
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The hydrolytic behavior of N,N′-(dimethylamino)ethyl acrylate-functionalized polymeric stars†
Marianne S.
Rolph
,
Anaïs
Pitto-Barry
 and
Rachel K.
O'Reilly
*
and
Rachel K.
O'Reilly
*
Department of Chemistry, University of Warwick, Gibbet Hill Road, Coventry, CV4 7AL, UK. E-mail: r.k.o-reilly@warwick.ac.uk
First published on 17th March 2017
Abstract
Well-defined N,N′-(dimethylamino)ethyl acrylate (DMAEA) functionalized polymeric stars have been synthesized via an arm-first approach. Utilizing reversible addition–fragmentation chain transfer polymerization, linear homopolymers (PEGA, PHEA) were chain extended with DMAEA and a divinyl crosslinker to produce a series of crosslinked polymeric stars. These stars were characterized using a range of techniques including NMR, SEC, DLS and TEM analysis. The hydrolytic behavior of the DMAEA when tethered within a micellar core was investigated by 1H NMR spectroscopy and was found to be strongly dependent on temperature. At elevated temperatures either a higher crosslinking density or a longer arm length was found to offer greater protection to the amine resulting in slower hydrolysis, with hydrolysis found to level off at a lower final percentage hydrolysis. In contrast, the composition and nature of the arm was found to have little impact on the hydrolysis, with the same trends relating to the effect of temperature and crosslinking density observed with a linear (HEA) and a brush (PEGA) arm. Additionally, the release of DMAE from the polymeric stars was successfully confirmed through the use of an enzymatic assay, producing a concentration of DMAE in good agreement with the theoretical concentration based on the 1H NMR spectroscopic analysis.
Introduction
Recent developments in controlled radical polymerization techniques have enabled the synthesis of polymers with well-defined molecular architectures, ranging from combs and brushes through to stars.1–5 Numerous studies report the formation of well-defined polymeric stars through the use of reversible addition–fragmentation chain transfer (RAFT) polymerization.4,6–10 A structurally well-defined star polymer has one defined branching point, or core, from which multiple arms extend, and thus exhibits a globular shape and, commonly, a core–shell microstructure.11–13 Due to the defined nature of their structure and associated properties (for example core density and arm length), there is a large number of applications for polymeric stars that range from drug delivery to nanoelectronics.14–20 Through varying the size, crosslinking density and arm composition, the physical properties of the stars can be tailored for various applications.21The hydrolysis of ester linkages is widely exploited in the synthesis of degradable polymers, with the majority of the work focused on hydrolyzing the polymer backbone.22–25 In contrast, relatively little work has been reported on the hydrolytic behavior of pendent ester functionalities, especially for either acrylate or methacrylate based materials. Despite the relatively limited literature available, it is still widely accepted that methacrylate-based polymers are significantly more stable to hydrolysis than their acrylate-based equivalents.26–28 Recently, Monteiro and co-workers carried out an in-depth study into the self-catalyzed hydrolysis of linear homopolymers of the amino-functionalized monomer, N,N′-(dimethylamino)ethyl acrylate (DMAEA).29 The self-catalyzed nature of the hydrolysis, in which the rate of catalysis is further accelerated by the carboxylic acid by-product, forms poly(acrylic acid) and a small molecule of N,N′-dimethylaminoethanol. The rate of hydrolysis was found to be independent of both the pH of the solution as well as the molecular weight of the polymer, further confirming the self-catalyzed nature of the process. More recently, our group reported the hydrolysis of DMAEA, within a DMAEA-methyl acrylate copolymer, to be independent of the amount of amino groups present.30 The lack of requirement for an internal or external stimulus in order to trigger degradation, coupled with the resultant change in environment from basic to acidic (attributed to the acrylic acid moieties), results in these materials having the potential to be used in a vast range of applications, for example in the release of siRNA complexes from cationic polymers as well as DNA release.31–34
Despite the hydrolysis of DMAEA being widely acknowledged in the literature, as well as the proposed multiple potential applications for these materials, the self-catalyzed hydrolysis of DMAEA within complex molecular architectures has not yet been extensively studied, with the majority of hydrolysis studies reporting hydrolysis of homopolymers or block copolymers. Indeed, based on related catalysis work,35,36 it would be expected that the rate of hydrolysis would be affected by localization within the confined core of a star copolymer. Sun et al. recently reported the synthesis and self-catalyzed hydrolysis of PHEA-g-PDMAEA graft copolymers, concluding that hydrolysis of the PDMAEA block was independent of both polymer molecular weight and solution pH,37 similar to the previously mentioned work on homopolymer hydrolysis by Monteiro and co-workers. Furthermore, Whitfield et al. reported that hydrolysis of a PDMAEA armed star, whose arms consisted of PDMAEA homopolymers, was found to undergo a similar rate of hydrolysis compared to those reported for both the linear homopolymers and graft copolymers,38 and Perrier and co-workers reported the hydrolysis of P(dimethyl siloxane-b-DMAEA) block copolymers, which exhibited the same effect of quaternization on the prevention of PDMAEA hydrolysis as noted by Monteiro and co-workers.39 Whilst the aforementioned studies indicate little effect of polymer structure on the hydrolysis behavior, hydrolysis of the PDMAEA occurs when the polymers are in a fully hydrated environment. To the best of our knowledge there are, so far, no studies reporting the impact on hydrolysis when the DMAEA is confined within a non-hydrated environment (e.g. the core of a micelle or a polymeric star). Indeed the outcomes of such studies would potentially have a significant impact on, for example, the self-assembly and functionality of DMAEA-containing polymers amongst other applications.39 Hence, in this work the hydrolytic behavior of DMAEA-containing polymeric stars is studied to determine the effect of temperature, crosslinking density, arm length, and arm type on the hydrolytic behavior of the DMAEA when confined within a core environment. We also demonstrate the release of a small molecule amine from the star copolymer upon hydrolysis, which highlights the potential utility of this hydrolysis reaction for controlled release applications.
Experimental
Materials
Monomers were received from Sigma-Aldrich and stored at 4 °C. Inhibitor was removed by passing through basic alumina. 2,2′-Azobis(isobutyronitrile) (AIBN) was received from Molekula, recrystallized from methanol and stored at 4 °C. The chain transfer agent cyanomethyl dodecyl trithiocarbonate was obtained from Sigma-Aldrich and used as received. Deuterated solvents were received from Apollo Scientific. All other chemicals were obtained from Sigma Aldrich, Fisher Chemicals, and Acros Chemicals and used as received.Instrumentation
Nuclear magnetic resonance (1H NMR) spectra were recorded on a Bruker DRX-400 spectrometer. Chemical shifts are reported at δ in parts per million and quoted downfield from the internal standard tetramethylsilane (δ = 0 ppm). Monomer conversion was calculated by comparing the resonances associated with the vinyl peaks to the resonances associated with CH2N (for DMAEA) and OCH2CH2 (for DEGDA). Size exclusion chromatography (SEC) measurements were carried out using an Agilent 390-MDS multi detector suite fitted with a viscometer, a refractive index (RI) and a light scattering (LS) detector, and equipped with a guard column (Varian PLGel) and two PLGel 5 μm mixed-D columns. The mobile phase was DMF with 5 mM NH4BF4. Data was analyzed using Cirrus v3.3 and Agilent GPC/SEC software v1.1 with calibration curves produced using Varian Polymer Laboratories linear PMMA standards. Dynamic light scattering was conducted using a Malvern Zetasizer NanoS instrument equipped with a 4 mW He–Ne 633 nm laser module at 25 and 50 °C, with data analysis using Malvern DTS 6.20 software. Measurements were carried out at a detection angle of 173° (backscattering). TEM solutions were made up at 2 mg mL−1 in 18.2 MΩ cm water. TEM samples were prepared on graphene oxide (GO)-coated carbon grids (Quantifoil R2/2).40 Generally, a drop of sample was pipetted onto a grid and left to dry overnight. Samples were analyzed with a JEOL-2100 microscope, operating at 200 keV. UV measurements were carried out in triplicate using a FLUO-star Optima plate reader, fitted with an excitation filter at λ = 405 nm, with data analyzed using MARS data analysis software v3.01.Synthetic methods and procedures
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 hexane/diethyl ether, affording a viscous pale yellow liquid (70%).
:1 hexane/diethyl ether, affording a viscous pale yellow liquid (70%).
20% crosslinked polymer (2-20). 1H NMR (CDCl3): δ (ppm) 4.16 (br s, OCH2CH2, OCH2CH2O(CH2CH2O)7 and CH2CH2N), 3.46–3.80 (m, O(CH2CH2O)8 and OCH2CH2O), 3.38 (s, CH2OCH3 and SCH2(CH2)9), 2.62 (br s, CH2N), 2.23 (br s, N(CH3)2), 1.24–2.06 (m, CH2 backbone, CH2(CH2)10 CTA), 0.88 (t, 3H, (CH2)10CH3, 3JH–H = 6.1 Hz). Mn, SEC = 23.8 kDa, ĐM = 2.25. Dh = 11 nm.
:1 hexane/diethyl ether, affording a viscous pale yellow liquid (27%).
20% crosslinked polymer (8-20). 1H NMR (DMSO-d6): δ (ppm) 4.75 (br s, CH2OH), 4.02 (br s, OCH2CH2OH and OCH2CH2O), 3.33 (br s, OCH2CH2OH, SCH2(CH2)9 and OCH2CH2O), 2.50 (br s, CH2N), 2.17 (br s, N(CH3)2), 1.60–1.80 (m, CH2 backbone, CH2(CH2)10 CTA), 0.88 (br s, 3H, (CH2)10CH3). Mn, SEC = 29.1 kDa, ĐM = 1.56. Dh = 23 nm.
Results and discussion
RAFT polymerization, with its compatibility towards amine functionalized monomers, allows for the synthesis of DMAEA-containing polymeric stars with defined arm lengths and crosslinking densities. Star polymers were synthesized via an arm-first approach,41 with initial synthesis of the polymeric arms and subsequent extension with DMAEA and the difunctionalized crosslinking monomer di(ethylene glycol) diacrylate (DEGDA).Synthesis and characterization of polymeric stars
The synthesis of poly(ethylene glycol) monomethyl ether acrylate (PEGA, Mn = 480 g mol−1) polymeric arms of varying arm length was carried out using RAFT polymerization at 70 °C with the radical initiator 2,2′-azobis(2-methylpropionitrile) (AIBN) in 1,4-dioxane, and in the presence of the chain transfer agent (CTA) cyanomethyl dodecyl trithiocarbonate (Scheme 1).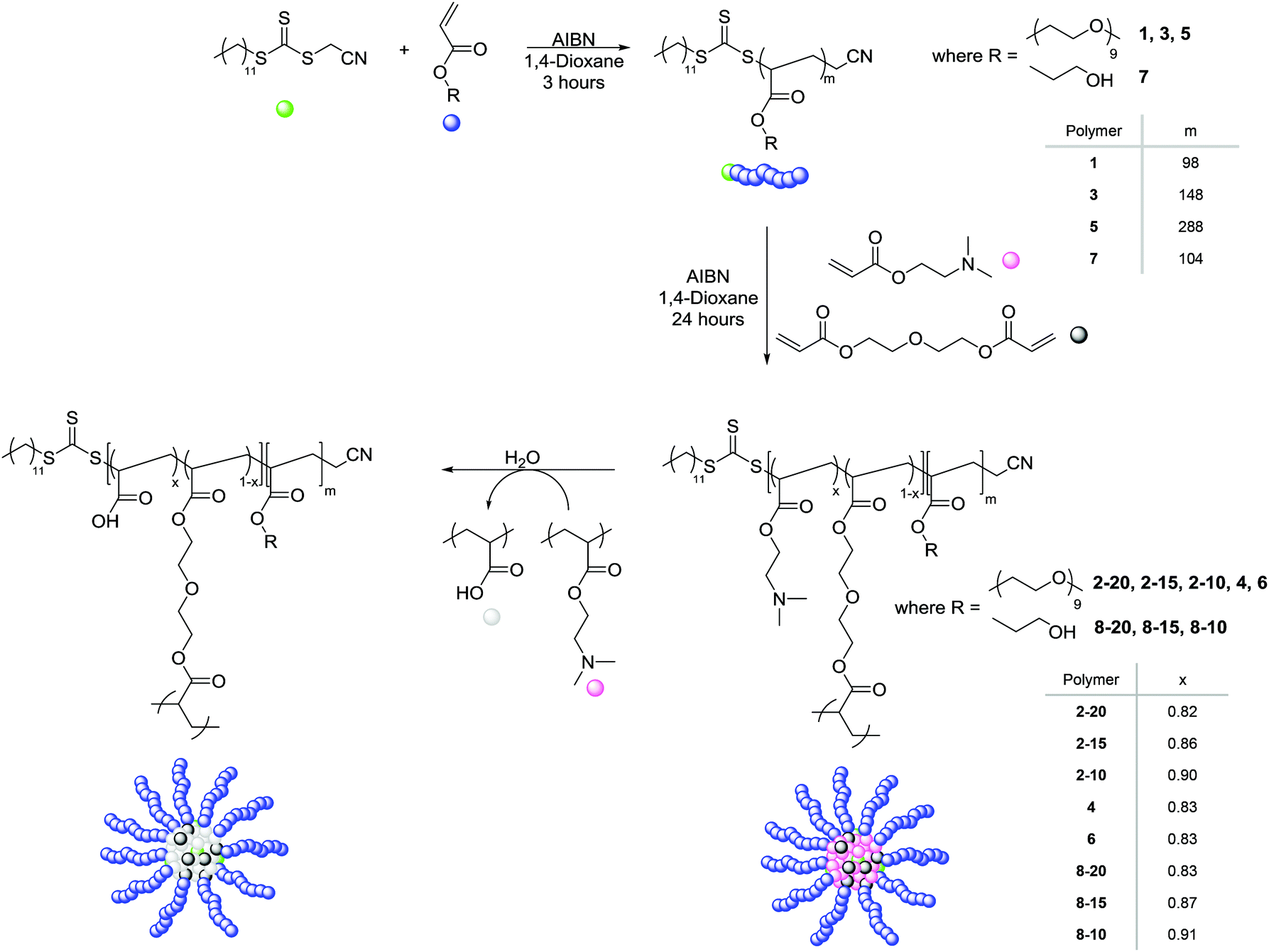 | ||
| Scheme 1 Synthesis of DMAEA functionalized polymeric stars (2-20, 2-15, 2-10, 4, 6, 8-20, 8-15 and 8-10) via an arm-first approach using RAFT polymerization, and hydrolysis of the amine functionality to acrylic acid moieties. Amine content determined by the polymer DPs calculated using 1H NMR spectroscopy. | ||
The three resulting macro-CTA arms had degrees of polymerization (DP) of 98 (1), 148 (3) and 288 (5) (Table 1). The theoretical number-average molecular weight (Mn,th), determined by conversion from the 1H NMR spectrum, was found to be in good agreement with the observed number-average molecular weight (Mn,obs.), as calculated by 1H NMR analysis through comparison of the integrals attributed to the CTA methyl end group (δ = 0.80 ppm) to the methyl protons in the PEGA repeat unit (δ = 3.30 ppm), indicating the controlled nature of the polymerization. Analysis of 1 by size exclusion chromatography (SEC, in DMF with PMMA standards) demonstrated a monomodal peak of moderate dispersity (ĐM = 1.51), as well as a good overlap between the refractive index (RI) trace and the UV trace at λ = 309 nm, the wavelength attributed to the trithiocarbonate of the CTA, indicative of the presence of the trithiocarbonate functionality in the polymer, confirming the ability of the PEGA arms to act as a macro-CTA for further extension to produce polymeric stars (ESI, Fig. S1†). The short PEGA arms (1) were chain extended with the amino-functionalized monomer DMAEA and the divinyl crosslinking monomer DEGDA. The monomer feed was altered to produce polymeric stars with approximately 20, 15, and 10% crosslinking density (2-20, 2-15, and 2-10 respectively, Table 1). 1H NMR spectroscopic analysis indicated the incorporation of the amine, with the characteristic peaks at δ = 2.15 and 2.50 ppm (N(CH3)2 and CH2N respectively), and SEC demonstrated a shift in molecular weight from the macro-CTA to the chain extended polymer, confirming successful chain extension (Fig. 1a). Analysis of the polymeric stars by SEC (with viscometry detection) enabled calculation of the Mark–Houwink characteristic constant a, derived from the linear fit of the Mark–Houwink curve generated by plotting log[η] vs. log[MW], where MW is the viscosity-average molecular weight calculated by universal calibration and η is the weight average intrinsic viscosity as measured by the viscometer.42–45 For polymers, a = 0.5 for a polymer chain in a theta solvent, a = 0.8 for a polymer in a good solvent, and therefore for a flexible polymer (for example a linear polymer) 0.5 ≤ a ≤ 0.8; for crosslinked /branched polymers, a < 0.5.43 Measuring the gradient, it was found that all the short-armed stars (2-20, 2-15, and 2-10) had a values ranging from 0.37–0.40, consistent with the crosslinked nature of the particles (Fig. S2†). Moreover, a was found to be less than the linear PEGA macro-CTA, where a = 0.52, confirming a structural change from a linear polymer to a branched architecture. To confirm that the introduction of the DMAEA had not caused the branched nature of the polymer, a linear analogue [PEGA98-b-(DMAEA60-co-MA13)] was synthesized through chain extension of the same PEGA macro-CTA with DMAEA and methyl acrylate (MA) in the place of the crosslinker to produce a polymer of approximately the same molecular weight. Comparison of the star polymers 2-20, 2-15 and 2-10 to the linear analogue indicated that all particles displayed lower intrinsic viscosities than the linear analogue, hence further confirming the structural change from a linear to a branched/crosslinked polymer (Fig. 1b). The similarity in the intrinsic viscosities of the particles is likely an indication of a similar number of arms per star. To further probe the structure of the polymers and confirm this theory, the star functionality, f, was calculated (Table 1). Star functionality was calculated using the branching function in the Agilent software,46 with the triple detection SEC data for the linear macro-CTA used as the linear reference for branching calculations. All particles were found to have a similar number of arms, ranging from 12–14 arms per star.
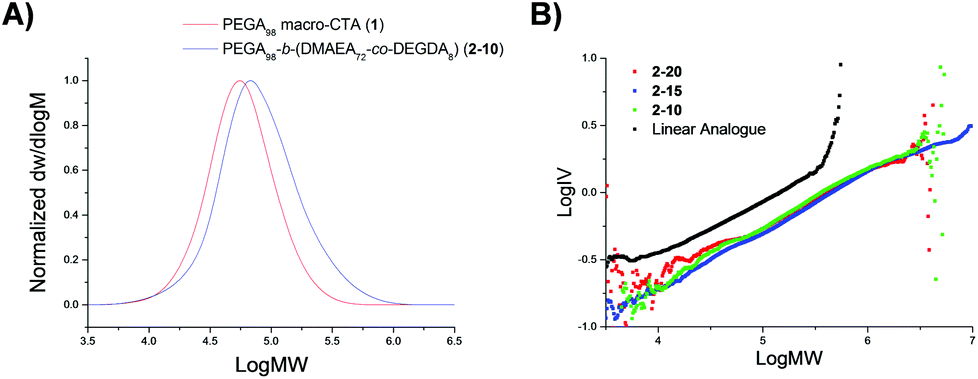 | ||
| Fig. 1 (a) RI traces of the linear PEGA98 macro CTA (1, red) and PEGA98-b-(DMAEA72-co-DEGDA8) (2-10, blue) obtained by SEC analysis. (b) Triple detection SEC Mark–Houwink curves for PEGA98 armed particles with varying crosslinking densities compared to the linear analogue PEGA98-b-(DMAEA60-co-MA13), in DMF with 5 mM NH4BF4 and PMMA as standards. | ||
| Entry | Polymera | Crosslinking density (%) |
M
n, SECb (kDa) |
Đ
Mb |
ac |
fd |
M
n, th.e (kDa) |
D
hf (nm) |
|---|---|---|---|---|---|---|---|---|
| a DPs calculated by 1H NMR spectroscopy in CDCl3 (PEGA) and DMSO-d6 (HEA). b Measured by SEC, DMF with 5 mM NH4BF4 and PMMA as standards. c Mark–Houwink parameter calculated using triple detection SEC (DMF with 5 mM NH4BF4 and PMMA standards). d Star functionality calculated using Agilent GPC/SEC software v1, with the branching model set to “star branched-regular” and a branching frequency of 1. e Theoretical molar mass calculated based on monomer conversion (1H NMR spectroscopy). f Hydrodynamic diameter, by number, determined by dynamic light scattering analysis (detection angle = 173°) at 5 mg mL−1 in chloroform at 25 °C. | ||||||||
| 1 | PEGA98 | — | 44.7 | 1.51 | 0.51 | — | 53.1 | — |
| 2-20 | PEGA98-b-(DMAEA76-co-DEGDA16) | 18 | 23.8 | 2.25 | 0.37 | 12 | 63.6 | 11 |
| 2-15 | PEGA98-b-(DMAEA67-co-DEGDA11) | 14 | 39.9 | 2.88 | 0.40 | 13 | 65.1 | 12 |
| 2-10 | PEGA98-b-(DMAEA72-co-DEGDA8) | 10 | 43.8 | 2.25 | 0.40 | 14 | 67.6 | 9 |
| 3 | PEGA148 | — | 46.2 | 1.64 | — | — | 89.4 | — |
| 4 | PEGA148-b-(DMAEA100-co-DEGDA20) | 17 | 66.3 | 1.73 | 0.40 | 16 | 96.9 | 11 |
| 5 | PEGA288 | — | 47.0 | 1.52 | — | — | 114.1 | — |
| 6 | PEGA288-b-(DMAEA81-co-DEGDA17) | 17 | 43.6 | 1.81 | 0.46 | — | 165.8 | 11 |
| 7 | PHEA104 | — | 20.2 | 1.10 | 0.50 | — | 7.8 | — |
| 8-20 | PHEA104-b-(DMAEA74-co-DEGDA14) | 17 | 29.1 | 1.56 | 0.34 | 12 | 32.0 | 23 |
| 8-15 | PHEA104-b-(DMAEA69-co-DEGDA10) | 13 | 27.8 | 1.25 | 0.32 | 11 | 31.8 | 14 |
| 8-10 | PHEA104-b-(DMAEA71-co-DEGDA7) | 9 | 24.9 | 1.31 | 0.40 | 14 | 35.7 | 25 |
Particle size was analyzed by dynamic light scattering (DLS) in chloroform at 25 °C, at 5 mg mL−1. All the particles were found to have similar hydrodynamic diameters (Dh), ranging in size from 9–11 nm (Table 1, Fig. 2, 3 and S3, and Table S1†). Light scattering analysis indicated the presence of larger entities alongside the small particles, potentially caused by aggregation. Examination of the particles by transmission electron microscopy (TEM) analysis using graphene oxide (GO) supported TEM grids, found to produce higher contrast images without staining,40 confirmed that these larger species were aggregated particles. The particle diameter produced by TEM analysis was significantly smaller than that obtained by DLS analysis, with a particle size of approximately 3 nm (Fig. 3). The smaller size produced by TEM analysis can be attributed to the dry-state of the TEM samples in which the PEGA shell is not hydrated, resulting in poor contrast between the shell of the polymer and the GO grid, and therefore only imaging the particle core.40,47
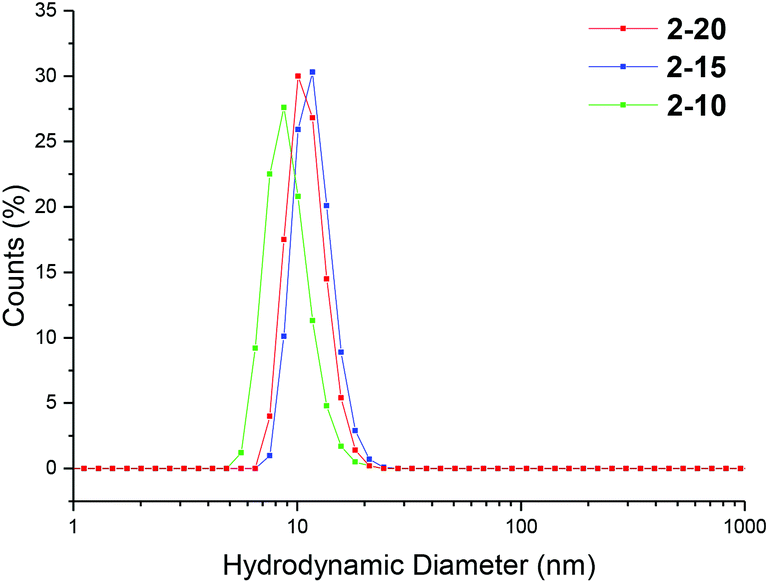 | ||
| Fig. 2 Size distributions, by number, for PEGA armed particles 2-20, 2-15, 2-10, obtained by DLS (detection angle = 173°) at 5 mg mL−1 carried out in chloroform at 25 °C. | ||
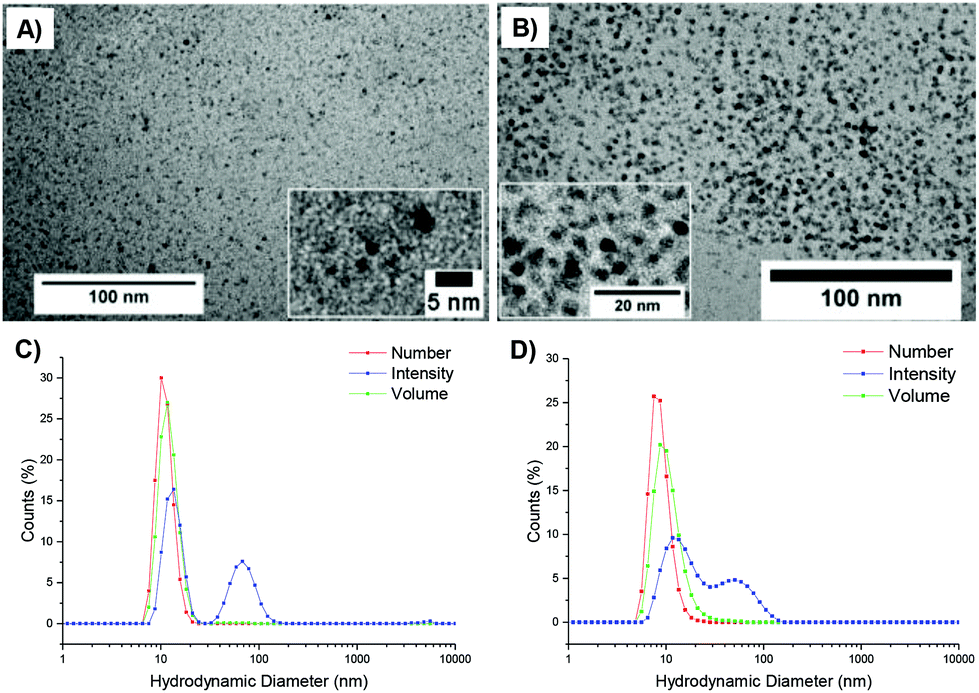 | ||
| Fig. 3 Representative TEM images and size distributions, as determined by DLS (5 mg mL−1 in chloroform), of PEGA98-b-(DMAEA76-co-DEGDA16), 2-20 (a and c), and PEGA98-b-(DMAEA72-co-DEGDA8), 2-10 (b and d) prepared by arm-first RAFT polymerization. | ||
In order to investigate the effect of arm type, the same approach was utilized to synthesize hydroxyethyl acrylate (HEA) arms (7), with subsequent chain extension with DMAEA and DEGDA, using the same conditions, to produce a series of PHEA armed polymeric stars with varying crosslinking densities (8-20, 8-15 and 8-10) and with similar arm lengths to the smallest PEGA homopolymer arm. Both the PHEA arms and resultant stars were characterized by the same techniques used for the PEGA armed particles (1H NMR spectroscopy and SEC, Table 1 and Fig. S4†). Similar to the PEGA98 armed particles, the PHEA104 polymeric stars were found to be crosslinked in nature (with Mark–Houwink a values ranging from 0.25–0.41, Fig. S5†), and displayed a similar number of arms, with a star functionality ranging from 11 to 14 (Table 1), determined using the macro CTA PHEA104 as the linear analogue. Particle size analysis by DLS yielded slightly larger particle sizes than the PEGA analogues (23–25 nm, Table 1 and Fig. S6†).
Evaluation of hydrolytic behavior
The hydrolysis of the DMAEA containing polymeric stars was investigated via1H NMR spectroscopic analysis of the particles in D2O (50 mg mL−1). The resulting spectrum of the particles (Fig. 4, representative of the PEGA armed polymers (2-20, 2-15, 2-10, 4, 6)), clearly displays the protons associated with the incorporated amine monomer, observed at δ = 2.65 ppm for the methyl groups bound to the nitrogen and at δ = 3.08 ppm for the methylene protons bound to the amine (Fig. 4, protons 3 and 2 respectively), with the intensity of two new signals at δ = 2.90 and 3.20 ppm, corresponding to the equivalent protons in the hydrolytically released small molecule N,N′-dimethylaminoethanol (Fig. 4, protons c and b respectively), increasing over time. From these signals, which confirm hydrolysis through the ester linkage within the amine-functionalized monomer in the polymer, the percentage hydrolysis can be calculated based on the ratio between hydrolyzed and non-hydrolyzed amine integrals. It should be noted that hydrolysis calculated using the ratio of polymer proton 3 (at δ = 2.65 ppm) to the small molecule proton c (at δ = 2.90 ppm) was found to produce the same percentage hydrolysis.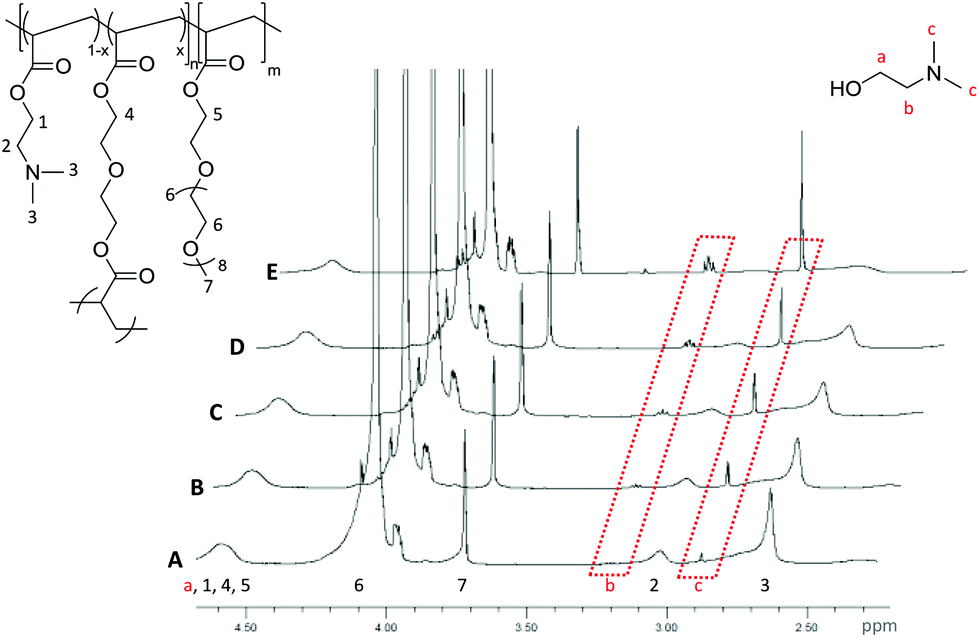 | ||
| Fig. 4 1H NMR spectra (D2O, 400 MHz) of PEGA98-b-(DMAEA76-co-DEGDA16) for (A) 1 h., (B) 2 h., (C) 4 h., (D) 6 h and (E) 24 h at 50 mg mL−1. Spectra normalized to the peak at δ = 3.72 ppm (protons 7). | ||
Influence of temperature on the rate of hydrolysis for different crosslinking densities
An initial set of hydrolysis experiments was carried out to investigate the influence of temperature on the rate of hydrolysis for different crosslinking densities (Fig. 5a). Reactions performed at 50 °C resulted in a significantly faster rate of hydrolysis, with 20% of the amino-functionalized repeat units in star copolymer 2-10 hydrolyzed after 21 minutes compared to 230 minutes at 25 °C to achieve the same degree of hydrolysis. Subsequent heating of the particles already hydrolyzed at 25 °C to the increased temperature of 50 °C resulted in an increase in the rate of hydrolysis, with the overall hydrolysis tailing off at the same level as those initially hydrolyzed at 50 °C (Fig. 5b). Through comparison of the hydrolysis at both 25 °C and 50 °C it is evident that at the lower temperature crosslinking density has little effect on the observed hydrolysis, with all crosslinking densities exhibiting approximately the same degradation rate and hydrolysis of all polymers falling within error regardless of the crosslinking density (Fig. 5c). In contrast, at the raised temperature there appears to be an influence of crosslinking density, with the most highly crosslinked particle (2-20) displaying the slowest rate of hydrolysis, followed by 2-15, and with the lowest crosslinking density (2-10) demonstrating the greatest hydrolysis over the period of 4 hours (Fig. 5d). The decrease in the rate of hydrolysis over time is likely a consequence of one of two effects. The first effect is that the decrease in the rate of hydrolysis over time is a result of the build-up of electrostatic repulsion throughout the reaction. At the beginning of the hydrolysis, the DMAEA units begin with no adjacent units hydrolyzed to form acrylic acid groups. As the reaction proceeds, the adjacent units begin to hydrolyze and the DMAEA repeat unit progresses from having no adjacent units hydrolyzed, to one adjacent unit hydrolyzed and then both hydrolyzed. The formation of polyacrylic acid groups within the core increases the electrostatic repulsion between the non-hydrolyzed ester linkages, the acrylic acid moieties, and the hydrolyzing water molecule, slowing the rate of hydrolysis. Moreover, as demonstrated by Higuchi and Senju,48 this electrostatic repulsion between the hydrolyzed groups and the water results in an increase in the activation free energy required to hydrolyze the ester bond, thus rendering further hydrolysis energetically unfavorable, with Higuchi et al. mathematically demonstrating a maximum hydrolysis of approximately 60% achievable.48 It is hypothesized that an increase in crosslinking density results in a less mobile core and subsequently less mobile pendent functionality on the polymer chain. Upon hydrolysis, the polyacrylic acid product is therefore in a more fixed position in the core giving rise to a build-up of localized negative charge, attributed to the carboxylic acid moieties. Therefore, in a more crosslinked particle, the build-up of a localized charge is greater owing to lower core mobility, resulting in both a slower rate of hydrolysis as well as a lower overall percentage hydrolysis.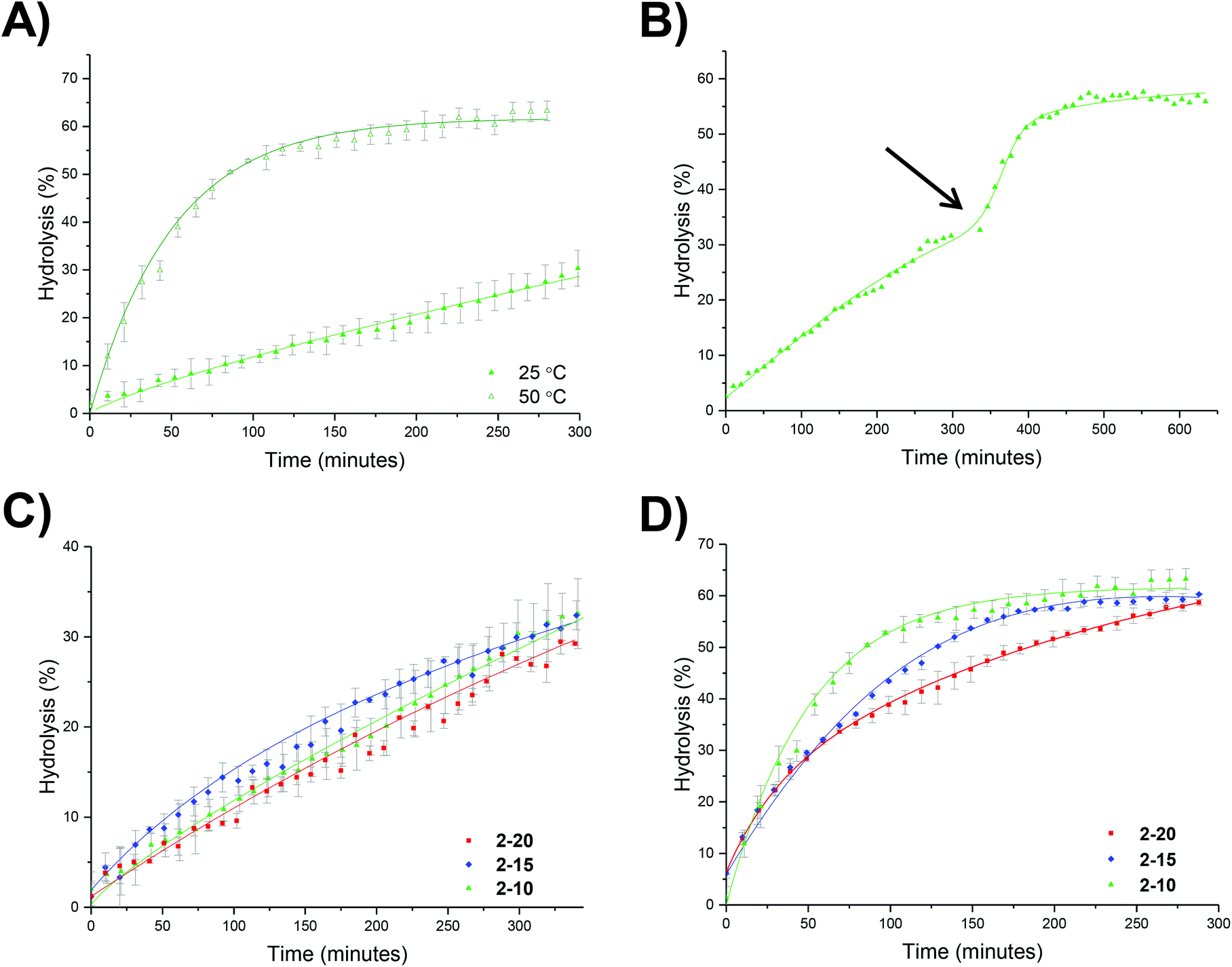 | ||
| Fig. 5 Hydrolysis kinetics of PEGA-b-(DMAEA-co-DEGDA) in D2O as determined by 1H NMR spectroscopy: (a) hydrolysis of 2-10 carried out at 25 °C and 50 °C; (b) 2-10 initially heated at 25 °C with an increase in temperature to 50 °C at 320 minutes; (c) hydrolysis at 25 °C with different crosslinking densities, and (d) hydrolysis at 50 °C with different crosslinking densities. Error bars produced from the standard deviation of 3 repeats. | ||
An alternative to the electrostatic argument is that at raised temperatures the particles swell resulting in a greater ingress of water thus producing a faster rate of hydrolysis. An increase in crosslinking density is thought to result in less swelling of the particle and as such less hydrolysis. To exclude the possibility of particle swelling having an impact on the rate of hydrolysis, DLS analysis of the particles (2-20) was carried out both before and after hydrolysis at 25 °C, and additionally measured at 50 °C (Fig. S7†). Particle size was found to remain approximately the same at both 25 °C and 50 °C regardless of the crosslinking density (Table S1†). Moreover, hydrolysis had no effect on the particle size, with no size change following 60% hydrolysis (Fig. S7†). Additionally, the integrals in the 1H NMR spectrum assigned to the crosslinker (Fig. 4, protons 4) remained unchanged throughout the hydrolysis confirming that only the amine is hydrolyzed and hence the observed effect is not due to crosslinker hydrolysis.
Influence of particle arm length
To further probe the effect of star composition on the protection afforded to the amine, hydrolysis kinetics at the raised temperature of 50 °C were measured for short, medium, and long armed PEGA particles all with approximately 20% crosslinking density. As an increase in arm length would result in lower core mobility, it was expected that an increase in arm length would result in lower hydrolysis. Polymeric stars with a crosslinking density of 20% and a similar DP for DMAEA were synthesized according to the previously described procedure: PEGA homopolymers with a medium arm length (3, DP = 148) and a long arm length (5, DP = 288) were first synthesized and subsequently chain extended with DMAEA and DEGDA. Both the medium and long arm homopolymers (3 and 5 respectively) and their corresponding star polymers (4 and 6 for medium and long arms respectively) were analyzed using 1H NMR spectroscopy and SEC analysis (Table 1 and Fig. S8†). Both 4 and 6 were found to be similar in size by DLS analysis (11 and 10 nm respectively, Table 1 and Fig. S9†). It was hypothesized that approximately the same particle size was produced regardless of the increase in arm length as a consequence of increased chain entanglement as the length of the arms increase, which has the effect of producing approximately the same star polymer size even as the arm length increases. It should also be noted, however, that the particle size may also be relatively similar for all arms' lengths owing to the relatively small size of the stars rendering them difficult to obtain accurate size information from the DLS instrument, and hence resulting in it being more difficult to observe small increases in size as a consequence of increasing arm length. Whilst an increase in arm length did result in a decrease in hydrolysis (Fig. 6), the initial rates of hydrolysis for all arm lengths are similar suggesting that arm length does not have such a great impact on hydrolysis compared to crosslinking density. This may be attributed to the properties of the star polymer: as the arm length increases, the density of the shell decreases which allows for greater influx of water into the core.12 Therefore, there is little difference between the initial rates of hydrolysis. However, longer arms result in a less mobile core, therefore there is a greater buildup of electrostatic repulsion in the polymers with larger PEGA DPs, thus resulting in a lower final percentage hydrolysis.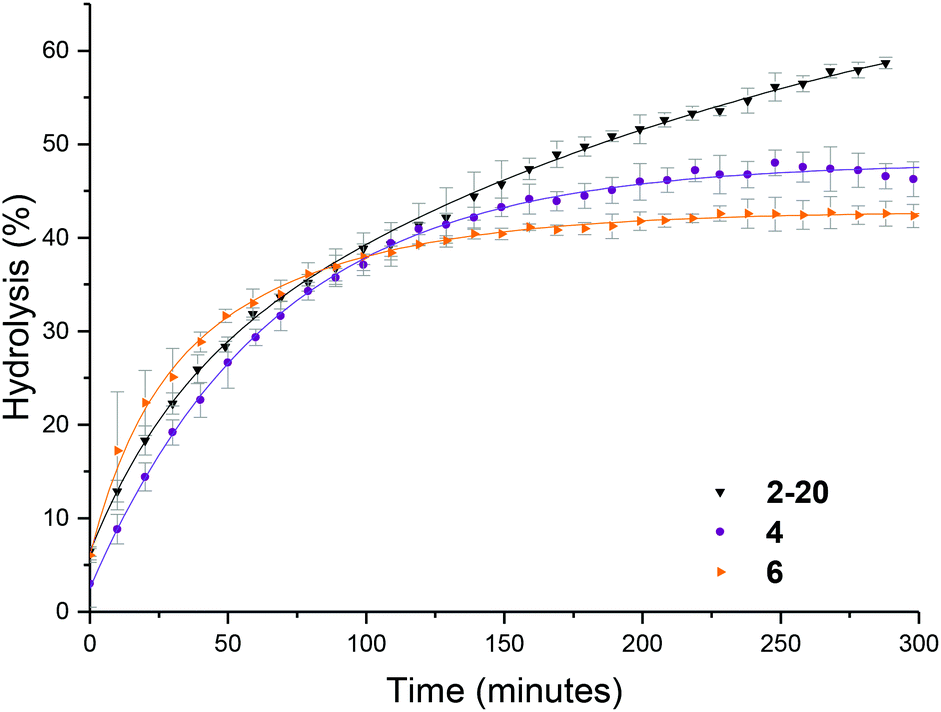 | ||
| Fig. 6 Hydrolysis kinetics of short, medium and long armed PEGA stars (2-20, 4, and 6 respectively), at 50 °C in D2O as determined by 1H NMR spectroscopy. Error bars produced from the standard deviation of 3 repeats. | ||
Effect of polymer architecture
The hydrolysis rate observed for the polymeric particles utilized in this study were found to be higher than that previously reported, with Monteiro and co-workers reporting only 13% hydrolysis in a linear PDMAEA homopolymer (with a similar DP with respect to the amine) after 7 hours at 25 °C,29 and Whitfield et al. reporting 36% hydrolysis after 1 day for a four-armed star with homopolymer PDMAEA arms.38 To ascertain whether tethering the amine functionality in a confined environment within a polymeric particle had changed the hydrolysis rate, a linear analogue was synthesized, in which the 20% crosslinking monomer was replaced with non-crosslinking methyl acrylate (MA). Hydrolysis at room temperature showed little difference in the rate of hydrolysis between the linear and star hydrolysis kinetics, with the star polymer reaching 26% hydrolysis over 300 minutes compared to 34% hydrolysis of the linear polymer (Fig. 7a). At 50 °C, however, the rate of hydrolysis for the linear polymer begins to deviate from the star polymer, with a much faster hydrolysis rate observed for the linear copolymer (Fig. 7b). It was hypothesized that the rigid core of the stars gives rise to a more confined build-up of acrylic acid moieties as hydrolysis proceeds, resulting in increased electrostatic repulsion thus generating both a lower overall hydrolysis and a slower rate of hydrolysis for the star polymer in comparison to the linear analogue. Nonetheless, whilst the star appears to afford protection, the linear MA based analogue was still found to have higher hydrolysis than the DMAEA homopolymers and the DMAEA-armed stars previously reported. This can be attributed to the MA group acting as a spacer: in the copolymer there is less electrostatic repulsion between the hydrolyzed and non-hydrolyzed groups as the MA lies between them on the polymer backbone, disrupting the build-up of electrostatic repulsion along the backbone thus increasing the hydrolysis rate. In contrast, both the homopolymer and the four-armed star (reported previously) do not have this spacer to disrupt the electrostatic charge, resulting in the build-up of electrostatic repulsion along the backbone and a slower hydrolysis rate. Indeed, previous work by our group which investigated the hydrolysis rate of a series of P(MA-co-DMAEA) copolymers with different amine contents indicated 15% hydrolysis over a period of 50 minutes (with the temperature ramping at 1 °C per minute) regardless of spacer (MA) content. This is in good agreement with the 22% hydrolysis observed in this study, with the slightly higher percentage hydrolysis in this study to be expected as a consequence of the constant heating vs. ramped heating in our earlier study. Furthermore, both of these studies had noticeably faster hydrolysis rates than the previous homopolymer hydrolysis studies, consistent with both the MA in the linear analogue, and the DEGDA in the crosslinked stars, disrupting the build-up of electrostatic repulsion allowing for faster hydrolysis rates.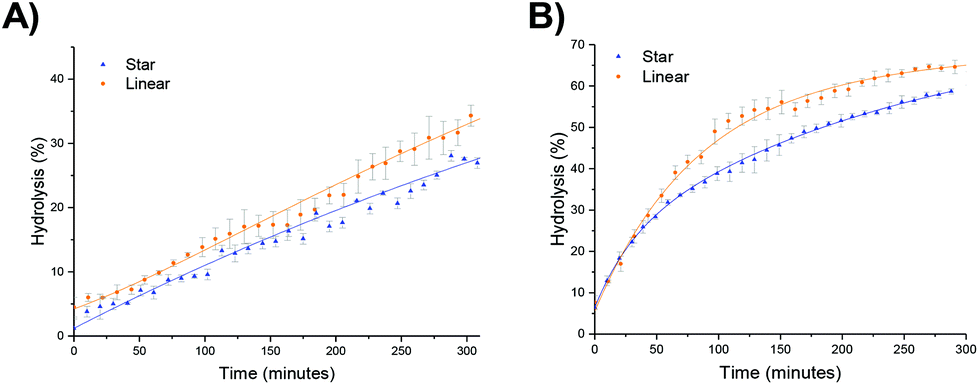 | ||
| Fig. 7 Hydrolysis kinetics of 2-20 and the linear analogue PEGA98-b-(DMAEA60-co-MA13) at (a) 25 °C and (b) 50 °C, in D2O as determined by 1H NMR spectroscopy. Error bars produced from the standard deviation of 3 repeats. | ||
Influence of the polymer arm type at different crosslinking densities
We hypothesized that the brush-like character of the PEGA arms may influence the hydrolysis rate, for example by affecting diffusion of water into the particle. To this end, a series of analogous particles using PHEA104 (that did not have brush-like character) with varying crosslinking densities of approximately 20, 15 and 10% were synthesized (8-20, 8-15 and 8-10, Table 1). Whilst investigating the effect of temperature, at 25 °C it was observed that crosslinking density has little effect on the hydrolysis rate (Fig. S10†), similar to the PEGA armed counterparts. At raised temperatures (50 °C) the same trend as for the PEGA armed particles was observed: increasing the crosslinking density lowered the hydrolysis rate (Fig. 8). Direct comparison to the PEGA analogues at different crosslinking densities confirmed that the chemical nature of the polymer arm had little effect on the hydrolysis rate (Fig. 9). At 25 °C both PEGA and HEA arms produced similar profiles for the hydrolysis kinetics. Further analysis suggests that at 50 °C, whilst both arms produced similar profiles, the PEGA armed particles achieved a slightly higher overall hydrolysis in 300 minutes. Whilst there is almost no difference for the 10% crosslinked particle (Fig. 9c), there is a small difference at 15% crosslinking density (2-15 and 8-15, Fig. 9b), and a more significant difference (ca. 6%) at 20% crosslinking density (2-20 and 8-20, Fig. 9a). Whilst the difference for the 15% crosslinked particle could be within error, the error associated with the PEGA armed particles at 50 °C is ±1.3%, and for the HEA particles it is ±0.6%, suggesting that the difference between the HEA and PEGA particle hydrolysis is significant in the high crosslinking density polymer stars. This could be attributed to the more dense PEGA shell slowing diffusion of the N,N′-dimethylaminoethanol out of the core in comparison to the HEA shell. Moreover, an increase in crosslinking density will create a more densely packed core which would further prevent the release of the small molecule N,N′-dimethylaminoethanol, which may be able to buffer the hydrolysis in the core which would further slow down hydrolysis.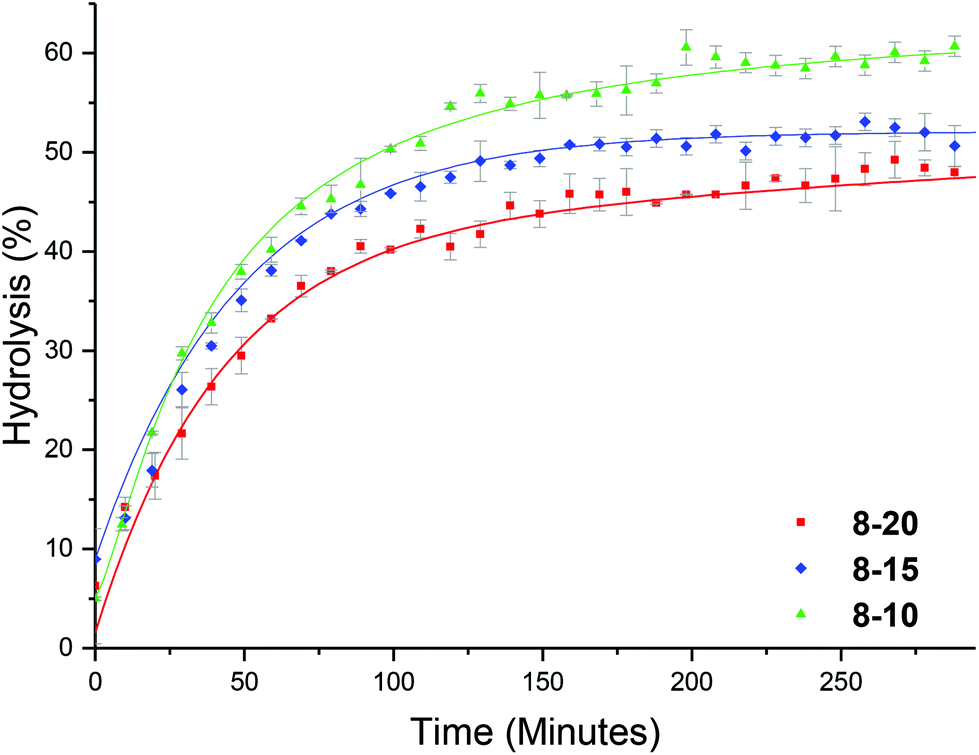 | ||
| Fig. 8 Hydrolysis kinetics of PHEA-b-(DMAEA-co-DEGDA) of varying crosslinking densities, in D2O at 50 °C as determined by 1H NMR spectroscopy. Error bars produced from the standard deviation of 3 repeats. | ||
 | ||
| Fig. 9 Hydrolysis kinetics of both HEA (8-x) and PEGA (2-x) armed particles as (a) 20% crosslinking density (2-20, 8-20), (b) 15% crosslinking density (2-15, 8-15) and (c) 10% crosslinking density (2-10, 8-10), in D2O as determined by 1H NMR spectroscopy. | ||
Enzymatic confirmation of DMAE release
Whilst 1H NMR spectroscopy confirms the successful hydrolysis of PDMAEA it does not, however confirm release of the small molecule DMAE from within the polymeric star. Indeed, the changing integrals attributed to the PDMAE (at δ = 3.08 ppm) observed by 1H NMR spectroscopy may not accurately reflect the degree of hydrolysis, as the resonances may be distorted through shielding of the protons as a consequence of their location within the core of the polymeric star. To this end, a release study was undertaken for comparison to the hydrolysis study to enable calculation of the concentration of DMAE, and comparison to the theoretical concentration based on the conversion values obtained by 1H NMR spectroscopy, both confirming release of the small molecule and validating the 1H NMR spectroscopy results.Choline oxidase catalyzes the oxidation of choline to produce betaine aldehyde and hydrogen peroxide.49,50 Additionally, it has been previously reported that the enzyme also catalyzes the oxidation of DMAE, with the Michaelis–Menten constant (Km) reported to be approximately 10 times greater for choline oxidase (Km = 1.3 mM) than DMAE (Km = 14 mM).51 The production of hydrogen peroxide from the process can then be monitored using a widely accepted colorimetric test,52 in which p-nitrophenyl boronic acid (p-NPBA) is reacted with hydrogen peroxide resulting in the production of p-nitrophenol, which has an intense UV absorption at λ = 405 nm. To enable calculation of the concentration of released DMAE, a calibration curve was first generated, relating the absorbance at λ = 405 nm to the concentration of DMAE. Plotting the initial rate of rise for the absorbance vs. concentration of DMAE resulted in a calibration curve which was fitted using an exponential equation (Fig. S11, eqn (S2)†).
Following establishment of the calibration, analysis was carried out on the 20% crosslinked PEGA armed star copolymer (2-20). The star copolymer was dissolved in 18.2 MΩ cm water (50 mg mL−1) and stirred slowly at room temperature. Aliquots were removed at regular time intervals, the polymer removed from solution using a spin concentrator (5 kDa MWCO) and the resultant supernatant was stored in the freezer until analysis (see ESI† for details). Using the calibration curve, it can be seen that the concentration of DMAE released is in relatively good agreement with the theoretical concentration based on 1H NMR spectroscopy (Fig. 10). This confirms the release of the small molecule from the core of the star copolymer. Moreover, the similarity between the theoretical and enzymatically determined values ratifies the hydrolysis results produced by 1H NMR spectroscopic analysis.
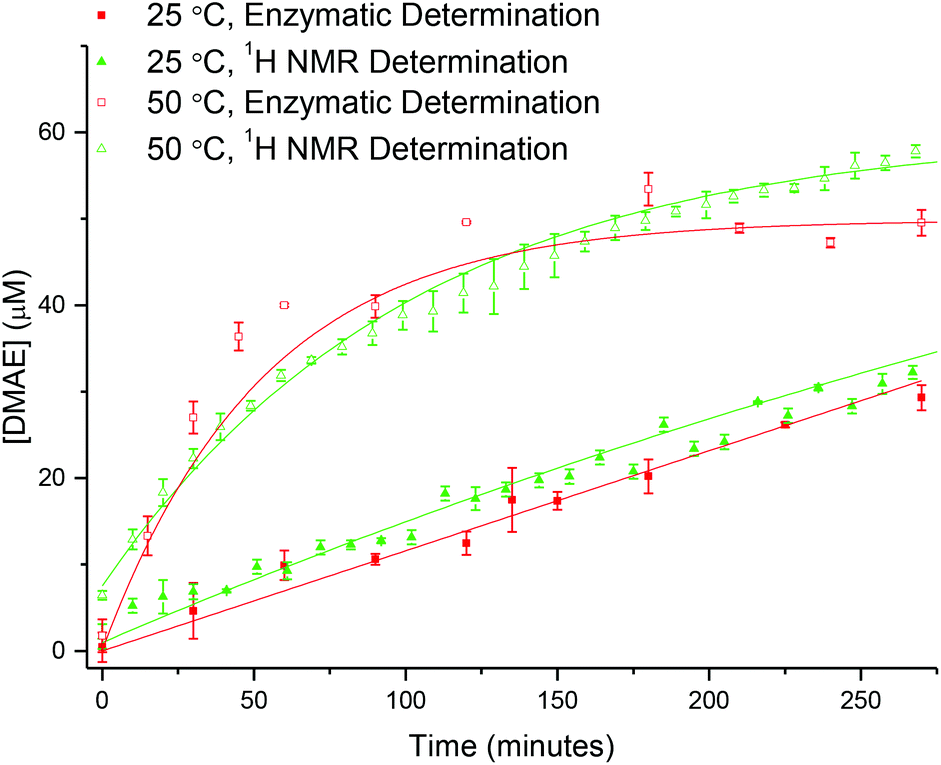 | ||
| Fig. 10 Comparison between the theoretical concentration of DMAE released based on 1H NMR spectroscopic analysis (green) and enzymatically determined (red) at 25 and 50 °C. Error bars produced from the standard deviation of three repeats. | ||
Conclusions
RAFT polymerization has been successfully applied to the synthesis of amino-functionalized polymeric stars with differing arm lengths, arm types and varying crosslinking densities in the core of the particles. To understand the hydrolytic behavior of the amine tethered within the core of the particle, the polymers were studied in situ by 1H NMR spectroscopy through monitoring of the signals attributed to both the polymer and the released N,N′-dimethylaminoethanol. Results indicated that at 25 °C there is little effect of crosslinking density but at the raised temperature of 50 °C an increase in crosslinking density results in lower overall hydrolysis. Moreover, increasing the length of arm was found to demonstrate the same effect, though not as significantly as increasing the crosslinking density. It was also indicated that the type of arm had little effect on hydrolysis, suggesting therefore that amines tethered within other architectures would be expected to exhibit similar hydrolytic behavior. We have also demonstrated that the small molecule released upon hydrolysis is available to undergo further reaction, which highlights the potential application of this monomer system in controlled release applications.Acknowledgements
AWE are thanked for funding a PhD studentship to M. S. R. EPSRC are also acknowledged for funding to support R. K. O. (Career Acceleration Fellowship). Dr Rebecca Williams is thanked for corrections and suggestions.Notes and references
- C. Boyer, M. H. Stenzel and T. P. Davis, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 551–595 CrossRef CAS.
- N. V. Tsarevsky and B. S. Sumerlin, Fundamentals of Controlled/Living Radical Polymerization, Royal Society of Chemistry, 2012 Search PubMed.
- J. Ferreira, J. Syrett, M. Whittaker, D. Haddleton, T. P. Davis and C. Boyer, Polym. Chem., 2011, 2, 1671–1677 RSC.
- C. Barner-Kowollik, T. P. Davis and M. H. Stenzel, Aust. J. Chem., 2006, 59, 719–727 CrossRef CAS.
- J. Jagur-Grodzinski, Living and Controlled Polymerization: Synthesis, Characterization, and Properties of the Respective Polymers and Copolymers, Nova Science Publishers, 2006 Search PubMed.
- R. T. A. Mayadunne, J. Jeffery, G. Moad and E. Rizzardo, Macromolecules, 2003, 36, 1505–1513 CrossRef CAS.
- M. Stenzel-Rosenbaum, T. P. Davis, V. Chen and A. G. Fane, J. Polym. Sci., Part A: Polym. Chem., 2001, 39, 2777–2783 CrossRef CAS.
- J. F. Quinn, R. P. Chaplin and T. P. Davis, J. Polym. Sci., Part A: Polym. Chem., 2002, 40, 2956–2966 CrossRef CAS.
- G. Zheng and C. Pan, Polymer, 2005, 46, 2802–2810 CrossRef CAS.
- H. Chaffey-Millar, M. H. Stenzel, T. P. Davis, M. L. Coote and C. Barner-Kowollik, Macromolecules, 2006, 39, 6406–6419 CrossRef CAS.
- H. Gao and K. Matyjaszewski, Prog. Polym. Sci., 2009, 34, 317–350 CrossRef CAS.
- W. Wu, W. Wang and J. Li, Prog. Polym. Sci., 2015, 46, 55–85 CrossRef CAS.
- G. Lapienis, Prog. Polym. Sci., 2009, 34, 852–892 CrossRef CAS.
- B. Jeong, Y. K. Choi, Y. H. Bae, G. Zentner and S. W. Kim, J. Controlled Release, 1999, 62, 109–114 CrossRef CAS PubMed.
- L. Qiu and Y. Bae, Pharm. Res., 2006, 23, 1–30 CrossRef CAS PubMed.
- G. R. Whittell, M. D. Hager, U. S. Schubert and I. Manners, Nat. Mater., 2011, 10, 176–188 CrossRef CAS PubMed.
- B. Helms, S. J. Guillaudeu, Y. Xie, M. McMurdo, C. J. Hawker and J. M. J. Frechet, Angew. Chem., Int. Ed., 2005, 44, 6384–6387 CrossRef CAS PubMed.
- S. Kobayashi and R. Akiyama, Chem. Commun., 2003, 449–460 RSC.
- J. Liu, H. Duong, M. R. Whittaker, T. P. Davis and C. Boyer, Macromol. Rapid Commun., 2012, 33, 760–766 CrossRef CAS PubMed.
- J. M. Ren, T. G. McKenzie, Q. Fu, E. H. Wong, J. Xu, Z. An, S. Shanmugam, T. P. Davis, C. Boyer and G. G. Qiao, Chem. Rev., 2016, 116, 6743–6836 CrossRef CAS PubMed.
- N. Hadjichristidis, A. Hirao, Y. Tezuka and F. Du Prez, Complex Macromolecular Architectures: Synthesis, Characterization, and Self-Assembly, Wiley-VCH Verlag GmbH & Co. KGaA, 2011 Search PubMed.
- Y. S. Jo, J. Gantz, J. A. Hubbell and M. P. Lutolf, Soft Matter, 2009, 5, 440–446 RSC.
- L. S. Nair and C. T. Laurencin, Prog. Polym. Sci., 2007, 32, 762–798 CrossRef CAS.
- R. Chandra and R. Rustgi, Prog. Polym. Sci., 1998, 23, 1273–1335 CrossRef CAS.
- K. E. Uhrich, S. M. Cannizzaro, R. S. Langer and K. M. Shakesheff, Chem. Rev., 1999, 99, 3181–3198 CrossRef CAS PubMed.
- J. C. Bevington, D. E. Eaves and R. L. Vale, J. Polym. Sci., Part A: Polym. Chem., 1958, 32, 317–322 CAS.
- K. L. Mallik and M. N. Das, Naturwissenschaften, 1964, 51, 37–37 CrossRef CAS.
- A. P. Freidig, H. J. M. Verhaar and J. L. M. Hermens, Environ. Toxicol. Chem., 1999, 18, 1133–1139 CAS.
- N. P. Truong, Z. Jia, M. Burges, N. A. J. McMillan and M. J. Monteiro, Biomacromolecules, 2011, 12, 1876–1882 CrossRef CAS PubMed.
- P. Cotanda, D. B. Wright, M. Tyler and R. K. O'Reilly, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 3333–3338 CrossRef CAS.
- N. P. Truong, W. Gu, I. Prasadam, Z. Jia, R. Crawford, Y. Xiao and M. J. Monteiro, Nat. Commun., 2013, 4, 1902 CrossRef PubMed.
- W. Gu, Z. Jia, N. P. Truong, I. Prasadam, Y. Xiao and M. J. Monteiro, Biomacromolecules, 2013, 14, 3386–3389 CrossRef CAS PubMed.
- S. B. Hartono, N. T. Phuoc, M. Yu, Z. Jia, M. J. Monteiro, S. Qiao and C. Yu, J. Mater. Chem. B, 2014, 2, 718–726 RSC.
- M. Gillard, Z. Jia, P. P. Gray, T. P. Munro and M. J. Monteiro, Polym. Chem., 2014, 5, 3372–3378 RSC.
- A. C. Evans, A. Lu, C. Ondeck, D. A. Longbottom and R. K. O′Reilly, Macromolecules, 2010, 43, 6374–6380 CrossRef CAS.
- A. D. Ievins, X. Wang, A. O. Moughton, J. Skey and R. K. O′Reilly, Macromolecules, 2008, 41, 2998–3006 CrossRef CAS.
- F. Sun, C. Feng, H. Liu and X. Huang, Polym. Chem., 2016, 7, 6973–6979 RSC.
- R. Whitfield, A. Anastasaki, N. P. Truong, P. Wilson, K. Kempe, J. A. Burns, T. P. Davis and D. M. Haddleton, Macromolecules, 2016, 49, 8914–8924 CrossRef CAS.
- W. Zhao, P. Fonsny, P. Fitzgerald, G. G. Warr and S. Perrier, Polym. Chem., 2013, 4, 2140–2150 RSC.
- J. P. Patterson, A. M. Sanchez, N. Petzetakis, T. P. Smart, T. H. Epps III, I. Portman, N. R. Wilson and R. K. O'Reilly, Soft Matter, 2012, 8, 3322–3328 RSC.
- H. Gao and K. Matyjaszewski, J. Am. Chem. Soc., 2007, 129, 11828–11834 CrossRef CAS PubMed.
- W. Zeng, Y. Du, Y. Xue and H. L. Frisch, in Physical Properties of Polymers Handbook, ed. J. Mark, Springer, New York, 2007, ch. 17 Search PubMed.
- P. C. Hiemenz and T. P. Lodge, Polymer Chemistry, Taylor & Francis, 2nd edn, 2007 Search PubMed.
- A. B. James, M. H. David and C. R. Becer, in Progress in Controlled Radical Polymerization: Materials and Applications, American Chemical Society, 2012, ch. 6, vol. 1101, pp. 81–98 Search PubMed.
- W. Radke, in Macromolecular Engineering, Wiley-VCH Verlag GmbH & Co. KGaA, 2007 Search PubMed.
- K. McEwan, R. K. Randev and D. M. Haddleton, Analysis of Star Polymers Using the Agilent 1260 Infinity Multi-Detector GPC/SEC System, Agilent Technologies, Inc., USA, 2013 Search PubMed.
- J. P. Patterson, M. P. Robin, C. Chassenieux, O. Colombani and R. K. O'Reilly, Chem. Soc. Rev., 2014, 43, 2412–2425 RSC.
- M. Higuchi and R. Senju, Polym. J., 1972, 3, 370–377 CrossRef CAS.
- F. Fan and G. Gadda, J. Am. Chem. Soc., 2005, 127, 2067–2074 CrossRef CAS PubMed.
- S. Ikuta, S. Imamura, H. Misaki and Y. Horiuti, J. Biochem., 1977, 82, 1741–1749 CrossRef CAS PubMed.
- H. Yamada, N. Mori and Y. Tani, Agric. Biol. Chem., 1979, 43, 2173–2177 CAS.
- C.-P. Lu, C.-T. Lin, C.-M. Chang, S.-H. Wu and L.-C. Lo, J. Agric. Food Chem., 2011, 59, 11403–11406 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Characterization and additional hydrolysis analysis of all polymers. See DOI: 10.1039/c7py00219j |
| This journal is © The Royal Society of Chemistry 2017 |