
Double-networks based on pH-responsive, amphiphilic “core-first” star first polymer conetworks prepared by sequential RAFT polymerization
Elina N.
Kitiri
a,
Costas S.
Patrickios
*a,
Chrysovalantis
Voutouri
b,
Triantafyllos
Stylianopoulos
b,
Ingo
Hoffmann
cd,
Ralf
Schweins
d and
Michael
Gradzielski
c
aDepartment of Chemistry, University of Cyprus, P. O. Box 20537, 1678 Nicosia, Cyprus. E-mail: kitiri.eleni@ucy.ac.cy; costasp@ucy.ac.cy
bDepartment of Mechanical and Manufacturing Engineering, University of Cyprus, P. O. Box 20537, Nicosia 1678, Cyprus. E-mail: valantis.vo@gmail.com; stylianopoulos.triantafyllos@ucy.ac.cy
cStranski Laboratorium für Physikalische und Theoretische Chemie, Institut für Chemie Technische Universität Berlin, Strasse des 17. Juni 124, 10623 Berlin, Germany. E-mail: ingo.hoffmann@tu-berlin.de; michael.gradzielski@tu-berlin.de
dInstitut Max von Laue-Paul Langevin (ILL), F-38042 Grenoble Cedex 9, France. E-mail: schweins@ill.eu
First published on 19th September 2016
Abstract
This manuscript presents the preparation and study of a new double-network hydrogel system, comprising an amphiphilic, pH-responsive first polymer conetwork synthesized via reversible addition–fragmentation chain transfer (RAFT) polymerization, and a second hydrophilic polymer network prepared via free radical photopolymerization. The amphiphilic character of the first conetwork led to its phase separation on the nanoscale, as indicated by small-angle neutron scattering (SANS) in deuterium oxide, whereas the presence of segments consisting of tertiary amine-bearing monomer repeating units resulted in pH-dependent equilibrium swelling in water. Finally, the introduction of a second, reinforcing network into the structure of the first conetwork produced a double-network hydrogel system with mechanical properties (compressive stress and strain at break, and low-strain elastic modulus) superior to those of the first conetwork. Thus, the present complex hydrogel system bears three important functions: high mechanical properties to endure an environment with high stresses, hydrophobic pockets to solubilize non-polar substances within an overall aqueous milieu, and an ability to respond to changes in pH. Such multi-functional water-swollen polymer systems can pave the way toward next-generation biomaterials.
Introduction
Amphiphilic polymer conetworks (APCN) are important materials attracting increasing attention due to their properties and potential applications.1 These polymeric materials comprise hydrophilic and hydrophobic segments, covalently interconnected to a network. When placed in water, the hydrophobic segments rearrange themselves in order to minimize contact with the aqueous environment, leading to phase separation on the nanoscale. This internal organization makes APCNs ideal materials for use in antifouling coatings, modern soft contact lenses, matrices for phase transfer reactions with applications in bio- and organocatalysis, and the fabrication of gas and optical sensors.The increased interest in APCNs by the polymer community becomes apparent from the numerous publications that recently appeared in high impact factor journals exploring new applications and broadening existing ones. For example, Müllen and co-workers prepared polysulfone-based APCNs and studied their drug loading behavior, their structure, and also their swelling in water and methanol both gravimetrically and by solid-state NMR spectroscopy.2 Bruns et al. developed a self-sealing composite membrane comprising APCN and poly(ether ester) (PEE) layers, whose APCN water-swelling imparted self-sealing, whereas the PEE layers conferred a waterproof but breathable nature.3 The same team also prepared a photosensitive APCN-based membrane whose permeability to caffeine solution could be adjusted by visible light irradiation.4 Tew's team prepared bicontinuous APCNs in which both ion conductivity and storage modulus were high in a range of compositions, a result of the judicious choice of the two polymer components and the morphological co-continuity within a large polymer composition window.5 Kennedy and Cakmak prepared APCNs for pancreatic cell immunoisolation6 and developed an efficient optical method to monitor segment surface rearrangement in these materials.7 He et al., prepared tough APCNs on which L929 cells were cultured with a high viability.8 Agarwal and co-workers prepared enzymatically degradable APCNs synthesized by controlled radical ring opening copolymerization,9 Tiller et al. employed oxazoline-based APCNs10 to activate the enzymatic activity of Lipase Cal B in organic solvents,11 whereas Jewrajka and colleagues prepared biocompatible and biodegradable APCNs for the high loading and sustained release of both hydrophobic and hydrophilic drugs.12 In an interesting approach, Lin and Gitsov prepared and characterized APCNs bearing covalently attached drugs and fluorescent markers.13 Finally, Okay and colleagues employed scattering and microscopy techniques to follow the nanostructural evolution and unveil the self-healing mechanism in micellar APCNs,14 Wooley and co-workers used APCNs based on hyperbranched fluoropolymers and poly(ethylene glycol) (PEG) as anti-icing materials,15 while Kali and Iván prepared APCNs based on polyisobutylene and poly(methacrylic acid), with the methacrylic acid monomer being introduced via a novel chemical protection method.16
Due to the presence of the cross-links, and the inhomogeneity of the polymer chains, the morphologies exhibited by all known APCNs are not perfect, displaying only short-range order and blurred interfaces. To improve the order in the morphologies of nanophase separated APCNs, we optimized their synthesis by employing block copolymer building blocks and relatively low loadings of cross-linker. However, a non-ideality in this system was the broad distribution in the core functionality. Despite this defect, the system internally self-assembled, resulting in a structure comprising lamellae with relatively long-range order as confirmed via both scattering and microscopy techniques, and also exhibiting superior mechanical properties (high compressive stress at break, ∼14 MPa).17 In a most recent effort, we were able to enhance the mechanical properties (maximum compressive stress at break of 8 MPa) of APCNs nanophase separating only with short-range order by applying the concept of interpenetration and making them double-network hydrogels (DN, see one of next paragraphs reviewing DNs).18
Thus, there is much scope for further studies on well-defined APCNs, in order to afford self-assembled morphologies with long-range order, and also to endow upon them superior mechanical strength either via their well-defined morphologies or via reinforcement by the presence of a second network; herein, we pursued the development of mechanically robust APCNs via their interpenetration using a second, reinforcing network (DN approach).
Double-network hydrogels (DN)19–25 are interpenetrating hydrogels, presenting extraordinary mechanical properties (stress and strain at break and Young's modulus), and most efficiently addressing the problem of hydrogel fragility compared to other approaches, including slide-ring networks26 and nanocomposite networks.27 Developed in 2003,19 DNs comprised a first chemically cross-linked polyelectrolyte network and a second chemically cross-linked electrically neutral network. With a small number of exceptions,28 the originally optimized DNs display better mechanical properties than most subsequently developed DNs, with a record of compression stress and strain at break of 17.2 MPa and 92%, respectively,19 and tensile stress and strain at break of 0.9 MPa and 800%, respectively.29,30
Since the original development of DNs, it was proved that the principle of mechanical property-enhancement through network interpenetration is more general, and also applies when the positions of the polyelectrolytic and neutral hydrogels are reversed,31 and when one or both hydrogels are physically (rather than chemically) cross-linked too.28,32 It was most recently proven that the DN principle can also be applied to enhance the mechanical properties of organogels.33 Finally, our Research Team has applied the concept of DNs in well-defined, end-linked hydrophilic networks34 and in APCNs.18
Thus the DN principle is an important new concept, readily employable for the mechanical reinforcement of polymeric hydrogels, and it will be applied for the development of the materials in the present investigation.
Reversible addition–fragmentation chain transfer (RAFT) polymerization emerges as a powerful tool for macromolecular engineering, affording the preparation of (co)polymers with controlled molecular weight and molecular weight distribution, composition and architecture, from a wide range of monomer types.35 To the above must be added recent developments in the field, where RAFT polymerization can be activated by visible light or mild UV irradiation, without the need for conventional photo-initiators, metal-catalysts, or dye sensitizers.36 Compared to group transfer polymerization (GTP),37 our long-time synthetic work-horse, RAFT polymerization is tolerant to moisture and can afford polymers of higher molecular weight.
The aim in this investigation is to prepare and characterize DNs based on APCN first networks and hydrophilic second networks. Unlike our previous studies18,34 where the first networks were synthesized using GTP,37 the present work employs the more robust and timely technique of RAFT polymerization.35 Similar to our previous studies,18 the building blocks for the APCNs in this work are also star block copolymers,38 albeit of longer arms afforded by RAFT polymerization. The present DN materials are pH-responsive, a result of the utilization of a tertiary amine monomer as the hydrophilic component in the APCNs. Another property of these materials is the presence of sizable hydrophobic pockets, arising from the self-organization of the hydrophobic blocks in the APCNs. Finally, the DN hydrogels are mechanically stronger than their parent APCNs, due to the presence of the second, reinforcing, hydrophilic network.
Experimental section
Materials
The hydrophilic monomer 2-(dimethylamino)ethyl methacrylate (DMAEMA, 99%), the hydrophobic monomer lauryl methacrylate (LauMA, 96%), the cross-linker ethylene glycol dimethacrylate (EGDMA, 98%), and the radical initiator 2,2′-azobis(isobutyronitrile) (AIBN, 95%) were used for the synthesis of the first networks, while the hydrophilic monomer acrylamide (AAm, ≥98%), the cross-linker N,N′-methylenebisacrylamide (MBAAm, 99%), and the photoinitiator 2-oxoglutaric acid (OA, ≥99%) were used for the synthesis of the second networks. All the above-mentioned reagents, as well as 2,2-diphenyl-1-picrylhydrazyl hydrate (DPPH, 95%), calcium hydride (CaH2, 90–95%), basic alumina (≥97%), magnesium (98%), iodine flakes (99+%), bromobenzene (99%), hydrochloric acid (HCl, ≥37%), and 1,4-dioxane (99.8%) were purchased from Aldrich, Germany. Dichloromethane (DCM, 99%) was purchased from Labscan, Ireland. Tetrahydrofuran (THF, 99.8%, both HPLC and reagent grade), and ethanol (96%) were purchased from Scharlau, Spain. n-Hexane (≥96%), and carbon disulfide (≥99.5%) were purchased from Merck, Germany. Dimethylsulfoxide (DMSO, 99.9%) and ethyl acetate (EtAc, 99.96%) were purchased from Fisher Scientific, UK. Fig. 1 shows the chemical structures, names and abbreviations for the monomers, cross-linkers, initiators, and the chain transfer agent (CTA) used for the preparation of the amphiphilic “core-first” star polymer first conetworks, and the randomly cross-linked hydrophilic second networks.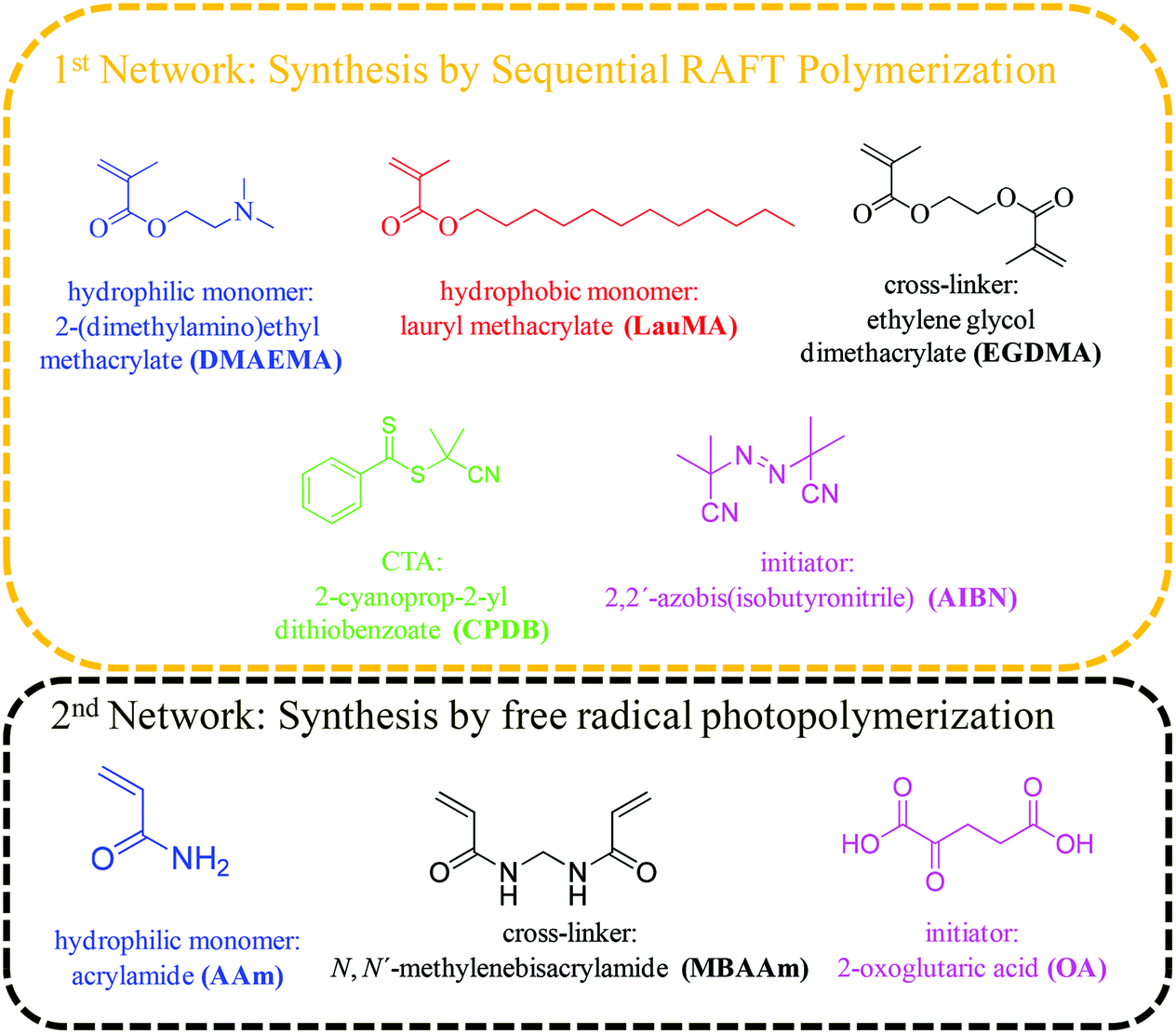 | ||
| Fig. 1 Chemical structures, names and abbreviations of the main reagents used for the synthesis of the “core-first” star first polymer conetworks and the second randomly cross-linked polymer networks. | ||
Methods
The DMAEMA and LauMA monomers and the EGDMA cross-linker were passed through basic alumina columns to remove inhibitors and any other acidic impurities. Subsequently, they were stirred over CaH2 for 72 h, in the presence of the free-radical inhibitor DPPH to avoid thermal polymerization. Finally, they were freshly distilled prior to use. The AIBN radical initiator and the MBAAm cross-linker were recrystallized from ethanol, whereas the AAm monomer was recrystallized from chloroform. The polymerization solvent 1,4-dioxane was dried over CaH2, and was freshly distilled prior to use.Synthesis of 2-cyanoprop-2-yl dithiobenzoate
The RAFT chain transfer agent (CTA) 2-cyanoprop-2-yl dithiobenzoate (CPDB) was synthesized in two stages. The first stage was a Grignard reaction for the preparation of the dimer of dithiobenzoic acid, following the procedure developed by Wager et al.39 To this end, 1.1 g of Mg (45.8 mmol) and 130 mL of THF were transferred into a two-necked round-bottomed flask, under an inert argon atmosphere, and 4.8 mL of bromobenzene (7.1 g, 45.5 mmol) was added dropwise into the mixture. The reaction mixture was warmed to 40 °C, until Mg was consumed, and then 2.7 mL of carbon disulfide (3.5 g, 45.4 mmol) was added dropwise. The system was left to stir for 30 min at −5 °C before removing the solvent using a rotary evaporator. The residue was dissolved in 60 mL DCM, transferred into a separating funnel and treated with fuming hydrochloric acid until the organic phase turned from orange to purple. The organic layer was concentrated and dried over anhydrous magnesium sulfate. Removal of the DCM under reduced pressure yielded dithiobenzoic acid. Subsequently, 15 mL of ethanol, a few flakes of iodine, and 3.55 mL of DMSO were added to the pure dithiobenzoic acid. The resulting dimer of dithiobenzoic acid was purified via recrystallization in ethanol. In the second stage, 3 g of dithiobenzoyldisulfide (9.8 mmol) and 2.4 g of 2,2′-azobis(isobutyronitrile) (14.7 mmol) were added to a round-bottomed flask with 40 mL of ethyl acetate and the mixture was left to react for 19 h at 90 °C. After this time, the solvent was removed under vacuum and the crude product was purified by column chromatography (ethyl acetate![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :hexane, 9:1) to yield CPDB as a dark red oil. The purity of the CTA was confirmed by 1H and 13C NMR spectroscopy.
:hexane, 9:1) to yield CPDB as a dark red oil. The purity of the CTA was confirmed by 1H and 13C NMR spectroscopy.
Preparation of “core-first” star homopolymer first networks
Two “core-first” star homopolymer-based first networks were synthesized in this study by sequential RAFT polymerization of monomer and cross-linker, one based on LauMA and the other on DMAEMA. The procedure followed for the preparation of the “core-first” star LauMA homopolymer first network is described below (network no. 2 in Table 1). To a 25 mL Schlenk tube, 80 μL of freshly distilled EGDMA (84 mg, 0.423 mmol), 0.094 g of CPDB (0.423 mmol), 43 mg of AIBN (0.264 mmol), and 4.15 mL of freshly distilled 1,4-dioxane were added. Subsequently, the mixture was degassed by three freeze–vacuum–thaw cycles, and the Schlenk was placed in an oil bath at 70 °C for 20 h. After sampling for gel permeation chromatography (GPC) and 1H NMR spectroscopy to characterize the produced star polymer core (cross-linker conversion by 1H NMR spectroscopy = 100%; GPC molecular weight = 8470 g mol−1; Đ = molecular weight dispersity = 2.34), 12.4 mL of LauMA (10.8 g, 42.3 mmol) and 0.45 mL of 1,4-dioxane were added and left to polymerize for 24 h. A sample of the synthesized “core-first” star LauMA homopolymer was also obtained for GPC and 1H NMR spectroscopy analyses (LauMA conversion by 1H NMR spectroscopy = 85%; GPC molecular weight = 33100 g mol−1; Đ = 1.15), 0.48 mL of EGDMA (0.504 g, 2.54 mmol) was added to the polymerization mixture, and gel formation occurred within 24 h.
| No. | Polymer structurea | Monomer conversion (%) | LauMA content in polymer (mol%) | GPC results | |||||
|---|---|---|---|---|---|---|---|---|---|
| DMAEMA | LauMA | EGDMA | 1H NMR | Theory | Molecular weight | Đ | No. of armsb | ||
| a EGDMA: ethylene glycol dimethacrylate; DMAEMA: 2-(dimethylamino)ethyl methacrylate; LauMA: lauryl methacrylate. b Determined as the ratio of the star (co)polymer molecular weight divided by the theoretical molecular weight of the arm calculated from the molecular weight(s) of the monomer(s) and the targeted degree(s) of polymerization, also taking into account monomer conversion, and adding the contributions from the residues of the CTA (221 g mol−1) and EGDMA (198 g mol−1). | |||||||||
| 1 | EGDMA1 | — | — | 100 | 0 | 0 | 8290 | 2.23 | — |
| EGDMA1-b-DMAEMA100 | 87.7 | — | — | 0 | 0 | 64300 |
1.04 | 4.5 | |
| 2 | EGDMA1 | — | — | 100 | 0 | 0 | 8470 | 2.34 | — |
| EGDMA1-b-LauMA100 | — | 84.7 | — | 100 | 100 | 33100 |
1.15 | 1.5 | |
| 3 | EGDMA1 | — | — | 100 | 0 | 0 | 7930 | 2.11 | — |
| EGDMA1-b-DMAEMA50 | 86.2 | — | — | 0 | 0 | 65000 |
1.22 | 9.1 | |
| EGDMA1-b-DMAEMA50-b-LauMA50 | — | 72.5 | — | 44.1 | 50.0 | 91300 |
1.10 | 5.2 | |
| 4 | EGDMA1 | — | — | 100 | 0 | 0 | 8520 | 2.23 | — |
| EGDMA1-b-LauMA50 | — | 92.6 | — | — | — | 55500 |
1.14 | 4.6 | |
| EGDMA1-b-LauMA50-b-DMAEMA50 | 70.6 | — | — | 41.8 | 50.0 | 129000 |
1.26 | 6.9 | |
| 5 | EGDMA1 | — | — | 100 | 0 | 0 | 6440 | 2.84 | — |
| EGDMA1-b-DMAEMA25 | 94.3 | — | — | 0 | 0 | 121500 |
1.75 | 29.5 | |
| EGDMA1-b-DMAEMA25-b-LauMA75 | — | 79.4 | — | 70.9 | 75.0 | 197760 |
1.10 | 10.2 | |
| 6 | EGDMA1 | — | — | 100 | 0 | 0 | 7480 | 2.34 | — |
| EGDMA1-b-LauMA75 | — | 80.6 | — | 0 | 0 | 39100 |
1.16 | 2.5 | |
| EGDMA1-b-LauMA75-b-DMAEMA25 | 84.6 | — | — | 75.2 | 75.0 | 173000 |
1.30 | 7.6 | |
| 7 | EGDMA1 | — | — | 100 | 0 | 0 | 7780 | 2.33 | — |
| EGDMA1-b-(DMAEMA50-co-LauMA50) | 79.4 | 77.2 | — | 48.7 | 50.0 | 46600 |
1.26 | 2.8 | |
Preparation of “core-first” star block copolymer first conetworks
All amphiphilic “core-first” star polymer first conetworks were prepared by the sequential RAFT polymerization of the cross-linker, the two comonomers and the cross-linker again. As an example, we describe below the experimental procedure followed for the preparation of the EGDMA1-b-LauMA50-b-DMAEMA50-b-EGDMA6 conetwork (conetwork no. 4 in Table 1). To a 25 mL Schlenk tube, 80 μL of freshly distilled EGDMA (84 mg, 0.423 mmol), 0.094 g of CPDB (0.423 mmol), 43 mg of AIBN (0.264 mmol), and 4.15 mL of freshly distilled 1,4-dioxane were added. After complete dissolution of the reagents, three freeze–vacuum–thaw cycles were performed, and the Schlenk was placed in an oil bath at 70 °C for 20 h. The produced core was sampled for GPC and 1H NMR spectroscopy (cross-linker conversion by 1H NMR spectroscopy = 100%; GPC molecular weight = 8520 g mol−1; Đ = 2.23). Afterwards, 6.2 mL of LauMA (5.38 g, 21.0 mmol) and 0.4 mL of 1,4-dioxane were added and left to polymerize for 24 h. After sampling again for GPC and 1H NMR spectroscopy analyses (LauMA conversion by 1H NMR spectroscopy = 93%; GPC molecular weight = 55500 g mol−1; Đ = 1.14), 3.6 mL of DMAEMA (3.32 g, 21.0 mmol) and 0.4 mL of 1,4-dioxane were added and left to polymerize for 92 h. A sample of the synthesized “core-first” star block copolymer was also obtained for GPC and 1H NMR spectroscopy analyses (DMAEMA conversion by 1H NMR spectroscopy = 71%; GPC molecular weight = 129000 g mol−1; Đ = 1.26), and 0.48 mL of EGDMA (0.504 g, 2.54 mmol) was added to the polymerization mixture, leading to conetwork formation within 24 h.
Preparation of second networks
The final double-networks (DNs) were synthesized according to the following procedure. Pieces from each first network (equilibrated in THF) were cut, placed in vials and washed several times with deionized water. Then, these pieces were left to equilibrate for one week in deionized water. Subsequently, each sample was acidified by adding a precalculated amount of fuming HCl in order to achieve full ionization of the DMAEMA units. Next, all samples were placed in a vacuum oven and dried for 72 h at room temperature. Finally, the samples were immersed in an aqueous solution of 2 M AAm, containing 0.1 mol% MBAAm and 0.1 mol% OA relative to AAm for two days until equilibrium was reached. The composite mixture was then irradiated using a commercial UV system from Vivo Ltd. model NW107RG-T3296 with four UV lamps of total power of 36 W, leading to the photopolymerization of the AAm-MBAAm components, the formation of the second network, and the preparation of the final DNs.Determination of the sol fraction in the conetworks and in the DNs
The resulting amphiphilic conetworks were placed in 200 mL of THF for 1 week to remove the sol fraction. After this time, the extract was recovered by filtration and the solvent was removed using a rotary evaporator. The remaining polymer was further dried for three days in a vacuum oven at room temperature. The sol fraction was determined by the following equation: | (1) |
Characterization of the conetwork precursors and conetwork sol fraction
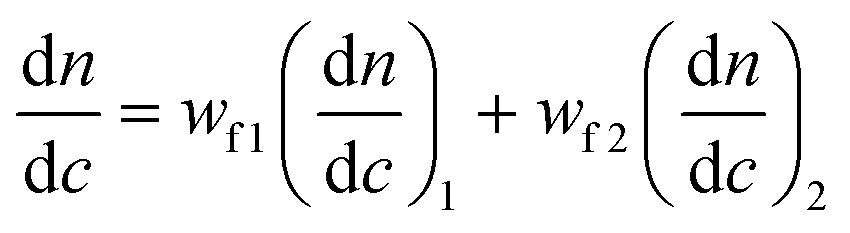 | (2) |
Measurements of the degrees of swelling (DS)
Small pieces from each conetwork were cut, dried under vacuum at room temperature for 72 h, and finally weighed. Subsequently, the degrees of swelling (DS) of the conetworks were measured in THF, in pure water (pH ∼8), and in aqueous solutions in different pHs (2–12) as described in the following lines. In the case of THF and pure water, dry samples were immersed in the appropriate solvent (THF or pure water) and allowed to equilibrate for two weeks and reweighed. For the measurements of the DSs at different pHs, ten preweighed dry samples were immersed in water and then the appropriate volume of a 0.5 M HCl or a 0.5 M NaOH standard solution was added, so that the pH be adjusted within the range between 2 and 12, and the samples were again left to equilibrate for two weeks. In all cases, the DSs were calculated as the ratio of the swollen divided by the dry conetwork mass.In order to study the swelling behavior of the DNs in the pH range from 2 to 12, small pieces from each DN were cut, dried, weighed and finally immersed in aqueous buffer solutions, each maintained at a certain pH value. A phosphoric acid (H3PO4) solution was used for pH ∼2, a solution of a mixture of citric acid and sodium phosphate dibasic (Na2HPO4) for pH ∼3, a mixed solution of sodium acetate and acetic acid for pH from ∼4 to ∼6, a solution mixture of Na2HPO4 and sodium phosphate monobasic (NaH2PO4) for pH from ∼7 to ∼8, and a solution of a mixture of sodium carbonate (Na2CO3) and sodium bicarbonate (NaHCO3) for pHs from ∼9 and above. Then, the samples were left to equilibrate for two weeks and the DSs of each sample were calculated as described for the case of the first networks.
Small-angle neutron scattering (SANS)
All the first (co)networks of this study were characterized using small-angle neutron scattering (SANS) in D2O. SANS measurements were performed on the D11 instrument at the Institut Laue-Langevin (ILL) in Grenoble, France. The wavelength was λ = 6 Å. Three sample-to-detector distances of 1.2, 8, and 34 m were employed, using a collimation length of 8 m for the first two configurations and 34 m for the third. The sensitivity of the detector elements was taken into account by comparison with the scattering of a 1 mm sample of H2O. With the aid of this measurement together with the measured sample transmissions, the SANS data were put on an absolute scale.Mechanical properties
Characterization of the mechanical properties of the water-swollen first and DNs in compression was performed using the high precision mechanical testing system Instron (Norwood, MA, USA) model 5944. Compression tests were performed on rectangular specimens (length × width × thickness = 4 × 4 × 5 mm3) to a final strain of 95%, with an applied strain rate set at 0.05 mm min−1. Each measurement was repeated at least twice. The initial compressive elastic modulus, E (Young's modulus) was determined from the slope of the measured stress–strain curve in the range of 0–10% of strain. The fracture stress and strain were determined from the point of discontinuity in the stress–strain curve. The 1st Piola Kirchhoff (engineering) stress and the infinitesimal strain were used as measures of stress and strain, respectively.Results and discussion
Synthesis of “core-first” star polymer first networks
The synthesis of the first networks based on “core-first” star polymer (co)networks was accomplished by three- or four-step sequential RAFT polymerization. Seven first polymer (co)networks were synthesized in this work, two based on the star homopolymers of DMAEMA and LauMA, and five based on LauMA-DMAEMA star copolymers, four of which being star block copolymers and one a star polymer with statistical copolymer arms. The total degree of polymerization of the arms of all star (co)polymers was fixed at 100. The content in hydrophobic monomer was adjusted to be relatively high, from 50–75 mol%, corresponding to hydrophobic weight fractions of 0.614–0.827, which would substantially lower aqueous swelling, but would also strongly drive self-assembly, with the particular compositions favoring the formation of anisotropic morphologies (cylinders and lamellae) in water. The loading of EGDMA cross-linker in the first step (forming the first or primary core) was one EGDMA equivalent relative to the CPDB CTA, which represented the highest EGDMA loading (the higher the EGDMA loading, the higher the number of arms of the stars, expected to confer superior mechanical properties to both the first and DNs) which did not induce macroscopic gelation at this first addition step in preliminary experiments. The loading of EGDMA cross-linker in the last step (yielding the secondary core) was six EGDMA equivalents relative to the CPDB CTA, which was just above the minimum loading of EGDMA capable of inducing macroscopic gelation in preliminary experiments (determined to be between four and five EGDMA equivalents).Scheme 1 illustrates the synthetic sequence followed for the preparation of one of the “core-first” star block copolymer first networks, and, in particular, the synthesis of EGDMA1-b-LauMA50-b-DMAEMA50-b-EGDMA6. For this particular conetwork, the procedure included four steps. In the first step, EGDMA was oligomerized, giving the star polymer core. Second, LauMA was polymerized by growing it from the EGDMA core outward, giving the “core-first” star homopolymer of LauMA. This was followed in the third step by the addition of DMAEMA, which was polymerized by growing it from the tips of the LauMA star homopolymer, yielding an amphiphilic LauMA-DMAEMA “core-first” star block copolymer. Finally, the synthesis was completed in the fourth step by the addition of EGDMA cross-linker for a second time, which led to gelation and formation of the amphiphilic “core-first” star block copolymer first conetwork. Afterward, this first network was rinsed in THF (to remove the sol fraction), transferred into water (by daily replacing the water for several days), acidified by adding the appropriate volume of HCl, then immersed in an aqueous solution of 2 M AAm (this concentration was found optimal in our previous investigation18), 0.002 M MBAAm and 0.002 M OA, and finally photopolymerized with the aid of UV irradiation.
 | ||
| Scheme 1 Schematic representation of the synthetic procedure followed for the preparation of the “core-first” star block copolymer first conetwork EGDMA1-b-LauMA50-b-DMAEMA50-b-EGDMA6 and the corresponding DN. The LauMA units are colored red, the DMAEMA units are painted light blue, and the EGDMA units are shown in grey. | ||
Characterization of the conetwork precursors
Table 1 shows the chemical structures of all the precursors to the first (co)networks, their molecular weights and molecular weight dispersities, the polymer compositions, and the monomer and cross-linker conversions as determined by GPC and 1H NMR spectroscopy, respectively. Furthermore, Fig. 2 shows the GPC traces for one of the “core-first” star block copolymers, EGDMA1-b-LauMA50-b-DMAEMA50, and its two precursors, the “core-first” star LauMA homopolymer and the EGDMA core. Table 1 indicates that the conversions for the two comonomers were fairly high and ranged between 71 and 94% for DMAEMA, and between 73 and 93% for LauMA, whereas the EGDMA conversion was always 100%. The molecular weights of the EGDMA cores ranged from 6400 and 8500 g mol−1, while their Đ values were, in most cases, between 2.2 and 2.3, relatively high, and as expected for a polydisperse species resulting from the multiple and uncontrolled addition of EGDMA units. However, the subsequently produced “core-first” star polymers were much more homogeneous in their size, exhibiting Đ values between 1.2–1.3 (also see Fig. 2) for most samples. Their molecular weights exhibited values between 39100 and 198000 g mol−1, corresponding to number of arms between 3 and 29. The lowest star polymer molecular weights and arm numbers were displayed by star polymers whose first block was the more sterically-hindered LauMA, with the polymers bearing longer LauMA blocks presenting even lower molecular weights and arm numbers. Conversely, the highest star polymer molecular weights and arm number were displayed by star polymers whose first block was the less sterically-hindered DMAEMA, with the polymers bearing shorter DMAEMA blocks presenting even higher molecular weights and arm numbers. Finally, Table 1 shows a good agreement between the copolymer composition determined using 1H NMR spectroscopy and the one calculated on the basis of the comonomer feed ratio.
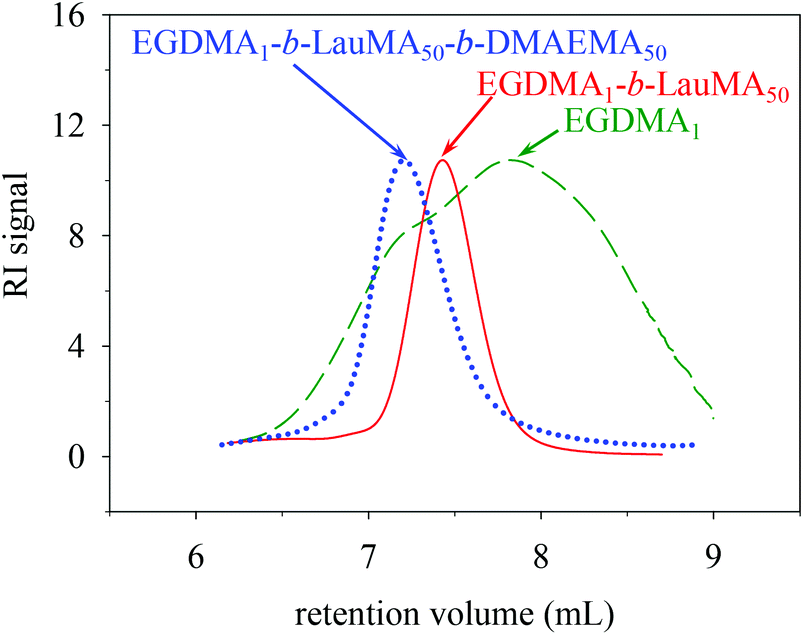 | ||
| Fig. 2 GPC traces of the core and the star precursors to the APCN first network with the structure EGDMA1-b-LauMA50-b-DMAEMA50-b-EGDMA6. | ||
Percentage, molecular weights and composition of the sol fraction of the conetworks
Table 2 shows the mass percentage, the molecular weight and Đ values, and the composition of the extractables from each first conetwork as measured by gravimetry, GPC and 1H NMR spectroscopy. The sol fractions of the (co)networks were lower than 19% w/w, indicating satisfactory interconnection of the star polymers to a (co)network. Highest percentages of extractables were obtained from the conetworks in which LauMA was added as the second monomer, right before the final addition of EGDMA cross-linker. This can be attributed to the inferior cross-reactivity between LauMA and EGDMA compared to DMAEMA and EGDMA, due to the steric hindrance arising from the long hydrocarbon side-chain in LauMA. The molecular weights of the (co)polymers in the extractables were lower than those of the final star precursors to the (co)networks, indicating that deactivation occurred before the polymerization of the last monomer was completed. The sol fraction in all DNs was lower than 1% w/w, suggesting near-quantitative polymerization of AAm in the second networks. The 1H NMR spectra of the extractables from the DNs indicated that their sol fraction mainly consisted of AAm homopolymer, 85–95%, and secondarily of AAm monomer, 5–15%.| No. | Polymer network structurea | Mass (%) | PolyLauMA (mol%) | GPC results | ||
|---|---|---|---|---|---|---|
| 1H-NMR | Theory | MW | Đ | |||
| a E is further abbreviation for EGDMA. | ||||||
| 1 | E1-DMAEMA100-E6-network | 16.3 | 0.0 | 0.0 | 10200 |
1.38 |
| 2 | E1-LauMA100-E6-network | 12.8 | 100.0 | 100.0 | 24900 |
1.18 |
| 3 | E1-(DMAEMA50-b-LauMA50)-E6-network | 18.7 | 54.40 | 50.0 | 12300 |
1.20 |
| 4 | E1-(LauMA50-b-DMAEMA50)-E6-network | 11.6 | 53.2 | 50.0 | 17800 |
1.27 |
| 5 | E1-(DMAEMA25-b-LauMA75)-E6-network | 13.4 | 71.3 | 75.0 | 27100 |
1.07 |
| 6 | E1-(LauMA75-b-DMAEMA25)-E6-network | 12.6 | 85.0 | 75.0 | 23500 |
1.25 |
| 7 | E1-(DMAEMA50-co-LauMA50)-E6-network | 14.5 | 51.3 | 50.0 | 14100 |
1.22 |
pH-Dependence of the degrees of swelling of the APCN first networks in water
Fig. 3 shows the pH-dependence of the DSs in water and the degrees of ionization (DI) of the six DMAEMA-bearing “core-first” star-based first (co)networks. In all cases, low DSs in water were obtained at high pH values (pH > 7) where the DMAEMA units were not ionized. In contrast, at pH below 7 the aqueous DS values increased due to the ionization of the DMAEMA units. Ionization of the DMAEMA units led to an increase in the osmotic pressure within the gels, due to accumulated chloride counteranions to the positively charged DMAEMA units. Furthermore, this charged state of the DMAEMA units resulted in electrostatic repulsions between the charged polyDMAEMA segments in the networks. Thus, the responsiveness of the DMAEMA units to (low) pH and concomitant ionization promoted gel swelling through two mechanisms, osmotic pressure and repulsive Coulombic interactions. In each of the graphs in Fig. 3, the swelling vs. pH curves followed the ionization vs. pH curves, confirming the importance of the electrostatics for the swelling of the conetworks. At very low pH, a high ionic strength, arising from the high hydrochloric acid concentration, caused a decrease in the DSs due to the screening of the electrostatic interactions.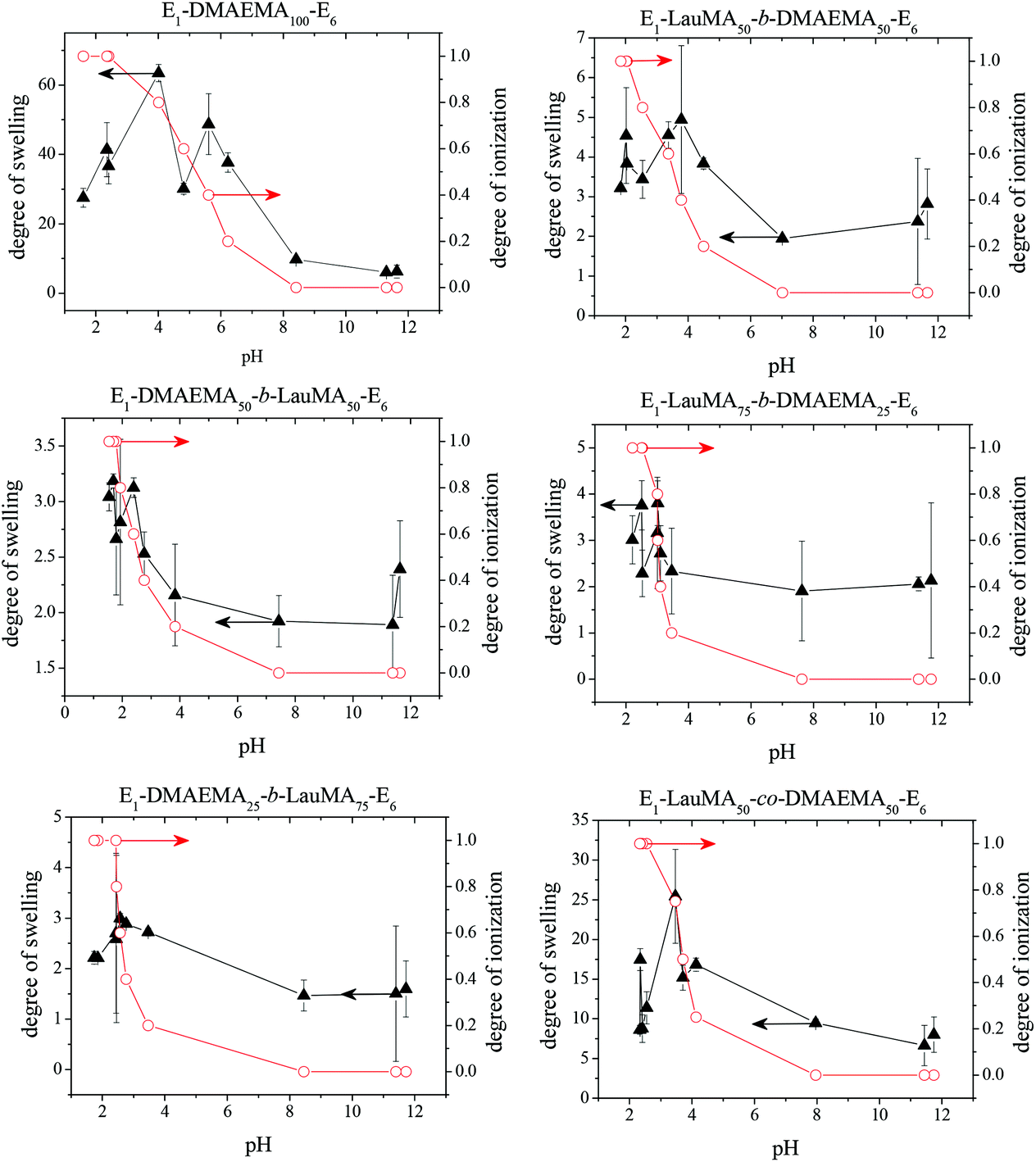 | ||
| Fig. 3 Degrees of swelling and ionization of all DMAEMA-containing first (co)networks as a function of pH. | ||
Fig. 4 displays the graphs for the pH-dependence of the DSs for all DMAEMA-bearing single (first) and double networks. The DSs of most DNs (four, in particular) were higher than those of their parent first networks over the whole pH range investigated, from pH 2 to 12, whereas for two DNs, their DSs were higher than those of their corresponding first networks only in the alkaline pH range. The generally higher DSs of the DNs were due to the increase in their hydrophilic character arising from the incorporation of the hydrophilic polyAAm second network. The two DNs which exhibited lower DSs in the acidic pH region compared to those of their parent first networks were the one based on the DMAEMA homopolymer first network and the one based on the statistical copolymer first network. Due to the absence of LauMA hydrophobic units, the DMAEMA homopolymer first network could present very high DSs in the acidic pH range where the DMAEMA units became ionized, thus exceeding the DSs of its daughter DN also bearing the very hydrophilic but nonionic AAm units in its second network. The reason why the DSs in acidic pH of the statistical copolymer-based first network (marginally) exceeded those of its daughter DN might be related to the difficulty/inability of the segments in the first network to microphase separate, as a result of the random distribution of the hydrophobic LauMA and hydrophilic DMAEMA units in the arms of its the star building blocks (see SANS section in a following paragraph); thus, for this first network, and unlike its two star block copolymer-based conetwork isomers, both monomer components contributed to its maximum swelling at acidic pH.40
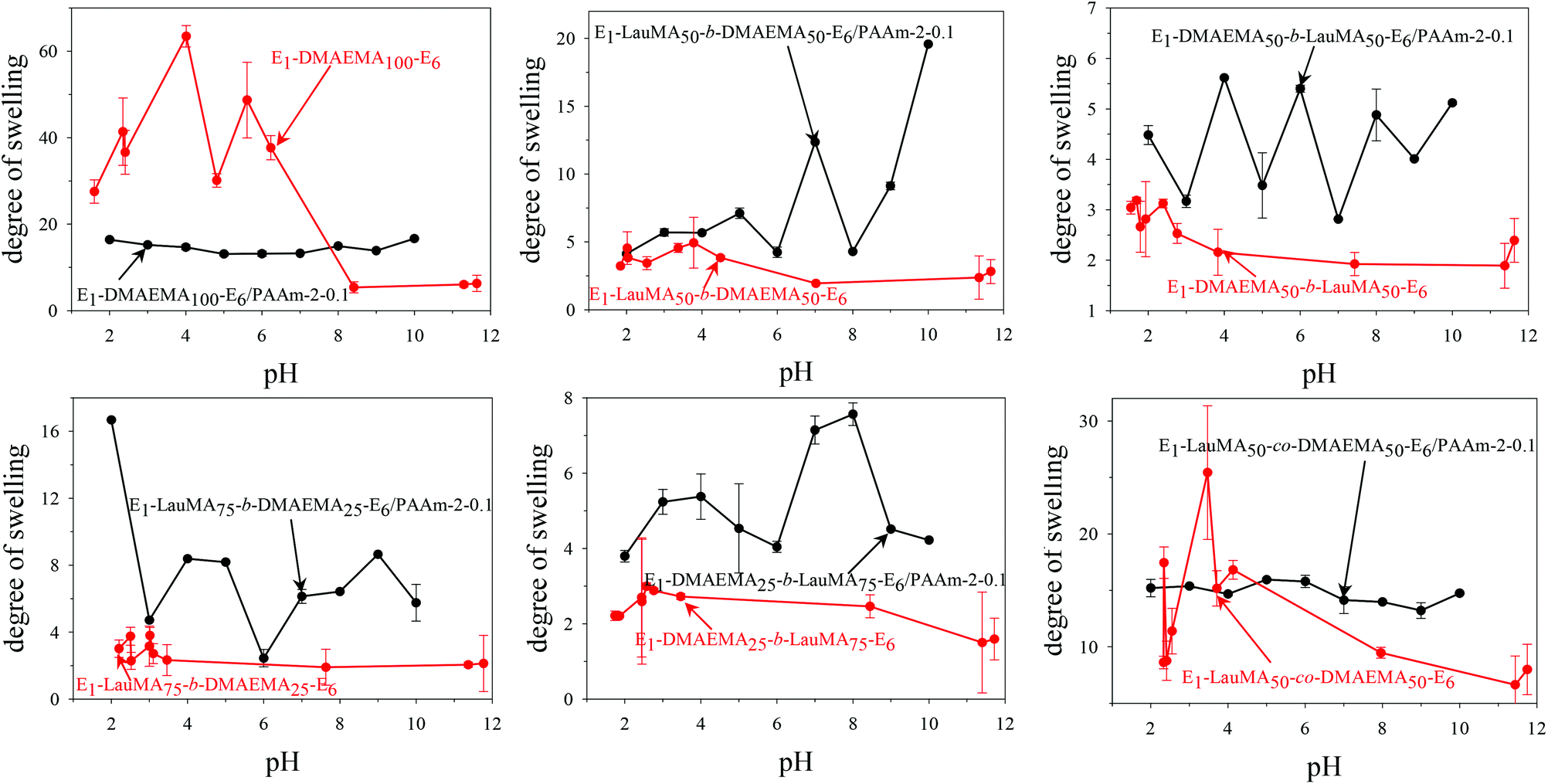 | ||
| Fig. 4 Degrees of swelling as a function of pH for all DMAEMA-bearing first and double networks. | ||
Degrees of swelling of the first (co)networks in THF, in pure water and in low pH water
The DSs in pure water, in low pH water and in THF of all the first (co)networks are presented in Fig. 5. Parts (a) and (b) of the figure show the effect of conetwork composition on the DSs of APCNs with EGDMA1-LauMAx-b-DMAEMA100−x-EGDMA6 and EGDMA1-DMAEMA100−x-b-LauMAx-EGDMA6 architectures, respectively, while part (c) illustrates how the DSs of equimolar conetworks are affected by conetwork architecture. Examining first parts (a) and (b), it appears that the network aqueous DSs, both in low pH (∼2) water and pure (∼8) water, decreased with increasing content in LauMA hydrophobic units, as expected. For all samples, the low-pH aqueous DS was higher than the corresponding DS in pure water, due to the ionization of the DMAEMA units under acidic conditions for the reasons explained previously. The largest difference in the aqueous DSs at these two different pH conditions was expectedly exhibited by the network richest in DMAEMA, i.e., the DMAEMA homopolymer first network. In contrast, the DSs in THF of the (co)networks in parts (a) and (b) of Fig. 5 displayed little variation with the LauMA content, because THF is a non-selective solvent for LauMA and DMAEMA, i.e., THF dissolves equally well these two types of monomer repeating units. Examining finally part (c) of Fig. 5, it is apparent that the two equimolar conetworks based on star block copolymers exhibited similar DSs which did not vary with conetwork architecture or solvent conditions. On the other hand, the equimolar network based on star polymers comprising statistical copolymer arms presented higher DSs in all three solvents as compared to its star block counterparts. The higher DSs in low-pH water of the network based on statistical copolymer stars can be attributed to its inability to self-assemble due to the random distribution of hydrophobic and hydrophilic monomer repeating units.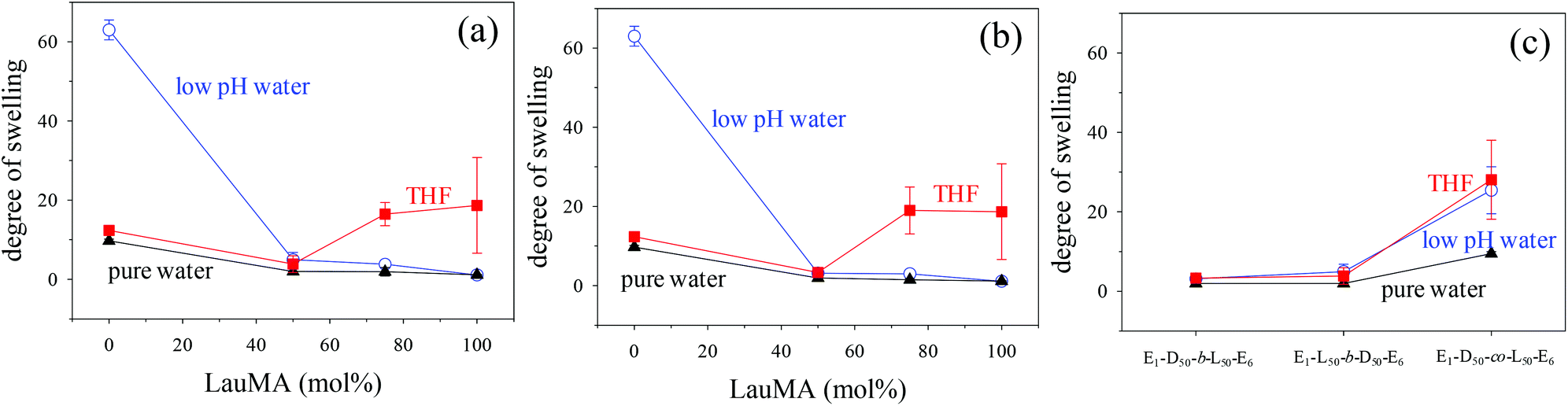 | ||
| Fig. 5 Dependence of the degrees of swelling (DSs) of the first (co)networks in pure water (pH ∼8), in low-pH (∼2) water, and in THF on their structure. Effect of LauMA content on the DSs of (co)networks with (a) EGDMA1-LauMAx-b-DMAEMAy-EGDMA6 architecture, and (b) EGDMA1-DMAEMAy-b-LauMAx-EGDMA6 architecture. (c) Effect of network architecture on the DSs of the three equimolar networks. E, D and L in part (c) are further abbreviations for EGDMA, DMAEMA and LauMA, respectively. | ||
Phase separation on the nanoscale
A detailed insight of the mesoscopic structures present in the first networks was obtained by small-angle neutron scattering (SANS), which was done for the gels swollen in D2O, i.e., at a pH ∼ 7. Fig. 6 shows the SANS profiles of all the “core-first” star first polymer conetworks in D2O, whereas Table 3 lists the separation distance, d, between the hydrophobic domains in the swollen gels, as simply calculated from the position of the intensity maximum, qmax (d = 2π/qmax) located in the SANS profiles in Fig. 6, as well as an estimation of the radius of the hydrophobic LauMA cores, R, calculated using the d values and the volume fraction of the hydrophobic cores, Φ. Parts (a) and (b) in the figure show the effect of the composition of conetworks with EGDMA1-LauMAx-b-DMAEMA100−x-EGDMA6 and EGDMA1-DMAEMA100−x-b-LauMAx-EGDMA6 architectures, respectively, while part (c) presents the SANS profiles for the two homopolymer networks and that of the statistical DMAEMA-LauMA copolymer network.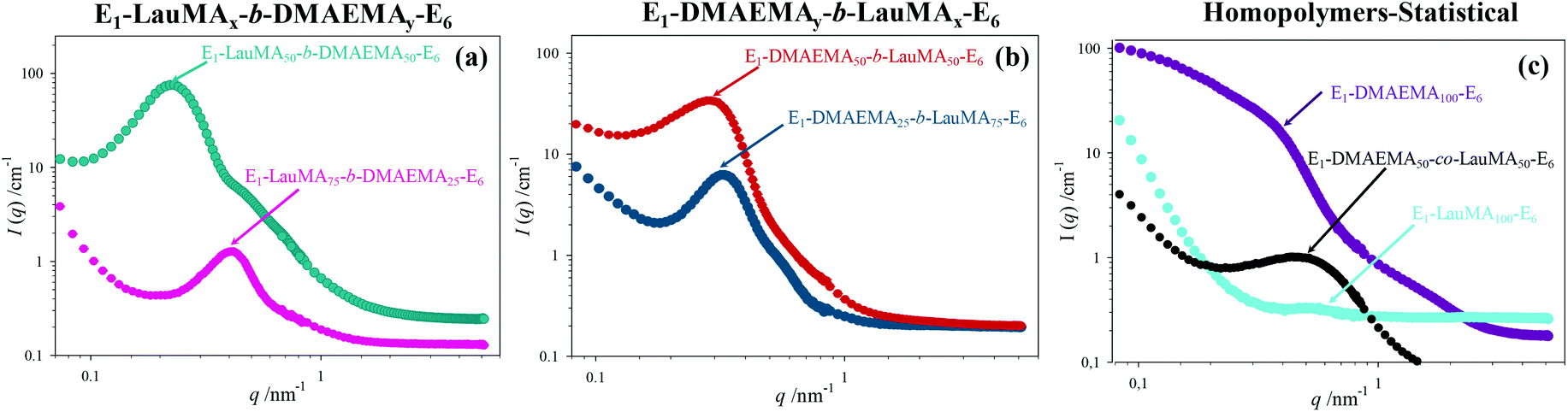 | ||
| Fig. 6 SANS profiles of all the D2O-swollen first networks. Effects of conetwork composition on conetworks with (a) EGDMA1-LauMAx-b-DMAEMA100−x-EGDMA6 and (b) EGDMA1-DMAEMA100−x-b-LauMAx-EGDMA6 architectures. (c) SANS profiles of the two homopolymer networks, plus the statistical network. In the graphs, E is further abbreviation for EGDMA. | ||
| No. | Polymer network structure | q max (nm−1) | d (nm) | Φ | R (nm) |
|---|---|---|---|---|---|
| a Calculated as: d = 2π/qmax. b Calculated from the conetwork composition and the aqueous degree of swelling. c Estimated as: R = d × (3Φ/4π)1/3, based on the assumption of a simple cubic lattice. | |||||
| 4 | EGDMA1-LauMA50-b-DMAEMA50-EGDMA6 | 0.24 | 26.2 | 0.30 | 10.9 |
| 3 | EGDMA1-DMAEMA50-b-LauMA50-EGDMA6 | 0.29 | 21.7 | 0.33 | 9.3 |
| 6 | EGDMA1-LauMA75-b-DMAEMA25-EGDMA6 | 0.42 | 15.0 | 0.36 | 6.6 |
| 5 | EGDMA1-DMAEMA25-b-LauMA75-EGDMA6 | 0.32 | 19.6 | 0.49 | 9.6 |
| 7 | EGDMA1-DMAEMA50-co-LauMA50-EGDMA6 | 0.49 | 12.8 | — | — |
The SANS profiles for all conetworks based on star block copolymers showed pronounced correlation peaks and a shoulder on the right of the correlation peak. The observation of a correlation peak in these samples indicates a rather high degree of internal organization within the conetworks, and, in particular, the presence of large hydrophobic domains placed at well-defined distances. The degree of ordering is highest for the case of the EGDMA1-LauMA50-b-DMAEMA50-EGDMA6 APCN whose SANS profile displayed the most pronounced correlation peak and most pronounced shoulder. This could be attributed to the location of the hydrophobic polyLauMA block right next to the first EGDMA core, and a rather large, flexible swollen polyDMAEMA block next to the second EGDMA core, allowing with its high flexibility for a very well-ordered arrangement. The presence of shoulders can partly be attributed to the second-order correlation peak, but also to the form factor of the cross-linkers and the densely packed hydrophobic blocks around them. The pronounced scattering in the case of the DMAEMA homopolymer network can be attributed to the scattering by EGDMA domains that are arranged in a large network. In contrast, the low scattering and the absence of a peak in the case of the LauMA homopolymer network is due to the fact that this network was in a collapsed state because it could not absorb D2O, a result of its high hydrophobicity. Interestingly, a correlation peak is also seen for the statistical DMAEMA-LauMA copolymer network. However, this peak is shifted to higher q values, thereby indicating the formation of much smaller hydrophobic domains, which is also confirmed by the much lower scattering intensity.
The EGDMA1-LauMA50-b-DMAEMA50-EGDMA6 sample not only has the most pronounced (abrupt) correlation peak (with the highest absolute scattering intensity), but that peak is also located at the lowest q position. Both of these facts indicate that within this sample the largest hydrophobic domains of all samples are formed. Table 3 shows that the estimated separation distance (spacing), d, between the hydrophobic domains within this sample was 26.2 nm, as compared to the corresponding distance of 21.6 nm estimated for the case of its isomer, EGDMA1-DMAEMA50-b-LauMA50-EGDMA6. These values for the spacing can be compared with the contour length of the block copolymer chain between the cross-links of 50.4 nm (equal to twice the contour length of the arm of the star; arm contour length = 25.2 nm = arm degree of polymerization × contribution of one monomer repeating unit = 100 × 0.252 nm41), which suggests rather stretched chains. In the case of the two more hydrophobic conetworks, EGDMA1-LauMA75-b-DMAEMA25-EGDMA6 and EGDMA1-DMAEMA25-b-LauMA75-EGDMA6, the estimated spacing between the scattering domains was 15.0 and 19.6 nm, respectively, consistent with the shorter segments of the more flexible and swollen polyDMAEMA. From the spacing, d, one can estimate the size of the hydrophobic domains via their effective volume fraction Φ (which again can be calculated using the degree of swelling as given in Fig. 5). Assuming a primitive cubic packing, one can calculate their radius, R, via: R = d × (3Φ/4π)1/3, which, for instance, for EGDMA1-LauMA50-b-DMAEMA50-EGDMA6, this would be ∼11 nm, which means that the domain size is given by the length of the hydrophobic block in a similar fashion to that typically observed for micelles. These calculated values for the radius R are given in the last column in Table 3. Interestingly, for the long LauMA75 blocks in EGDMA1-LauMA75-b-DMAEMA25-EGDMA6, the hydrophobic domains are the smallest. While at first glance this may seem counterintuitive, it may be attributed to the fact that in this copolymer the LauMA75 block is constrained at the first core and the hydrophilic part is too small to allow for a flexible and relaxed arrangement of the hydrophobic chains and, therefore, only smaller domains can be formed.
Mechanical properties
The mechanical properties in compression of all single (first) and double networks in water were measured in the equilibrium swollen state. These properties included the compressive stress (σmax) and strain (εmax) at break, and the compressive low-strain Young's modulus (E), and were extracted from the stress–strain curves. Typical stress–strain curves are plotted in Fig. 7, in which the failure points were at the end (top-right tip) of the two curves. These concern the first and DN based on the APCN with the structure EGDMA1-DMAEMA25-b-LauMA75-EGDMA1. Inspection of the two curves reveals that, while the two networks exhibited similar (and relatively low) low-strain Young's moduli, the strain at break of the DN (εmax = 78%) was fairly improved compared to that of its parent first conetwork (εmax = 68%), whereas the stress at break of the DN (σmax = 3.29 MPa) was significantly enhanced compared to that of its parent conetwork (σmax = 0.95 MPa). In fact, the particular DN displayed the highest stress at break of all DNs of this study. The present optimal DN stress at break of 3.3 MPa was lower than that determined in our previous study18 and found to be around 6 MPa for the LauMA-based DNs. This may be attributed to the greater number of arms in the primary cores of the stars constituting that sample in the previous study of about 50,18 as compared to a corresponding number of about 10 in the best sample in the present study. However, we wish to stress the significant improvement in the stress at break of this DN in the present study compared to its parent first APCN, and the ease with which this first APCN was prepared via RAFT polymerization in relation to those prepared by GTP. | ||
| Fig. 7 Compressive stress–strain curves for the EGDMA1-DMAEMA25-b-LauMA75-EGDMA6-based single and double networks in water. E in the figure labels is further abbreviation for EGDMA. | ||
The determined mechanical properties for all networks of this study, both first and double, are listed in Table 4, and are plotted for the conetworks based on the star block copolymers and for the networks based on the two homopolymers in Fig. 8.
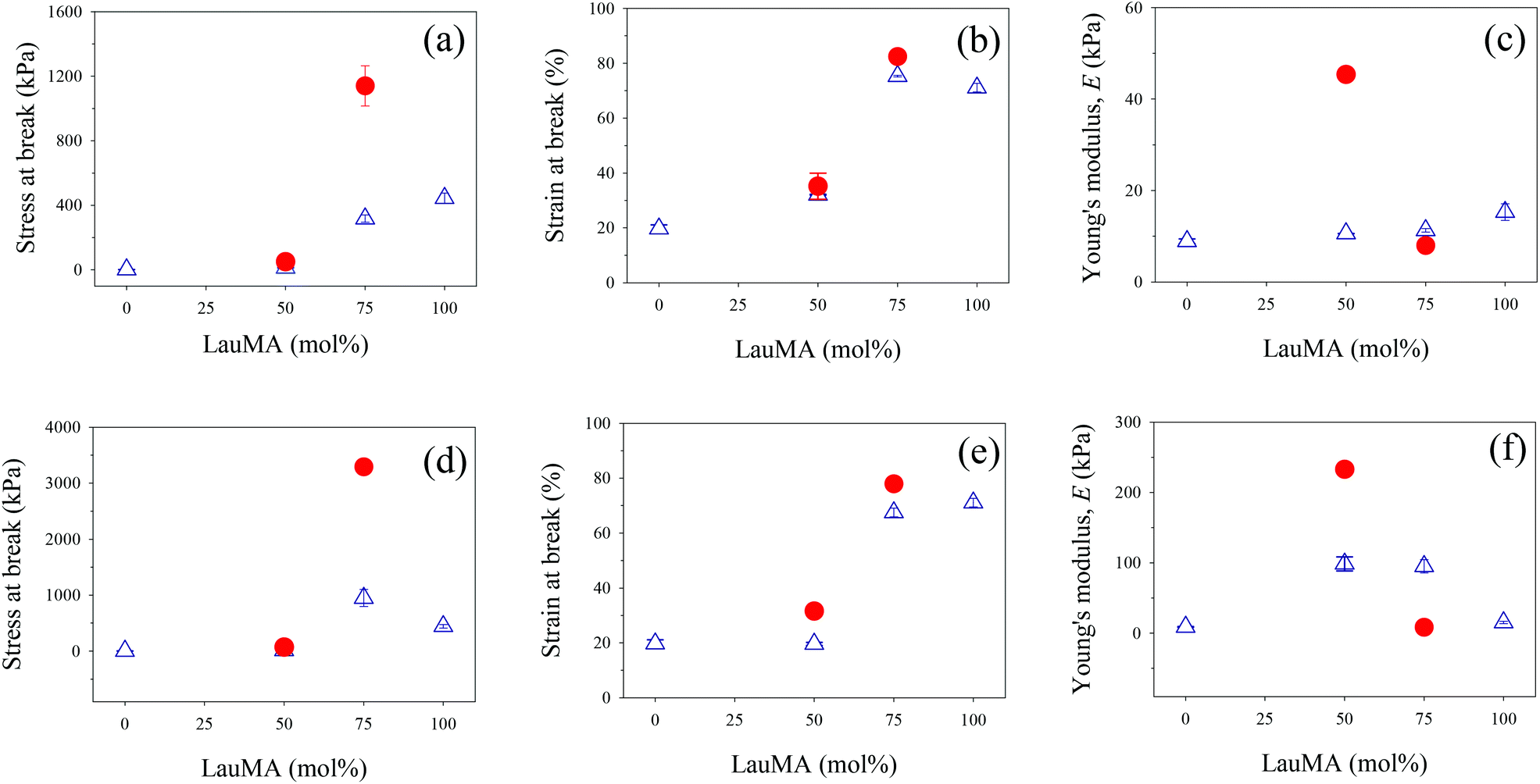 | ||
| Fig. 8 Effect of LauMA content in the first (co)network on the mechanical properties of the single (first) networks (SN, open blue triangles) and double networks (DN, closed red circles). Parts (a)–(c): first network with EGDMA1-LauMAx-b-DMAEMA100–x-EGDMA6 architecture; parts (d)–(f): first network with EGDMA1-DMAEMAx-b-LauMA100–x-EGDMA6 architecture. | ||
| Polymer network structurea | σ max (kPa) | ε max (%) | E (kPa) | |||
|---|---|---|---|---|---|---|
| SN | DN | SN | DN | SN | DN | |
| a E: further abbreviation for EGDMA. b Not measurable, very fragile network. | ||||||
| E1-DMAEMA100-E6-network | 2.29 ± 0.05 | — | 19.7 ± 1.4 | — | 8.9 ± 0.5 | — |
| E1-LauMA100-E6-network | 440 ± 30 | — | 71.1 ± 1.6 | — | 15.3 ± 1.8 | — |
| E1-(DMAEMA50-b-LauMA50)-E6-network | 14.4 ± 1.6 | 70.6 ± 0.06 | 19.6 ± 0.6 | 31.6 ± 0.4 | 98 ± 10 | 233 ± 5 |
| E1-(LauMA50-b-DMAEMA50)-E6-network | 13.0 ± 1.5 | 50 ± 12 | 32.1 ± 0.16 | 35.2 ± 4.8 | 10.6 ± 0.02 | 45.4 ± 0.07 |
| E1-(DMAEMA25-b-LauMA75)-E6-network | 950 ± 150 | 3290 ± 20 | 67.5 ± 1.6 | 77.9 ± 2.7 | 95 ± 9 | 8.3 ± 1.4 |
| E1-(LauMA75-b-DMAEMA25)-E6-network | 320 ± 22 | 1140 ± 130 | 75.3 ± 0.3 | 82.4 ± 0.3 | 11.3 ± 0.4 | 8.0 ± 0.8 |
| E1-(DMAEMA50-co-LauMA50)-E6-network | —b | 77 ± 7 | —b | 39 ± 3 | —b | 11 ± 2 |
Parts (a)–(c) in Fig. 8 plot the results for the mechanical properties of the SNs and DNs with EGDMA1-LauMAx-b-DMAEMA100−x-EGDMA6 architecture in the first network against the LauMA content in the first network, while parts (d)–(f) in the same figure present the corresponding results for the SNs and DNs with EGDMA1-DMAEMA100−x-b-LauMAx-EGDMA6 architecture in the first network.
Examining first parts (a) and (d) in Fig. 8, plotting the stress at break for the SNs and DNs against the composition of the SNs, it appears that the greatest values of stress at break occurred for the LauMA-rich DNs, comprising 75 mol% LauMA in the SNs. These values of stress at break were 1.14 and 3.29 MPa for the DNs based on APCNs first networks with EGDMA1-LauMA75-b-DMAEMA25-EGDMA6 and EGDMA1-DMAEMA25-b-LauMA75-EGDMA6 structures, respectively. The higher σmax value for the latter DN system compared to the former could be attributed to the higher σmax value for its parent SN compared to the corresponding value for its isomeric SN with the reverse block architecture. This, in turn, could be related to the greater number of arms (see Table 1) in the “core-first” star precursor to the first network in the case of the SN with the higher σmax value. It is noteworthy that the enhancement factor in stress at break (ratio of the σmax value for the DN divided by that of its parent SN) for the two systems is approximately the same, and equal to ca. 3.5. Interestingly, the enhancement factor in stress at break for the two other DN systems, whose first networks also possess a star block architecture but have a LauMA content of only 50 mol%, are even higher, and equal to 3.8 and 4.9 for the DNs with SN structures EGDMA1-LauMA50-b-DMAEMA50-EGDMA6 and EGDMA1-DMAEMA50-b-LauMA50-EGDMA6, respectively. It appears, therefore, that for the materials in the present study, the stress at break increases by a factor between 3.5 and 5, when going from the SNs to the DNs. Thus, the lower σmax values for the DNs based on SNs with only a 50 mol% LauMA content as compared to their counterparts richer in LauMA may be attributed to the lower σmax values for the SNs poorer in LauMA, which, in turn, it can be attributed to a reduced capability to dissipate energy in the case of the materials carrying less of the rubbery polyLauMA component and more of the fragile polyDMAEMA constituent. Despite the large improvement in the stress at break of the DNs of this study compared to their parent SNs, these DN values are still lower by a factor of about 5 relative to the original DNs developed by Gong.19 The mechanical properties of the APCN-based DNs can be improved by improving the corresponding properties of the single APCNs. This could be accomplished by substituting the rather brittle polymethacrylate components of the single APCNs for polyacrylates or polyacrylamides, exhibiting lower glass transition temperatures than the corresponding polymethacrylates. In the case of polyacrylamides, the improvement would be even greater due to the hydrogen-bonding capabilities in this type of polymers.
Examining now parts (b) and (e) in Fig. 8, displaying the strain at break for the SNs and DNs against the composition of the SNs, a moderate enhancement factor in the εmax values (ratio of the εmax value for the DN divided by that of its parent SN) is observed, ranging from 10 to 60%. The highest εmax values are recorded for DNs based again on LauMA-rich SNs, with the particular values being 82 and 78% for the DNs based on APCNs first networks with EGDMA1-LauMA75-b-DMAEMA25-EGDMA6 and EGDMA1-DMAEMA25-b-LauMA75-EGDMA6 structures, respectively. The εmax values recorded for the DNs based on SNs with only 50 mol% LauMA content were lower, and equal to 35 and 32% for the DNs based on APCNs first networks with EGDMA1-LauMA50-b-DMAEMA50-EGDMA6 and EGDMA1-DMAEMA50-b-LauMA50-EGDMA6 structures, respectively.
Finally, examining parts (c) and (f) in Fig. 8, presenting the low-strain Young's modulus, E, for the SNs and DNs plotted against the composition of the SNs, an enhancement factor in the E values (ratio of the E value for the DN divided by that of its parent SN) greater than one was now observed only for systems with a 50 mol% LauMA content (values of that factor were equal to 2.4 and 4.3), whereas the LauMA-rich systems displayed values of those factors lower than one, equal to 0.1 and 0.7. The highest E values recorded for the DNs were 230 and 45 kPa, corresponding to those materials whose first networks had structures EGDMA1-DMAEMA50-b-LauMA50-EGDMA6 and EGDMA1-LauMA50-b-DMAEMA50-EGDMA6, respectively. This order can be traced back to the same order in E values in the parent SN materials.
Conclusions
Sequential RAFT polymerization was employed to prepare pH-responsive, amphiphilic polymer conetworks (APCN) based on end-linked “core-first” star copolymers. These APCNs were used as templates (first, single networks, SN) and were converted to double networks (DN) through interpenetration with a second, hydrophilic polyacrylamide network prepared by photopolymerization within the APCNs. Characterization of the aqueous swelling behavior of the networks confirmed their pH-responsiveness. A structural study via small-angle neutron scattering (SANS) indicated that the networks in D2O phase separated on the nanoscale, forming regularly-spaced (15–26 nm) hydrophobic domains. Finally, the mechanical properties of all networks in water were explored, and an enhancement was recorded for the DNs compared to their parent SNs, thus proving that the DN principle is also operational in the present RAFT polymerized system. Significant improvements were measured for the stress at break, for which the ratio of this property for the DN divided by that for its SN counterpart ranged between 340 and 490%. Moderate but measurable improvements were also recorded for the strain at break, where the DN-over-SN ratios span a range from 110 to 160%. In some cases, the low-strain Young's modulus was also improved in the daughter DNs, with calculated DN-over-SN enhancement factors of 240 and 430%. The strongest DN, having the structure EGDMA1-DMAEMA25-b-LauMA75-EGDMA6/PAAm-2-0.1, displayed remarkable mechanical properties, and, in particular, stress and strain at break of 3.29 MPa and 78%, respectively. The present study shows that the design and fabrication of polymer hydrogels with a multitude of useful properties, including pH-responsiveness, nanophase separation, and mechanical strength, is possible, paving the way to next-generation biomaterials.Acknowledgements
We thank the European Regional Development Fund and the Republic of Cyprus for co-funding the purchase of the 500 MHz NMR spectrometer through Cyprus Research Promotion Foundation in the form of a NEKYP Infrastructure Project (Code: NEKYP/0308/02). Furthermore, we gratefully acknowledge allocation of SANS beam time by the Institut Laue-Langevin (ILL).References
- (a) M. Rikkou-Kalourkoti, C. S. Patrickios and T. K. Georgiou, in Polymer Science: A Comprehensive Reference, ed. K. Matyjaszewski and M. Möller, Elsevier BV, Amsterdam, 2012, vol. 6, pp. 293–308 Search PubMed; (b) L. Mespouille, J. L. Hedrick and P. Dubois, Soft Matter, 2009, 5, 4878–4892 RSC; (c) G. Erdodi and J. P. Kennedy, Prog. Polym. Sci., 2006, 31, 1–18 CrossRef CAS; (d) C. S. Patrickios and T. K. Georgiou, Curr. Opin. Colloid Interface Sci., 2003, 8, 76–85 CrossRef CAS.
- C. Nardi Tironi, R. Graf, I. Lieberwirth, M. Klapper and K. Müllen, ACS Macro Lett., 2015, 4, 1302–1306 CrossRef.
- M. Rother, J. Barmettler, A. Reichmuth, J. V. Araujo, C. Rytka, O. Glaied, U. Pieles and N. Bruns, Adv. Mater., 2015, 27, 6620–6624 CrossRef CAS PubMed.
- K. Schöller, S. Küpfer, L. Baumann, P. M. Hoyer, D. de Courten, R. M. Rossi, M. Vetushka, M. Wolf, N. Bruns and L. J. Scherer, Adv. Funct. Mater., 2014, 24, 5194–5201 CrossRef.
- C. N. Walker, K. C. Bryson, R. C. Hayward and G. N. Tew, ACS Nano, 2014, 8, 12376–12385 CrossRef CAS PubMed.
- G. Guzman, T. Nugay, I. Nugay, N. Nugay, J. Kennedy and M. Cakmak, Macromolecules, 2015, 48, 6251–6262 CrossRef CAS.
- G. Guzman, T. Nugay, J. P. Kennedy and M. Cakmak, Langmuir, 2016, 32, 3445–3451 CrossRef CAS PubMed.
- J. Xu, M. Qiu, B. Ma and C. He, ACS Appl. Mater. Interfaces, 2014, 6, 15283–15290 CAS.
- Y. Shi, H. Schmalz and S. Agarwal, Polym. Chem., 2015, 6, 6409–6415 RSC.
- C. Krumm, S. Konieczny, G. J. Dropalla, M. Milbradt and J. C. Tiller, Macromolecules, 2013, 46, 3234–3245 CrossRef CAS.
- I. Sittko, K. Kremser, M. Roth, S. Kuehne, S. Stuhr and J. C. Tiller, Polymer, 2015, 64, 122–129 CrossRef CAS.
- A. K. S. Chandel, C. U. Kumar and S. K. Jewrajka, ACS Appl. Mater. Interfaces, 2016, 8, 3182–3192 CAS.
- C. Lin and I. Gitsov, Macromolecules, 2010, 43, 10017–10030 CrossRef CAS.
- V. Can, Z. Kochovski, V. Reiter, N. Severin, M. Siebenbürger, B. Kent, J. Just, J. P. Rabe, M. Ballauff and O. Okay, Macromolecules, 2016, 49, 2281–2287 CrossRef CAS.
- J. S. Zigmond, K. A. Pollack, S. Smedley, J. E. Raymond, L. A. Link, A. Pavía-Sanders, M. A. Hickner and K. L. Wooley, J. Polym. Sci., Part A: Polym. Chem., 2016, 54, 238–244 CrossRef CAS.
- G. Kali and B. Iván, Macromol. Chem. Phys., 2015, 216, 605–613 CrossRef CAS.
- E. J. Kepola, E. Loizou, C. S. Patrickios, E. Leontidis, C. Voutouri, T. Stylianopoulos, R. Schweins, M. Gradzielski, C. Krumm, J. C. Tiller, M. Kushnir and C. Wesdemiotis, ACS Macro Lett., 2015, 4, 1163–1168 CrossRef CAS.
- (a) M. Rikkou-Kalourkoti, E. N. Kitiri, C. S. Patrickios, E. Leontidis, M. Constantinou, G. Constantinides, X. Zhang and C. M. Papadakis, Macromolecules, 2016, 49, 1731–1742 CrossRef CAS; (b) X. Zhang, K. Kyriakos, M. Rikkou-Kalourkoti, E. N. Kitiri, C. S. Patrickios and C. M. Papadakis, Colloid Polym. Sci., 2016, 294, 1027–1036 CrossRef CAS.
- J. P. Gong, Y. Katsuyama, T. Kurokawa and Y. Osada, Adv. Mater., 2003, 15, 1155–1158 CrossRef CAS.
- Y. Tanaka, J. P. Gong and Y. Osada, Prog. Polym. Sci., 2005, 30, 1–9 CrossRef CAS.
- J. A. Johnson, N. J. Turro, J. T. Koberstein and J. E. Mark, Prog. Polym. Sci., 2010, 35, 332–337 CrossRef CAS.
- S. Naficy, H. R. Brown, J. M. Razal, G. M. Spinks and P. G. Whitten, Aust. J. Chem., 2011, 64, 1007–1025 CrossRef CAS.
- M. A. Haque, T. Kurokawa and J. P. Gong, Polymer, 2012, 53, 1805–1822 CrossRef CAS.
- J. P. Gong, Soft Matter, 2010, 6, 2583–2590 RSC.
- X. H. Zhao, Soft Matter, 2014, 10, 672–687 RSC.
- Y. Okumura and K. Ito, Adv. Mater., 2001, 13, 485–487 CrossRef CAS.
- K. Haraguchi and T. Takehisa, Adv. Mater., 2002, 14, 1120–1124 CrossRef CAS.
- Q. Chen, L. Zhu, C. Zhao, Q. Wang and J. Zheng, Adv. Mater., 2013, 25, 4171–4176 CrossRef CAS PubMed.
- Y.-H. Na, Y. Tanaka, Y. Kawauchi, H. Furukawa, T. Sumiyoshi, J. P. Gong and Y. Osada, Macromolecules, 2006, 39, 4641–4645 CrossRef CAS.
- Q. M. Yu, Y. Tanaka, H. Furukawa, T. Kurokawa and J. P. Gong, Macromolecules, 2009, 42, 3852–3855 CrossRef CAS.
- (a) D. Myung, D. Waters, M. Wiseman, P.-E. Duhamel, J. Noolandi, C. N. Ta and C. W. Frank, Polym. Adv. Technol., 2008, 19, 647–657 CrossRef CAS PubMed; (b) D. J. Waters, K. Engberg, R. Parke-Houben, L. Hartmann, C. N. Ta, M. F. Toney and C. W. Frank, Macromolecules, 2010, 43, 6861–6870 CrossRef CAS PubMed; (c) D. J. Waters, K. Engberg, R. Parke-Houben, C. N. Ta, A. J. Jackson, M. F. Toney and C. W. Frank, Macromolecules, 2011, 44, 5776–5787 CrossRef CAS.
- J.-Y. Sun, X. Zhao, A. R. K. Illeperuma, O. Chaudhuri, K. H. Oh, D. J. Mooney, J. J. Vlassak and Z. Suo, Nature, 2012, 489, 11409 CrossRef PubMed.
- E. Ducrot, Y. Chen, M. Bulters, R. P. Sijbesma and C. Creton, Science, 2014, 344, 186–189 CrossRef CAS PubMed.
- E. N. Kitiri, M. Rikkou-Kalourkoti, M. Sophocleous and C. S. Patrickios, Eur. Polym. J., 2015, 69, 573–583 CrossRef CAS.
- (a) J. Chiefari, Y. K. Chong, F. Ercole, J. Krstina, J. Jeffery, T. P. T. Le, R. T. A. Mayadunne, G. F. Meijs, C. L. Moad, G. Moad, E. Rizzardo and S. H. Thang, Macromolecules, 1998, 31, 5555–5562 CrossRef; (b) G. Moad, E. Rizzardo and S. H. Thang, Aust. J. Chem., 2012, 65, 985–1076 CrossRef CAS; (c) G. Moad, E. Rizzardo and S. H. Thang, Aust. J. Chem., 2009, 62, 1402–1472 CrossRef CAS; (d) G. Moad, E. Rizzardo and S. H. Thang, Aust. J. Chem., 2006, 59, 669–692 CrossRef CAS; (e) G. Moad, E. Rizzardo and S. H. Thang, Aust. J. Chem., 2005, 58, 379–410 CrossRef CAS.
- (a) Q. Fu, T. G. McKenzie, S. Tan, E. Nam and G. G. Qiao, Polym. Chem., 2015, 6, 5362–5368 RSC; (b) T. G. McKenzie, Q. Fu, E. H. H. Wong, D. E. Dunstan and G. G. Qiao, Macromolecules, 2015, 48, 3864–3872 CrossRef CAS.
- (a) M. Rikkou-Kalourkoti, O. W. Webster and C. S. Patrickios, in Encyclopedia of Polymer Science and Technology, Wiley, 2013, vol. 99, pp. 1–17 Search PubMed; (b) K. Fuchise, Y. Chen, T. Satoh and T. Kakuchi, Polym. Chem., 2013, 4, 4278–4291 RSC; (c) J. Raynaud, A. Ciolino, A. Baceiredo, M. Destarac, F. Bonnette, T. Kato, Y. Gnanou and D. Taton, Angew. Chem., Int. Ed., 2008, 47, 5390–5393 CrossRef CAS PubMed; (d) O. W. Webster, Adv. Polym. Sci., 2004, 167, 1–34 CrossRef CAS; (e) O. W. Webster, J. Polym. Sci., Part A: Polym. Chem., 2000, 38, 2855–2860 CrossRef CAS; (f) O. W. Webster, W. R. Hertler, D. Y. Sogah, W. F. Farnham and T. V. RajanBabu, J. Am. Chem. Soc., 1983, 105, 5706–5708 CrossRef CAS.
- (a) N. Hadjichristidis, M. Pitsikalis, S. Pispas and H. Iatrou, Chem. Rev., 2001, 101, 3747–3792 CrossRef CAS PubMed; (b) N. Hadjichristidis, H. Iatrou, M. Pitsikalis and J. Mays, Prog. Polym. Sci., 2006, 31, 1068–1132 CrossRef CAS; (c) A. Blencowe, J. F. Tan, T. K. Goh and G. G. Qiao, Polymer, 2009, 50, 5–32 CrossRef CAS; (d) Q. Chen, X. Cao, Y. Xu and Z. An, Macromol. Rapid Commun., 2013, 34, 1507–1517 CrossRef CAS PubMed; (e) J. M. Ren, T. G. McKenzie, Q. Fu, E. H. H. Wong, J. Xu, Z. An, S. Shanmugam, T. P. Davis, C. Boyer and G. G. Qiao, Chem. Rev., 2016, 116, 6743–6836 CrossRef CAS PubMed.
- C. M. Wager, D. M. Haddleton and S. A. F. Bon, Eur. Polym. J., 2004, 40, 641–645 CrossRef CAS.
- (a) M. Vamvakaki and C. S. Patrickios, J. Phys. Chem. B, 2001, 105, 4979–4986 CrossRef CAS; (b) T. K. Georgiou, M. Vamvakaki and C. S. Patrickios, Polymer, 2004, 45, 7341–7355 CrossRef CAS.
- P. C. Hiemenz, Polymer Chemistry: The Basic Concepts, Marcel Dekker, New York, 1984, pp. 48–65 Search PubMed.
| This journal is © The Royal Society of Chemistry 2017 |