
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAn ortho C-methylation/O-glycosylation motif on a hydroxy-coumarin scaffold, selectively installed by biocatalysis†
Alexander
Gutmann
a,
Margaretha
Schiller
a,
Mandana
Gruber-Khadjawi
bc and
Bernd
Nidetzky
 *ab
*ab
aInstitute of Biotechnology and Biochemical Engineering, Graz University of Technology, NAWI Graz, Petersgasse 12, 8010 Graz, Austria. E-mail: bernd.nidetzky@tugraz.at
bAustrian Centre of Industrial Biotechnology, Petersgasse 14, 8010 Graz, Austria
cInstitute of Organic Chemistry, Graz University of Technology, NAWI Graz, Stremayrgasse 9, 8010 Graz, Austria
First published on 5th September 2017
Abstract
Various bioactive natural products, like the aminocoumarin antibiotics novobiocin and coumermycin, exhibit an aromatic C-methyl group adjacent to a glycosylated phenolic hydroxyl group. Therefore, tailoring of basic phenolic scaffolds to contain the intricate C-methyl/O-glycosyl motif is of high interest for structural and functional diversification of natural products. We demonstrate site-selective 8-C-methylation and 7-O-β-D-glucosylation of 4,5,7-trihydroxy-3-phenyl-coumarin (1) by S-adenosyl-L-methionine dependent C-methyltransferase (from Streptomyces niveus) and uridine 5′-diphosphate glucose dependent glycosyltransferase from apple (Malus × domestica). Both enzymes were characterized and shown to react readily with underivatized 1. However, glucosylation of the ortho-hydroxyl group prevented C-methylation, probably by precluding an essential substrate activation through deprotonation of 7-OH. Therefore, dual modification was only feasible when C-methylation occurred strictly before O-glucosylation. The target product was synthesized in near quantitative yield (98% conversion) from 500 μM 1 and its structure was confirmed by NMR. Combination of C-methyltransferase and O-glycosyltransferase reactions for synthetic tailoring of a natural product through biocatalysis was demonstrated for the first time.
Introduction
The aromatic scaffold of various natural products exhibits a characteristic pattern of structural modification in which C-methylation and O-glycosylation occur at adjacent positions on the aromatic ring (Chart 1). The structural motif is most abundant in antibiotics derived from actinomycetes.1–3 Among these, the aminocoumarin antibiotics novobiocin and coumermycin A1 have attracted considerable attention, for they are highly potent inhibitors of bacterial DNA gyrase and thus hold promise for use in anti-infective therapies.1,4 Other important examples include a variety of aureolic acid type polyketides with antitumor activities,2 glycopeptide antibiotics of the actaplanin family5 and the pyrrolobenzodiazepine sibiromycin.3 The relevant C-methyl/O-glycosyl motif is also found in different plant natural products. Prominent examples are flavanones of the matteuorienate group, which have drawn substantial interest for their ability to inhibit aldose reductase in the context of managing diabetic complications.6 Another relevant group of natural compounds bearing the C-methyl/O-glycosyl motif are anthraquinones.7 | ||
| Chart 1 Various aromatic natural products are decorated with a characteristic structural motif that comprises C-methylation and O-glucosylation at adjacent positions on the aromatic ring. | ||
Methylation and glycosylation typically occur late in natural product biosynthesis to tailor an initially formed backbone structure.4,8 Frequently these scaffold decorations define the bioactivity of a compound or augment its potency.9–11 The transfer of methyl and sugar groups is catalyzed by S-adenosyl-L-methionine (SAM) dependent methyltransferases (MTs; EC 2.1.1) and nucleotide sugar dependent (Leloir) glycosyltransferases (GTs; EC 2.4), respectively.12,13 On transferring the methyl group from SAM to the acceptor substrate, MTs release S-adenosyl-L-homocysteine (SAH) as their second product.14,15 GTs catalyze glycosylation of the acceptor substrate while releasing the nucleotide moiety from the sugar donor.13,16 MTs and GTs exhibiting a relatively flexible substrate specificity were exploited in previous studies to further the diversity in natural product structures.17,18 Ability of the enzymes to react with different acceptor substrates was utilized to graft a naturally occurring chemical modification onto new aromatic scaffolds.19–22 Alternatively, it was possible to vary the chemical groups transferred by MTs on applying different SAM analogues in a synthetic “alkylrandomization” approach.18,23,24 Similarly, a somewhat relaxed specificity of certain GTs for the sugar donor substrate was exploited in the synthesis of different glycosides using an approach referred to as “glycorandomization”.16,17,25
The aim of the current study was the synthetic instalment of the prominent C-methyl/O-glycosyl motif by using a one-pot biotransformation involving the reactions of MT and GT combined (Scheme 1). Despite the broad use of MTs and GTs in the preparation of natural product analogues, both types of modification were typically not combined. Two recent in vivo biotransformations coupling O-glycosylation to ortho O-methylation present notable exceptions.26,27 However, there is a lack of comparable studies involving C-methylations, which are perceived as chemically more challenging.9,28,29 The applied model substrate 4,5,7-trihydroxy-3-phenyl-coumarin (1) possesses a representative hydroxy-coumarin scaffold which resembles structurally the backbones of aminocoumarin antibiotics and matteuorienates (Chart 1).1,6 Site-selective 8-C-methylation of 1 by the methyltransferase NovO from Streptomyces niveus was already reported previously.22 However, to achieve the intended dual derivatization, we had to identify a suitable GT for selective 7-O-β-D-glucosylation of 1. Kinetic and thermodynamic characterization of both enzymatic conversions was critically important to establish a reaction setup allowing near quantitative conversion of 1 to the desired C-methylated and O-glucosylated final product. Most importantly, we recognized that an ill-timed application of the enzymes had to be avoided to prevent synthesis of the glucosylated dead-end product 3.
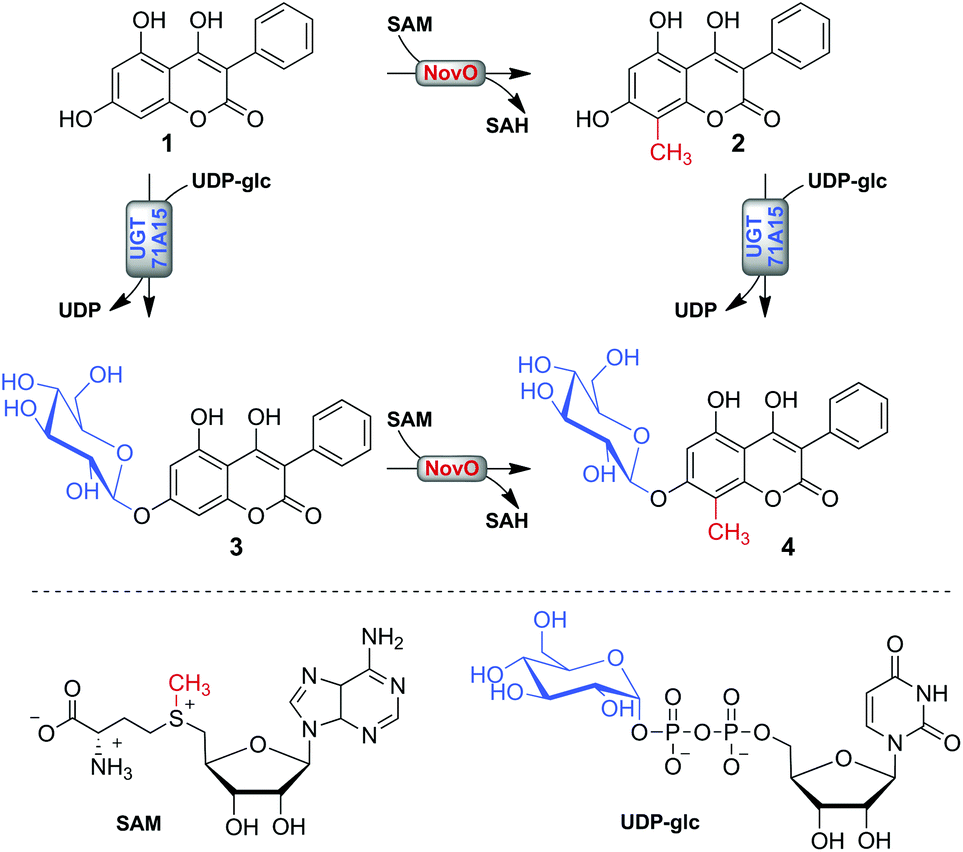 | ||
| Scheme 1 Possible paths of enzymatic methylation and glucosylation of substrate 1 for synthesis of 4 are shown. | ||
Results and discussion
Selection of enzymes for methylation and glucosylation of 1
We used NovO from the novobiocin biosynthesis pathway of Streptomyces niveus as methylation catalyst.4,30 NovO accepts a broad spectrum of donor and acceptor substrates and it was previously shown that it methylates 1 site-selectively at the desired C-8 position.22,31 However, a matching GT for ortho O-glucosylation still had to be identified. Although glucosylation of a broad range of (poly)phenolic acceptors by various promiscuous GTs was reported,13,19,20,32 the selective modification of a single hydroxyl group of a polyphenolic substrate remains a particular challenge. Frequently these promiscuous GTs lacked the required site-selectivity and tended to decorate acceptor substrates with multiple sugar moieties. Here we used UGT71A15, a flavonoid GT from apple (Malus × domestica), for uridine 5′-diphosphate glucose (UDP-glc) dependent O-glucosylation of 1.32 Although UGT71A15 forms mixtures of mono- and diglucosides of polyphenolic acceptors like phloretin and resveratrol,32,33 we showed here that conversion of 1 yielded the required 7-O-β-D-glucoside as a single product. As demonstrated later, the positions of glucosylation and methylation were confirmed by NMR spectroscopy. Because in addition to its perfect regio- and stereoselectivity for glucosylation of 1 the UGT71A15 also displayed an excellent rate of conversion of this substrate, the enzyme was particularly well suited for the intended biocatalytic transformation. Under optimized conditions, enzymatic glucosylation rates above 1 μmol min−1 mg−1 were achieved. Interestingly, the activity of UGT71A15 with 1 was significantly higher than with previously used acceptors like resveratrol (<0.4 μmol min−1 mg−1).33 To establish the in vitro bioconversion, NovO and UGT71A15 were recombinantly expressed in Escherichia coli and purified by Strep-tag affinity chromatography (Fig. S1†). Enzymatic conversions were monitored by reversed-phase HPLC (Fig. S2†). Baseline separation of compounds 1–4 was achieved within 10 min long isocratic runs.Establishing conditions for joint methylation and glucosylation
To establish synthesis of 4 in a one-pot conversion, conditions for the dual modification of the acceptor 1 had to be identified. Therefore, the glucosylation and methylation reactions were initially characterized separately to identify potential kinetic and thermodynamic restrictions. Note that previous evidence for 8-C-methylation of 1 by NovO resulted exclusively from single time point measurements.22 To avoid precipitation of the poorly water-soluble 1, DMSO was applied as cosolvent. We showed previously that UGT71A15 was well active in the presence of 20% DMSO.33 However, the activity of NovO was strongly affected by the DMSO content (Fig. S3†). A decrease in DMSO content from 20 to 5% resulted in a 6-fold increased methylation rate. Interestingly, within the observed time span of 50 min the DMSO concentration had no effect on the NovO stability. The DMSO content was fixed to 5% in further conversions involving NovO. This was sufficient to dissolve at least 2 mM 1.Reaction rates of NovO and UGT71A15 were determined at various concentrations of 1 to identify the applicable range of acceptor concentrations for the combined conversion (Fig. 1). Both enzymes displayed substrate inhibition with IC50 values of 0.4 mM (NovO) and 7.8 mM (UGT71A15). As a result, NovO and UGT71A15 showed distinct activity optima at 0.1 and 1 mM 1, respectively. Benchmarked against the NovO reaction, glucosylation of 1 by UGT71A15 benefited not only from a less pronounced substrate inhibition but also from generally higher reaction rates. The maximum glucosylation rate of 1.3 μmol min−1 mg−1 was about 10-fold higher than the maximum methylation rate of 0.12 μmol min−1 mg−1. Therefore, identification of optimal methylation conditions was crucial for efficient coupling of both enzymatic conversions. Because inhibition of NovO restricted the usable concentration of 1 in batch conversions, we applied 500 μM 1 to establish synthesis of 4.
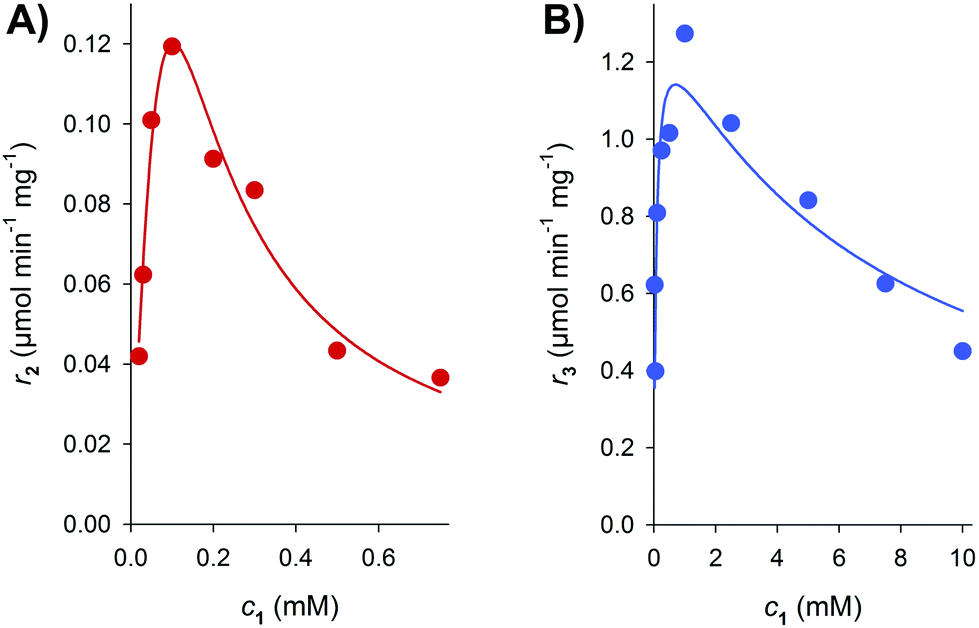 | ||
| Fig. 1 The influence of the 1 concentration on initial reaction rates at pH 6.5 is shown. (A) Rates of the formation of 2 by NovO (r2) were determined in presence of 1 mM SAM. (B) Rates of the formation of 3 by UGT71A15 (r3) were measured in presence of 2 mM UDP-glc. Substrate inhibition kinetics was used to fit the data (solid lines). | ||
We then evaluated the pH dependencies of NovO and UGT71A15 with the idea that through proper choice of pH kinetic limitations from the methylation might be reduced (Fig. S4†). Both enzymes were somewhat inhibited by HEPES. UGT71A15 maintained high reaction rates of 1.0–1.2 μmol min−1 mg−1 throughout the observed pH range of 6.5–8.5. However, NovO clearly profited from slightly alkaline conditions. Benchmarked against previously used pH of 6.5,22,34 the methylation rate roughly doubled to 0.23 μmol min−1 mg−1 on raising the pH above 7.5. Consequently pH 8.0 was used in further conversions.
The donor concentration dictates the final conversion of 1
The concentration of the donor substrates SAM and UDP-glc had to be optimized to overcome potential thermodynamic restrictions and to achieve the targeted near quantitative conversion of 1. Because C-methylations are perceived as irreversible reactions,35,36 only stoichiometric amounts or a slight excess of SAM should be required. On the other hand it was shown that various O-glycosylations are readily reversible.37,38 Therefore at least a moderate excess of UDP-glc would be needed to drive the synthesis of 3. Methylation and glucosylation of 500 μM 1 were tested in the presence of 500–1000 μM SAM and 750–2000 μM UDP-glc, respectively (Fig. 2). To account for differences in enzyme activity, 150 μg mL−1 NovO and 4 μg mL−1 UGT71A15 were used. Within 7 h of reaction, a near complete methylation of 1 by NovO was only possible in the presence of the highest applied SAM concentration of 1000 μM. Under these conditions, 497 μM 2 were formed by converting 99.4% of the initially applied 1. Using 500 and 750 μM SAM, the conversions stopped already at 310 μM 2 (62% conversion) and 440 μM 2 (88% conversion), respectively. Excess of SAM required in the reaction was probably reflected in the limited stability of the donor substrate.39 In the enzymatic glucosylation of 1, an increase in the UDP-glc donor concentration from 0.75 mM to 2 mM raised the final concentration of 3 from 457 μM (91% conversion) to 492 μM (98.4% conversion). Excess of the donor substrate most likely helped to overcome thermodynamic restrictions. Based on final yields after 24 h (data not shown), an equilibrium constant (Keq) of 25 ± 3 was estimated for the glucosylation of 1. This was in good agreement with the expectation of a slightly reversible glucosylation reaction. Also glucosylation rates benefited somewhat from increased UDP-glc concentrations (Fig. 2). However, overall improvements from raising UDP-glc above 1.5 mM were only marginal. Therefore 1.5 mM UDP-glc and 1 mM SAM were selected as optimum donor concentrations for the synthesis of 4. | ||
| Fig. 2 Time courses of formation of 2 by NovO (A) and of 3 by UGT71A15 (B) are shown. The concentrations of the donor substrates SAM (A) and UDP-glc (B) were varied: 500 μM (red), 750 μM (black), 1000 μM (grey), 1500 μM (blue), 2000 μM (green). Reaction conditions: 500 μM 1, 150 μg mL−1 NovO (A) or 4 μg mL−1 UGT71A15 (B), pH 8.0. | ||
Formation of 4 requires sequential methylation and glycosylation
To translate fundamental insights on the conversion of 1 by NovO and UGT71A15 to an actual synthesis of 4 we had to understand if the order of glucosylation and methylation impacts the overall efficiency of the two-step reaction (Scheme 1). Based on results of individual transformations (Fig. 2), near quantitative synthesis of 4 should be achievable if both reactions are done independent from each other. When methylation and glucosylation of 500 μM 1 were combined in one pot, formation of 4 was well feasible but stopped at a concentration of 390 μM (Fig. 3A). The rather high NovO loading of 150 μg mL−1 triggered fast accumulation of 2. Already after 5 min 113 μM 2 were formed but the concentrations of 3 and 4 were only 28 and 2 μM, respectively. At 30 min a maximum of 250 μM 2 accumulated. Upon prolonged incubation 2 quickly diminished until it was no longer detectable after 23 h. Formation of 3 was much slower and after 2 h the maximum concentration of 132 μM was reached. However, unlike the short-lived methylated intermediate, 3 was hardly further converted. Even after 47 h, 104 μM of 3 remained. In contrast all 1 and 2 were consumed within 2 and 23 h, respectively. This result indicated that glucosylation of 2 was well feasible whereas 3 was not, or only very slowly, methylated. | ||
| Fig. 3 Simultaneous and sequential methylation and glucosylation were tested for conversion of 1 to 4. (A) Simultaneous addition of NovO and UGT71A15; (B) NovO was added 23 h after UGT71A15; (C) UGT71A15 was added 23 h after NovO. Reaction conditions: 500 μM 1, 1 mM SAM, 1.5 mM UDP-glc, 150 μg mL−1 NovO, 4 μg mL−1 UGT71A15, pH 8.0. 1 (black squares), 2 (red circles), 3 (blue circles), 4 (green diamonds). | ||
To verify if successful synthesis of 4 required consecutive methylation and glucosylation, stepwise enzyme addition was tested. After 23 h of conversion with NovO or UGT71A15 the second enzyme and the corresponding donor substrate were added. Reactions were followed for another 24 h. When the reaction was initiated with UGT71A15 a stable level of 490 μM 3 (98% conversion) was reached after 4.5 h (Fig. 3B). In agreement with observations from the simultaneous conversion (Fig. 3A), formation of 4 from 3 was very slow. 24 h after the NovO addition, only 41 μM 4 were formed and 90% of initially applied 1 was still present as intermediate 3. The complementary approach relying on initial methylation by NovO was clearly best for synthesis of 4 (Fig. 3C). Again the first step of conversion was fast and around 98% of 1 was converted to intermediate 2 within 4.5 h. When glucosylation was performed as second step, near quantitative conversion to the final product 4 was achieved. Already 4 h after UGT71A15 addition 491 μM 4 were formed. Only minor amounts of 2 (9 μM) but no 1 or 3 were detectable. The concentration of 4 was enhanced from 390 μM (78% conversion) to 490 μM (98% conversion) by switching from simultaneous to stepwise addition of NovO and UGT71A15. Interestingly, glucosylation of methylated 2 was about 20% faster than conversion of 1 (Fig. 3B and C). It can be concluded that conditions initially selected for modification of 1 were also well suited for glucosylation of 2. On the other hand, when reacting with 3, NovO retained only 0.3% of the methylation rate achieved with 1. In conclusion, initial methylation was imperative for efficient one-pot methylation and glucosylation of 1.
The stringent order of reaction, methylation before glucosylation, may reflect requirements of the NovO mechanism on the substrate to become methylated. Recently reported crystal structures of NovO40 and the closely related CouO41 suggested that deprotonation of 7-OH by a catalytic His-Arg motif is required to activate C8 for methylation. An overall similar mechanism of aromatic C-methylation, assisted by a pair of active-site tyrosine residues however, was recently suggested for the 3-hydroxykynurenine methyltransferase from Streptosporangium sibiricum.42 Therefore, 7-O-glucosylation of 1 by UGT71A15 most likely abolished NovO activity for subsequent C-methylation by preventing hydroxyl deprotonation at the ortho position. We suggest that minor conversion of 3 to 4, as shown in Fig. 3B, was not achieved by direct methylation. It rather involved intermediary formation of 1 by reverse glucosylation of 3. The equilibrium of the UGT71A15 reaction (Keq ∼25) lies far on the product side. Therefore, reverse glucosylation was probably inefficient and limited formation of 4 from 3. We omitted the futile glucosylation–deglucosylation cycle by initial methylation of 1.
Upscaled synthesis, isolation and characterization of 2–4
Finally, synthesis of 4 by the coupled NovO–UGT71A15 reaction and production of 2 and 3 by NovO and UGT71A15, respectively, were upscaled to a reaction volume of 50 mL. Again around 98% conversion to the desired products was achieved. Compounds 2–4 were isolated by preparative reversed phase HPLC. Because product purification was not optimized, the isolated yields were only around 60%. Product identities were verified by NMR spectroscopy (Table 1 and Fig. S5–15†). Because conversion of 1 by NovO was reported previously,22 the identity of 2 was only verified by 1H-NMR. Compounds 3 and 4 were reported for the first time and therefore analyzed in detail by 1H-NMR, 13C-NMR, COSY, HSQC and HMBC spectroscopy.|
|
|
|
|||
|---|---|---|---|---|---|
| No. | 2 | 3 | 4 | ||
| δ H | δ C | δ H | δ C | δ H | |
| a 1H: 500 MHz; 13C: 126 MHz (CD3OD, δ in ppm). | |||||
| 2 | 167.67 | 165.97 | |||
| 3 | 100.91 | 102.92 | |||
| 4 | 175.70 | 167.09 | |||
| 4a | 103.87 | 100.89 | |||
| 5 | 161.63 | 155.45 | |||
| 6 | 6.28 (s, 1H) | 99.60 | 6.39 (s, 1H) | 99.22 | 6.65 (s, 1H) |
| 7 | 162.11 | 160.24 | |||
| 8 | 95.46 | 6.51 (s, 1H) | 107.76 | ||
| 8a | 156.21 | 153.34 | |||
| 1′ | 135.77 | 133.63 | |||
| 2′/6′ | 7.44 (d, J = 7.6 Hz, 2H) | 132.40 | 7.52 (d, J = 7.5 Hz, 2H) | 132.10 | 7.46 (d, J = 7.5 Hz, 2H) |
| 3′/5′ | 7.33 (t, J = 7.6 Hz, 2H) | 128.55 | 7.37 (t, J = 7.5 Hz, 2H) | 128.71 | 7.36 (t, J = 7.6 Hz, 2H) |
| 4′ | 7.22 (t, J = 7.4 Hz, 1H) | 126.81 | 7.23 (t, J = 7.4 Hz, 1H) | 127.94 | 7.26 (t, J = 7.4 Hz, 1H) |
| 1′′ | 101.84 | 5.02 (d, J = 7.0 Hz, 1H) | 102.25 | 4.95 (d, J = 7.6 Hz, 1H) | |
| 2′′ | 74.80 | 3.52 (m, 1H) | 74.76 | 3.53 (m, 1H) | |
| 3′′ | 77.91 | 3.53 (m, 1H) | 78.16 | 3.47 (m, 1H) | |
| 4′′ | 71.23 | 3.50 (m, 1H) | 71.25 | 3.42 (m, 1H) | |
| 5′′ | 78.21 | 3.54 (m, 1H) | 78.44 | 3.46 (m, 1H) | |
| 6′′ | 62.37 | 3.96 (dd, J = 12.2, 2.1 Hz, 1H) | 62.58 | 3.94 (dd, J = 12.1, 2.1 Hz, 1H) | |
| 3.79 (dd, J = 12.1, 5.1 Hz, 1H) | 3.73 (dd, J = 12.0, 5.5 Hz, 1H) | ||||
| α | 2.15 (s, 3H) | 8.06 | 2.26 (s, 3H) | ||
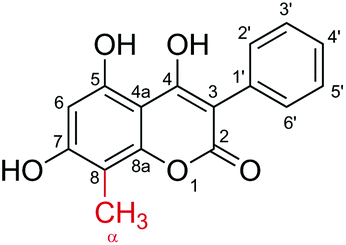


Conclusions
Using a representative hydroxy-coumarin model substrate, we demonstrated that a biocatalytic one-pot two-step MT–GT reaction allows for the installation of the intricate C-methylation and O-glycosylation structural motif present in various natural products. In contrast to the previously described 8-C-methylation of 1 by NovO,22 enzymatic ortho O-glucosylation still had to be established. UGT71A15 was identified as perfectly site-selective GT which only glucosylated the hydroxyl-group at position 7 without modifying the 4- and 5-OH. Because of the relaxed acceptor selectivity of both transferases, NovO and UGT71A15 should be useful for installing the same structural motif on various 7-hydroxy-coumarins.A detailed characterization of the individual enzymatic conversions of 1 was important to develop basis for proper use of NovO and UGT71A15 in combination. Therefore, near quantitative dual modification of 1 could be achieved. The slight reversibility of the O-glucosylation did not prohibit excellent overall conversions (≥98%) of 1 when UDP-glc was applied in 3-fold excess. Interestingly, the quasi irreversible C-methylation was revealed as the critically limiting step of the two-step conversion. Benchmarked against UGT71A15, NovO displayed 10 times lower reaction rates and a more pronounced inhibition by the acceptor 1. Most importantly, however, C-methylation had to occur strictly before ortho O-glucosylation. In agreement with recent mechanistic proposals,40–42 7-O-β-D-glucosylation precluded subsequent C-methylation at the adjacent position 8, probably by preventing acceptor activation through deprotonation of 7-OH. Therefore ortho C-methylation and O-glucosylation have to be performed sequentially, independent of the acceptor and the pair of enzymes used.
The improved understanding of one-pot MT–GT conversions facilitates the challenging diversification of natural product scaffolds.43,44 In order to use the biocatalytic principle for decoration of natural products at large scale, in situ regeneration of the rather expensive donor substrates will be required. Recycling of UDP-glc is well established and also regeneration of SAM was reported recently.14,33 Furthermore, we envision that one-pot MT–GT conversions facilitate the combination of glyco- and alkylrandomization approaches. This would boost the number of available natural product analogues.
Experimental
Chemicals and enzyme preparation
All chemicals were obtained in the highest purity available. UDP-glc and SAM were from Carbosynth (Berkshire, UK) and 1 was from Sigma-Aldrich (Vienna, Austria). Other chemicals were from Carl Roth (Karlsruhe, Germany) or Sigma-Aldrich.UGT71A15 (GenBank: NP_001315903.1) from apple (Malus × domestica) and NovO (GenBank: AAF67508.2) from Streptomyces niveus were recombinantly expressed in Escherichia coli BL21-Gold (DE3) cells and purified by Strep-tag affinity chromatography as described elsewhere in detail.22,33
Analysis of enzymatic conversions by reversed-phase HPLC
Enzymatic conversions were monitored by reversed-phase HPLC on a 100 × 4.6 mm Chromolith® HighResolution RP-18e column (Merck, Darmstadt, Germany). Baseline separation was achieved in 10 min long isocratic runs using a 9 mM ammonium acetate buffer (pH 5.5), which contained 10% acetonitrile. The flow rate was set to 1.5 mL min−1 and the temperature was 40 °C. Compounds were quantified by UV-detection at 318 nm.Unless mentioned otherwise, reaction mixtures were buffered by 50 mM sodium phosphate, pH 8.0 containing 1 mg mL−1 BSA and 5% DMSO. Conversions were performed at a scale of 0.5 mL in 1.5 mL reaction tubes on a thermomixer at 1000 rpm and 35 °C. Reactions were started by enzyme addition and stopped by mixing withdrawn aliquots with an equal volume of acetonitrile. Before HPLC analysis precipitated proteins were removed by centrifugation (10 min, 13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 200 rpm). Initial reaction rates were typically calculated from measurements after 4 distinct incubation times. Relative standard deviations of reaction rates and analyte concentrations, derived from independent triplicates, were less than 5%.
200 rpm). Initial reaction rates were typically calculated from measurements after 4 distinct incubation times. Relative standard deviations of reaction rates and analyte concentrations, derived from independent triplicates, were less than 5%.
Establishing methylation and glucosylation of 1
The influence of the DMSO concentration (5–20%) on initial NovO conversion rates was tested by reacting 100 μM 1 and 1 mM SAM with 5 μg mL−1 NovO in 50 mM sodium phosphate buffer, pH 6.5. Also the effect of 1 concentration on NovO and UGT71A15 reaction rates was studied in 50 mM sodium phosphate buffer at pH 6.5. 1–50 μg mL−1 NovO were used to convert 10–750 μM 1 with 1 mM SAM. Glucosylation of 25 μM – 10 mM 1 from 2 mM UDP-glc by 0.75–6 μg mL−1 UGT71A15 was performed in presence of 20% DMSO. The effects of buffer and pH on methylation and glucosylation rates were studied with 50 mM sodium phosphate (pH 6.3–8.3) and 50 mM HEPES buffer (pH 7.6–8.2). The pH of reaction mixtures was determined at the end of each conversion. 100 μM 1 were methylated from 1 mM SAM by 5 μg mL−1 NovO. 1 μg mL−1 UGT71A15 and 1.5 mM UDP-glc were used to glucosylate 1 mM 1 in presence of 20% DMSO. Conversion of 500 μM 1 was tested with various concentrations of donor substrate. 0.5–1 mM SAM were used for methylation with 150 μg mL−1 NovO and glucosylation by 4 μg mL−1 UGT71A15 was performed with 0.75–2 mM UDP-glc.Methylation and glucosylation of 500 μM 1 was combined in one pot. 150 μg mL−1 NovO and 1 mM SAM were applied for methylation. Glucosylation was performed with 4 μg mL−1 UGT71A15 and 1.5 mM UDP-glc. For simultaneous methylation and glucosylation both enzymes were added at once. Alternatively, NovO and SAM or UGT71A15 and UDP-glc were added after 23 h to study stepwise modification. All reactions were followed for 47 h.
Upscaled synthesis, isolation and characterization of 2–4
The above described optimized conditions for one-pot synthesis were applied for preparation of 2–4 at a scale of 50 mL. 4 was synthesized by consecutive methylation and glucosylation. Single step methylation and glucosylation of 1 was used to prepare 2 and 3, respectively.After conversions were complete, enzymes were removed by ultrafiltration using a molecular mass cut-off of 10 kDa. The products were purified by preparative reversed-phase C18 HPLC using a 250 × 10.0 mm SphereClone 5 μm ODS(2) column (Phenomenex, Aschaffenburg, Germany). Compounds were monitored by UV-detection at 318 nm. A flow rate of 3 mL min−1 was applied at 25 °C. Water and acetonitrile (0.1% formic acid each) were used as mobile phase A and B, respectively. Compounds were separated by a step gradient (3: 21% B, 4: 30% B; 2: 50% B). After evaporating acetonitrile under reduced pressure, water was removed by freeze-drying.
Compound identities were confirmed by NMR spectroscopy using a Varian Unity Inova 500 MHz spectrometer. 1H-NMR spectra were recorded for all products and for 3 and 4 also 13C-NMR, COSY, HSQC and HMBC spectra were recorded.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
Financial Support from the EU FP7 project SuSy (Sucrose Synthase as Cost-Effective Mediator of Glycosylation Reactions) is gratefully acknowledged. The authors would like to thank Dr Martin Tengg and Prof. Hansjoerg Weber, both from Graz University of Technology, for assisting methyltransferase expression and purification and for NMR analysis, respectively. B. N. acknowledges the EU COST action CM1303 Systems Biocatalysis.References
- L. Heide, Int. J. Med. Microbiol., 2014, 304, 31–36 CrossRef CAS PubMed.
- F. Lombó, N. Menéndez, J. A. Salas and C. Méndez, Appl. Microbiol. Biotechnol., 2006, 73, 1–14 CrossRef PubMed.
- W. Li, A. Khullar, S. Chou, A. Sacramo and B. Gerratana, Appl. Environ. Microbiol., 2009, 75, 2869–2878 CrossRef CAS PubMed.
- V. Dangel, J. Härle, C. Goerke, C. Wolz, B. Gust, J.-L. Pernodet and L. Heide, Microbiology, 2009, 155, 4025–4035 CrossRef CAS PubMed.
- M. Debono, K. E. Merkel, R. M. Molloy, M. Barnhart, E. Presti, A. H. Hunt and R. L. Hamill, J. Antibiot., 1984, 37, 85–95 CrossRef CAS PubMed.
- P. Basnet, S. Kadota, H. Koji and T. Namba, Chem. Pharm. Bull., 1995, 43, 1558–1564 CrossRef CAS PubMed.
- H. Itokawa, Y. Qiao and K. Takeya, Phytochemistry, 1989, 28, 3465–3468 CrossRef CAS.
- C. Olano, C. Méndez and J. A. Salas, Nat. Prod. Rep., 2010, 27, 571–616 RSC.
- H. Schönherr and T. Cernak, Angew. Chem., Int. Ed., 2013, 52, 12256–12267 CrossRef PubMed.
- A. C. Weymouth-Wilson, Nat. Prod. Rep., 1997, 14, 99–110 RSC.
- L. A. Wessjohann, J. Keim, B. Weigel and M. Dippe, Curr. Opin. Chem. Biol., 2013, 17, 229–235 CrossRef CAS PubMed.
- D. K. Liscombe, G. V. Louie and J. P. Noel, Nat. Prod. Rep., 2012, 29, 1238–1250 RSC.
- D. Bowles, E.-K. Lim, B. Poppenberger and F. E. Vaistij, Annu. Rev. Plant Biol., 2006, 57, 567–597 CrossRef CAS PubMed.
- S. Mordhorst, J. Siegrist, M. Müller, M. Richter and J. N. Andexer, Angew. Chem., Int. Ed., 2017, 56, 4037–4041 CrossRef CAS PubMed.
- A.-W. Struck, M. L. Thompson, L. S. Wong and J. Micklefield, ChemBioChem, 2012, 13, 2642–2655 CrossRef CAS PubMed.
- X. Fu, C. Albermann, J. Jiang, J. Liao, C. Zhang and J. S. Thorson, Nat. Biotechnol., 2003, 21, 1467–1469 CrossRef CAS PubMed.
- C. J. Thibodeaux, C. E. Melançon III and H.-w. Liu, Angew. Chem., Int. Ed., 2008, 47, 9814–9859 CrossRef CAS PubMed.
- S. Singh, J. Zhang, T. D. Huber, M. Sunkara, K. Hurley, R. D. Goff, G. Wang, W. Zhang, C. Liu, J. Rohr, S. G. Van Lanen, A. J. Morris and J. S. Thorson, Angew. Chem., Int. Ed., 2014, 53, 3965–3969 CrossRef CAS PubMed.
- R. W. Gantt, R. D. Goff, G. J. Williams and J. S. Thorson, Angew. Chem., Int. Ed., 2008, 47, 8889–8892 CrossRef CAS PubMed.
- D. Chen, R. Chen, R. Wang, J. Li, K. Xie, C. Bian, L. Sun, X. Zhang, J. Liu, L. Yang, F. Ye, X. Yu and J. Dai, Angew. Chem., Int. Ed., 2015, 54, 12678–12682 CrossRef CAS PubMed.
- K. Xie, R. Chen, D. Chen, J. Li, R. Wang, L. Yang and J. Dai, Adv. Synth. Catal., 2017, 359, 603–608 CrossRef CAS.
- H. Stecher, M. Tengg, B. J. Ueberbacher, P. Remler, H. Schwab, H. Griengl and M. Gruber-Khadjawi, Angew. Chem., Int. Ed., 2009, 48, 9546–9548 CrossRef CAS PubMed.
- T. D. Huber, B. R. Johnson, J. Zhang and J. S. Thorson, Curr. Opin. Biotechnol., 2016, 42, 189–197 CrossRef CAS PubMed.
- B. J. C. Law, A.-W. Struck, M. R. Bennett, B. Wilkinson and J. Micklefield, Chem. Sci., 2015, 6, 2885–2892 RSC.
- G. J. Williams, J. Yang, C. Zhang and J. S. Thorson, ACS Chem. Biol., 2011, 6, 95–100 CrossRef CAS PubMed.
- A. R. Brochado, C. Matos, B. L. Møller, J. Hansen, U. H. Mortensen and K. R. Patil, Microb. Cell Fact., 2010, 9, 84 CrossRef PubMed.
- N. Koirala, R. P. Pandey, P. Parajuli, H. J. Jung and J. K. Sohng, J. Biotechnol., 2014, 184, 128–137 CrossRef CAS PubMed.
- C. Sommer-Kamann, A. Fries, S. Mordhorst, J. N. Andexer and M. Müller, Angew. Chem., Int. Ed., 2017, 56, 4033–4036 CrossRef CAS PubMed.
- A. Schenk, Z. Xu, C. Pfeiffer, C. Steinbeck and C. Hertweck, Angew. Chem., Int. Ed., 2007, 46, 7035–7038 CrossRef CAS PubMed.
- M. Pacholec, J. Tao and C. T. Walsh, Biochemistry, 2005, 44, 14969–14976 CrossRef CAS PubMed.
- J. C. Sadler, L. D. Humphreys, R. Snajdrova and G. A. Burley, ChemBioChem, 2017, 18, 992–995 CrossRef CAS PubMed.
- C. Gosch, H. Halbwirth, B. Schneider, D. Holscher and K. Stich, Plant Sci., 2010, 178, 299–306 CrossRef CAS.
- A. Lepak, A. Gutmann, S. T. Kulmer and B. Nidetzky, ChemBioChem, 2015, 16, 1870–1874 CrossRef CAS PubMed.
- M. Tengg, H. Stecher, P. Remler, I. Eiteljörg, H. Schwab and M. Gruber-Khadjawi, J. Mol. Catal. B: Enzym., 2012, 84, 2–8 CrossRef CAS.
- J. C. Wu and D. V. Santi, J. Biol. Chem., 1987, 262, 4778–4786 CAS.
- M. Szyf, Trends Pharmacol. Sci., 2001, 22, 350–354 CrossRef CAS PubMed.
- C. Zhang, B. R. Griffith, Q. Fu, C. Albermann, X. Fu, I.-K. Lee, L. Li and J. S. Thorson, Science, 2006, 313, 1291–1294 CrossRef CAS PubMed.
- A. Gutmann, C. Krump, L. Bungaruang and B. Nidetzky, Chem. Commun., 2014, 50, 5465–5468 RSC.
- L. W. Parks and F. Schlenk, J. Biol. Chem., 1958, 230, 295–305 CAS.
- J. C. Sadler, C.-w. Chung, J. E. Mosley, G. A. Burley and L. D. Humphreys, ACS Chem. Biol., 2017, 12, 374–379 CrossRef CAS PubMed.
- T. Pavkov-Keller, K. Steiner, M. Faber, M. Tengg, H. Schwab, M. Gruber-Khadjawi and K. Gruber, PLoS One, 2017, 12, e0171056 Search PubMed.
- S.-C. Chen, C.-H. Huang, S.-J. Lai, J.-S. Liu, P.-K. Fu, S.-T. Tseng, C. S. Yang, M.-C. Lai, T.-P. Ko and Y. Chen, Sci. Rep., 2015, 5, 10100 CrossRef CAS PubMed.
- L. A. Wessjohann, Curr. Opin. Chem. Biol., 2000, 4, 303–309 CrossRef CAS PubMed.
- S. J. Kwon, M. Mora-Pale, M.-Y. Lee and J. S. Dordick, Curr. Opin. Chem. Biol., 2012, 16, 186–195 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures to determine 1 solubility, fit substrate inhibition kinetics and calculate Keq of the UGT71A15 reaction; SDS-PAGE of purified enzymes, HPLC-chromatogram, effect of DMSO on NovO activity, pH-activity profiles of NovO and UGT71A15 and NMR spectra of purified 2–4. See DOI: 10.1039/c7ob01513e |
| This journal is © The Royal Society of Chemistry 2017 |