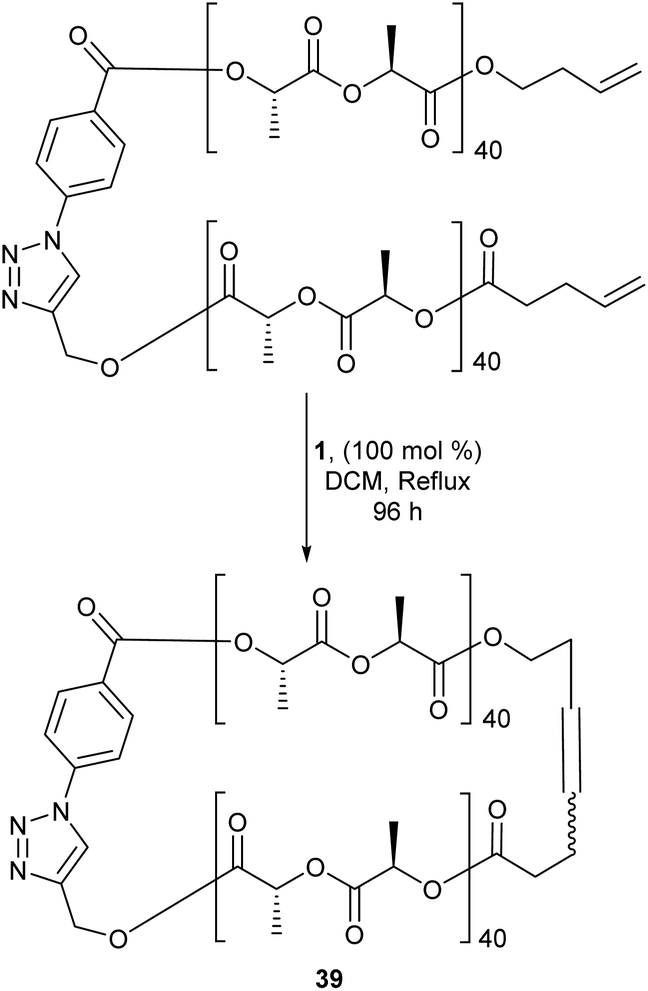 | ||
| Scheme 18 Synthetic scheme of cyclic stereoblock poly(lactide) acid via ring-closing metathesis. Reprinted (adapted) with permission from (N. Sugai, T. Yamamoto and Y. Tezuka, ACS Macro Lett., 2012, 1, 902–906). Copyright (2012) American Chemical Society. | ||
Later, Grubbs et al. used RCM to synthesise novel interlocked molecules. Starting with a cyclic polyammonium compound, a molecular charm bracelet structure was made possible by interlocking diolefin polyether fragments around the cyclic polyammonium backbone via RCM (40, Scheme 19).64 Another factor to consider in the success of RCM is the choice of solvent. Foster et al. investigated the effect of solvent in promoting RCM compared to intermolecular CM. They hypothesised that the efficiency of the intramolecular RCM could be enhanced in the presence of a solvent the polymer had unfavourable interactions with thereby forcing a more compact conformation of the polymer and increasing the probability of intramolecular over intermolecular metathesis. Foster et al. compared solvents cyclohexane to methylene chloride as an unfavourable and favourable solvent respectively. RCM was attempted in pure methylene chloride and a mixture of methylene chloride and cyclohexane. As hypothesised RCM in the solvent mixture lead to the formationof a polymer with lower hydrodynamic volume, an indication of successful RCM, whereas in pure methylene chloride an increase in molecular weight was observed which was indicative of CM.69 It is worth mentioning that tethered-alkylidene variants of standard metathesis catalysts open the door to high molecular weight cyclic polymers and high-concentration polymerisation reactions through ring-expansion metathesis polymerisation (REMP). Grubbs et al. reported a series of cyclic Ru-alkylidene catalysts resembling catalyst 1 as new catalysts systems that were able to mediate (REMP) of cyclic olefins to produce cyclic polymers.70,71
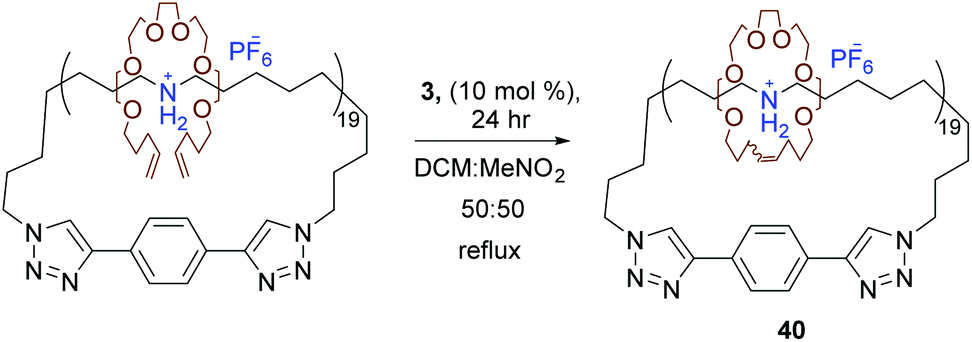 | ||
| Scheme 19 Ring-closing metathesis of polyether around cyclic polyammonium to generate a molecular charm bracelet. Reprinted (adapted) with permission from (P. G. Clark, E. N. Guidry, W. Y. Chan, W. Steinmetz and R. H. Grubbs, J. Am. Chem. Soc., 2010, 132, 3405–3412). Copyright (2010) American Chemical Society. | ||
4. Characterisation techniques
Identifying the difference between intramolecular RCM and intermolecular CM can be challenging, and although not a comprehensive list the following techniques can be helpful in identifying successful CM and RCM. NMR is the most common technique used to provide evidence of successful functional group incorporation in olefin CM (1H, 13C, COSY, HSQC and HMBC). As mentioned throughout, gel permeation chromatography (GPC) measures hydrodynamic volume of polymers which is then correlated to molecular weight. While CM functionalisation may alter the hydrodynamic volume, increasing the apparent molecular weight, this is not always representative. In the work of Shaver et al.25 no change in molecular weight was observed for the polymer post-metathesis, indicating a negligible difference in the hydrodynamic volume upon CM.In RCM, again 1H NMR spectroscopy can easily distinguish between the alkene methine proton of uncross-linked and cross-linked alkene (self-metathesis). In GPC, RCM is associated with a decrease in the hydrodynamic volume of the polymer displaying an associated lower retention time, while intermolecular CM should display an associated increase in molecular weight and broader dispersity due to the cross linking between the polymer chains and thus display a higher retention time.51,69 To give a more accurate assessment of the true molecular weight of a ring-closed polymer multi-angle laser light scattering (MALLS) can be used.57 Thermal analysis can also help aid the identification of RCM. Generally, as RCM occurs the glass transition temperature of the polymer should increase, a result of a decrease in chain mobility. Coates et al. studied the molecular weight and glass transition temperature of products of a RCM reaction at various time intervals. The results are shown in Table 1 and it can be seen that as the reaction time and conversion increase the molecular weight (observed by GPC) decreases while the Tg increases.58
| Entry | Time (h) |
M
nb (g mol−1) |
M
w/Mnc |
% Vinyls RCM |
T
gd (°C) |
|---|---|---|---|---|---|
| a All reactions were run with 2 mol% of Ru catalyst at 22 °C with 1.0 mg polymer per mL toluene. b Determined by GPC in THF at 40 °C versus polystyrene standards. c Determined by 1HNMRspectroscopy. d Determined by differential scanning calorimetry (second heat). Reprinted (adapted) with permission from (A. E. Cherian, F. C. Sun, S. S. Sheiko and G. W. Coates, J. Am. Chem. Soc., 2007, 129, 11350–11351). Copyright (2007) American Chemical Society. | |||||
| 1 | 0 | 54100 |
1.2 | 0 | 114 |
| 2 | 0.25 | 45700 |
1.34 | 42 | 157 |
| 3 | 0.50 | 39500 |
1.26 | 59 | 167 |
| 5 | 2.0 | 33000 |
1.19 | 70 | 185 |
| 6 | 4.0 | 31500 |
1.19 | 76 | 194 |
Moreover, matrix-assisted laser desorption ionization time-of-flight mass spectroscopy (MALDI-TOF-MS) is also a supporting technique to show the loss of ethylene mass and determining the end groups of the polymer after cyclization.72 Atomic-force microscopy (AFM) was also used to differentiate between starting linear polymers and the produced nanoparticles58 or macrocycles. Diffusion-ordered spectroscopy (DOSY) is a powerful technique to distinguish between different cycle sizes.64
5. Conclusions
This review highlights the importance of olefin metathesis as a tool to alter and improve polymer properties. It is key to understand the requirements for effective olefin CM and RCM in order to achieve and manipulate successful reactions to afford polymers of unique functionality and architectures. To summarise, in order to incorporate functional groups into polymers via olefin CM reacting alkenes of different types under dilute conditions is optimum. Moreover, polymers with long pendent olefin arms that are not sterically encumbered allow a wider scope of bulky cross-partners to be incorporated, giving polymers with tunable thermal properties. To favour RCM over intermolecular CM high dilutions and long reaction times are desired. RCM can enable architectures with enhanced biological activity and useful nanoparticles. With continued research into metathesis-driven polymer modification, the scope of new materials will further expand.Acknowledgements
FS, MA and MS thank the University of Edinburgh for a Principal's Career Development Scholarship and the Marie-Curie Actions Programme (FP7-PEOPLE-2013-CIG-618372) for financial support. JP and MA thank the University of Glasgow and the EPSRC (DTG EP/M506539/1) for financial support.References
- R. P. Brannigan and A. P. Dove, Biomater. Sci., 2017, 5, 9–21 RSC.
- A. W. Thomas and A. P. Dove, Macromol. Biosci., 2016, 16, 1762–1775 CrossRef CAS PubMed.
- J.-L. Hérisson and Y. Chauvin, Makromol. Chem., 1971, 141, 161–167 CrossRef.
- A.-C. Knall and C. Slugovc, in Olefin Metathesis, John Wiley & Sons, Inc., Hoboken, NJ, USA, 2014, pp. 269–284 Search PubMed.
- S. Hilf and A. F. M. Kilbinger, Nat. Chem., 2009, 1, 537–546 CrossRef CAS PubMed.
- C. W. Bielawski and R. H. Grubbs, Prog. Polym. Sci., 2007, 32, 1–29 CrossRef CAS.
- C. W. Bielawski and R. H. Grubbs, Angew. Chem., Int. Ed., 2000, 39, 2903–2906 CrossRef CAS PubMed.
- H. Mutlu, L. Montero de Espinosa and M. A. R. Meier, Chem. Soc. Rev., 2011, 40, 1404–1445 RSC.
- M. D. Schulz and K. B. Wagener, Macromol. Chem. Phys., 2014, 1936–1945 CrossRef CAS.
- A. J. Hall, P. Hodge, S. D. Kamau and A. Ben-Haida, J. Organomet. Chem., 2006, 691, 5431–5437 CrossRef CAS.
- J. Prunet, Eur. J. Org. Chem., 2011, 2011, 3634–3647 CrossRef CAS.
- M. Schuster and S. Blechert, Angew. Chem., Int. Ed. Engl., 1997, 36, 2036–2056 CrossRef.
- J. W. Herndon, Coord. Chem. Rev., 2016, 329, 53–162 CrossRef CAS.
- V. M. Marx, L. E. Rosebrugh, M. B. Herbert and R. H. Grubbs, Ruthenium in Catalysis, Springer International Publishing, 2014, pp. 1–19 Search PubMed.
- D. J. Nelson, S. Manzini, C. A. Urbina-Blanco and S. P. Nolan, Chem. Commun., 2014, 50, 10355–10375 RSC.
- A. H. Hoveyda and A. R. Zhugralin, Nature, 2007, 450, 243–251 CrossRef CAS PubMed.
- A. K. Chatterjee, T.-L. Choi, D. P. Sanders and R. H. Grubbs, J. Am. Chem. Soc., 2003, 125, 11360–11370 CrossRef CAS PubMed.
- X. Miao, C. Fischmeister, P. H. Dixneuf, C. Bruneau, J.-L. Dubois and J.-L. Couturier, Green Chem., 2012, 14, 2179 RSC.
- X. Miao, R. Malacea, C. Fischmeister, C. Bruneau and P. H. Dixneuf, Green Chem., 2011, 13, 2911–2919 RSC.
- A. Rybak, P. A. Fokou and M. A. R. Meier, Eur. J. Lipid Sci. Technol., 2008, 110, 797–804 CrossRef CAS.
- M. von Czapiewski, O. Kreye, H. Mutlu and M. A. R. Meier, Eur. J. Lipid Sci. Technol., 2013, 115, 76–85 CrossRef CAS.
- X. Miao, C. Fischmeister, C. Bruneau and P. H. Dixneuf, ChemSusChem, 2009, 2, 542–545 CrossRef CAS PubMed.
- M. Sacristán, J. C. Ronda, M. Galià and V. Cádiz, J. Appl. Polym. Sci., 2011, 122, 1649–1658 CrossRef.
- M. Winkler and M. A. R. Meier, Green Chem., 2014, 16, 3335–3340 RSC.
- F. Sinclair, L. Chen, B. W. Greenland and M. P. Shaver, Macromolecules, 2016, 49, 6828–6834 CrossRef.
- A. A. Nagarkar, A. Crochet, K. M. Fromm and A. F. M. Kilbinger, Macromolecules, 2012, 45, 4447–4453 CrossRef CAS.
- H.-K. Lee, K.-T. Bang, A. Hess, R. H. Grubbs and T.-L. Choi, J. Am. Chem. Soc., 2015, 137, 9262–9265 CrossRef CAS PubMed.
- Y.-X. Lu and Z. Guan, J. Am. Chem. Soc., 2012, 134, 14226–14231 CrossRef CAS PubMed.
- J. A. Neal, D. Mozhdehi and Z. Guan, J. Am. Chem. Soc., 2015, 137, 4846–4850 CrossRef CAS PubMed.
- R. T. Mathers and G. W. Coates, Chem. Commun., 2004, 422–423 RSC.
- L. M. De Espinosa, K. Kempe, U. S. Schubert, R. Hoogenboom and M. A. R. Meier, Macromol. Rapid Commun., 2012, 33, 2023–2028 CrossRef PubMed.
- H. Balcar, T. Shinde, M. Lamač, J. Sedláček and J. Zedník, J. Polym. Res., 2014, 21, 557 CrossRef.
- L. Fournier, C. Robert, S. Pourchet, A. Gonzalez, L. Williams, J. Prunet and C. M. Thomas, Polym. Chem., 2016, 7, 3700–3704 RSC.
- K. Malzahn, F. Marsico, K. Koynov, K. Landfester, C. K. Weiss and F. R. Wurm, ACS Macro Lett., 2014, 3, 40–43 CrossRef CAS.
- H.-Y. Chen, J. H. Lai, X. Jiang and J. Lahann, Adv. Mater., 2008, 20, 3474–3480 CrossRef CAS.
- N. Kolb and M. A. R. Meier, Eur. Polym. J., 2013, 49, 843–852 CrossRef CAS.
- O. Kreye, D. Kugele, L. Faust and M. A. R. Meier, Macromol. Rapid Commun., 2014, 35, 317–322 CrossRef CAS PubMed.
- C. Ornelas, D. Méry, J. C. Blais, E. Cloutet, J. R. Aranzaes and D. Astruc, Angew. Chem., Int. Ed., 2005, 44, 7399–7404 CrossRef CAS PubMed.
- C. Ornelas, D. Méry, E. Cloutet, J. R. Aranzaes and D. Astruc, J. Am. Chem. Soc., 2008, 130, 1495–1506 CrossRef CAS PubMed.
- C. O. Liang and J. M. J. Fréchet, Macromolecules, 2005, 38, 6276–6284 CrossRef CAS.
- R. H. Grubbs and S. Chang, Tetrahedron, 1998, 54, 4413–4450 CrossRef CAS.
- S. K. Armstrong, J. Chem. Soc., Perkin Trans. 1, 1998, 371–188 RSC.
- D. J. Nelson, I. W. Ashworth, I. H. Hillier, S. H. Kyne, S. Pandian, J. A. Parkinson, J. M. Percy, G. Rinaudo and M. A. Vincent, Chem. – Eur. J., 2011, 17, 13087–13094 CrossRef CAS PubMed.
- J. C. Conrad, M. D. Eelman, J. A. Duarte Silva, S. Monfette, H. H. Parnas, J. L. Snelgrove and D. E. Fogg, J. Am. Chem. Soc., 2007, 129, 1024–1025 CrossRef CAS PubMed.
- M. E. Maier, Angew. Chem., Int. Ed., 2000, 39, 2073–2077 CrossRef CAS PubMed.
- S. Monfette and D. E. Fogg, Chem. Rev., 2009, 109, 3783–3816 CrossRef CAS PubMed.
- K. Arakawa, T. Eguchi and K. Kakinuma, J. Org. Chem., 1998, 63, 4741–4745 CrossRef CAS.
- S. L. Elmer and S. C. Zimmerman, J. Org. Chem., 2004, 69, 7363–7366 CrossRef CAS PubMed.
- S. C. Zimmerman, M. S. Wendland, N. A. Rakow, I. Zharov and K. S. Suslick, Nature, 2002, 418, 399–403 CrossRef CAS PubMed.
- Y. Kim, M. F. Mayer and S. C. Zimmerman, Angew. Chem., Int. Ed., 2003, 42, 1121–1126 CrossRef CAS PubMed.
- M. S. Wendland and S. C. Zimmerman, J. Am. Chem. Soc., 1999, 121, 1389–1390 CrossRef CAS.
- L. G. Schultz, Y. Zhao and S. C. Zimmerman, Angew. Chem., Int. Ed., 2001, 40, 1962–1966 CrossRef CAS PubMed.
- N. G. Lemcoff, T. A. Spurlin, A. A. Gewirth, S. C. Zimmerman, J. B. Beil, S. L. Elmer and H. G. Vandeveer, J. Am. Chem. Soc., 2004, 126, 11420–11421 CrossRef CAS PubMed.
- J. B. Beil and S. C. Zimmerman, Macromolecules, 2004, 37, 778–787 CrossRef CAS.
- G. E. Southard, K. A. Van Houten and G. M. Murray, Macromolecules, 2007, 40, 1395–1400 CrossRef CAS.
- E. Burakowska, J. R. Quinn, S. C. Zimmerman and R. Haag, J. Am. Chem. Soc., 2009, 131, 10574–10580 CrossRef CAS PubMed.
- Y. Bai, H. Xing, G. A. Vincil, J. Lee, E. J. Henderson, Y. Lu, N. G. Lemcoff and S. C. Zimmerman, Chem. Sci., 2014, 5, 2862 RSC.
- A. E. Cherian, F. C. Sun, S. S. Sheiko and G. W. Coates, J. Am. Chem. Soc., 2007, 129, 11350–11351 CrossRef CAS PubMed.
- H.-K. Lee, K.-T. Bang, A. Hess, R. H. Grubbs and T.-L. Choi, J. Am. Chem. Soc., 2015, 137, 9262–9265 CrossRef CAS PubMed.
- E.-H. Kang, I.-H. Lee and T.-L. Choi, ACS Macro Lett., 2012, 1, 1098–1102 CrossRef CAS.
- E.-H. Kang, S. Y. Yu, I. S. Lee, S. E. Park and T.-L. Choi, J. Am. Chem. Soc., 2014, 136, 10508–10514 CrossRef CAS PubMed.
- G. W. Coates and R. H. Grubbs, J. Am. Chem. Soc., 1996, 118, 229–230 CrossRef CAS.
- N. Kanbayashi, T. A. Okamura and K. Onitsuka, Macromolecules, 2015, 48, 8437–8444 CrossRef CAS.
- P. G. Clark, E. N. Guidry, W. Y. Chan, W. Steinmetz and R. H. Grubbs, J. Am. Chem. Soc., 2010, 132, 3405–3412 CrossRef CAS PubMed.
- S. Matsumura, A. R. Hlil, C. Lepiller, J. Gaudet, D. Guay, Z. Shi, S. Holdcroft and A. S. Hay, Am. Chem. Soc. Polym. Prepr. Div. Polym. Chem., 2008, 49, 511–512 CAS.
- Y. Tezuka, Polym. Chem., 2012, 3, 1903–1909 RSC.
- L. Ding, R. Lu, J. An, X. Zheng and J. Qiu, React. Funct. Polym., 2013, 73, 1242–1248 CrossRef CAS.
- N. Sugai, T. Yamamoto and Y. Tezuka, ACS Macro Lett., 2012, 1, 902–906 CrossRef CAS.
- R. P. Quirk, S. F. Wang, M. D. Foster, C. Wesdemiotis and A. M. Yol, Macromolecules, 2011, 44, 7538–7545 CrossRef CAS.
- Y. Xia, A. J. Boydston, Y. Yao, J. A. Kornfield, I. A. Gorodetskaya, H. W. Spiess and R. H. Grubbs, J. Am. Chem. Soc., 2009, 131, 2670–2677 CrossRef CAS PubMed.
- A. J. Boydston, Y. Xia, J. A. Kornfield, I. A. Gorodetskaya and R. H. Grubbs, J. Am. Chem. Soc., 2008, 130, 12775–12782 CrossRef CAS PubMed.
- T. Terashima, M. Kawabe, Y. Miyabara, H. Yoda and M. Sawamoto, Nat. Commun., 2013, 4, 1–9 Search PubMed.
| This journal is © The Royal Society of Chemistry 2017 |