
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Phenothiazine-linked nucleosides and nucleotides for redox labelling of DNA†
Anna
Simonova
a,
Luděk
Havran
b,
Radek
Pohl
a,
Miroslav
Fojta
*bc and
Michal
Hocek
 *ad
*ad
aInstitute of Organic Chemistry and Biochemistry, Czech Academy of Sciences, Flemingovo namesti 2, CZ-16610 Prague 6, Czech Republic
bInstitute of Biophysics, Czech Academy of Sciences, Kralovopolska 135, 612 65 Brno, Czech Republic. E-mail: fojta@ibp.cz
cCentral European Institute of Technology, Masaryk University, Kamenice 753/5, CZ-625 00 Brno, Czech Republic
dDepartment of Organic Chemistry, Faculty of Science, Charles University in Prague, Hlavova 8, Prague-2 12843, Czech Republic. E-mail: hocek@uochb.cas.cz
First published on 26th July 2017
Abstract
Nucleosides and 2′-deoxyribonucleoside triphosphates (dNTPs) bearing phenothiazine (PT) attached to a nucleobase (cytosine or 7-deazaadenine) either directly or through an acetylene linker were prepared through Suzuki or Sonogashira cross-coupling and triphosphorylation, and were studied as building blocks for polymerase construction of modified DNA. The directly PT-substituted dNTPs were better substrates for polymerases than the alkyne-linked dNTPs but all of them were used in enzymatic synthesis of DNA using primer extension, nicking enzyme amplification, PCR or 3′-tail labelling by terminal deoxynucleotidyl transferase. The phenothiazine served as an oxidizable redox label (giving two analytically useful signals of oxidation on electrode) for nucleosides and DNA and was also used in orthogonal combination with previously developed benzofurazane or nitrophenyl labels for redox coding of DNA bases. Therefore, the title PT-linked dNTPs are useful additions to the portfolio of nucleotides for enzymatic synthesis of redox-labelled DNA for electrochemical analysis.
Introduction
Electrochemical DNA sensors1 are widely applied in diagnostics and DNA labelling by redox-active groups is of great importance.2 We and others have previously reported labelling of DNA by diverse redox labels, including ferrocene,3,4 amino- and nitrophenyl,5 Os(bpy)3-,6 anthraquinone,3,7,8 benzofurazane,9 azidophenyl,10 methylene-blue,11,12 polyoxometalates13 or methoxyphenol14 through polymerase incorporation of redox-labelled 2′-deoxyribonucleoside triphosphate (dNTP). Combination of several orthogonal redox labels could be used for multipotential redox-coding of DNA nucleobases for development of electrochemical minisequencing techniques. The orthogonality requires labelling of each nucleobase by a different redox-active group (having a different redox potential), and each label should be “readable” in the presence of all the other labels and give a ratiometric signal intensity. Our first generations of redox coding5,7 suffered from weak and overlapping signals, limited stability and difficult incorporation of some labels into DNA. More recently, we reported the first orthogonal and ratiometric set of two reducible labels (nitrophenyl and benzofurazane) useful for electrochemical minisequencing of short DNA stretches.9 However, we need a set of at least four fully orthogonal labels for diagnostic applications and, given the rather narrow window of potentials available for electrochemical analysis of DNA, we should be able to combine some reducible and some oxidizable labels. While previously developed reducible labels, i.e. nitrophenyl,5 benzofurazane9 or azidophenyl,10 give strong signals due to multi-electron reductions, most of the oxidizable labels3,14 typically gave weak signals due to one-electron oxidation. Therefore, there is a great need for other oxidizable labels for DNA and we turned our attention to phenothiazine (PT).PT derivatives are known redox-active molecules giving either two waves of single electron oxidations15 or one strong two-electron oxidation.16,17 They have been extensively used as redox and fluorescent label for polymers and other compounds.18,19 Some PT derivatives have also been used for labelling of nucleic acids, mostly for photosensitizing20 or charge-transfer study.21–24 One example of a PT-linked dNTP (through a flexible non-conjugate amide linker) was shown in a recent work12 for polymerase labelling of DNA and for redox coding in a single-nucleotide polymorphism study. However, the PT-linked dNTP was not fully characterized and the biochemistry of the enzymatic incorporation was not reported either.12 Therefore, we decided to prepare a set of PTZ-modified nucleosides and dNTPs either directly linked or tethered through a conjugate acetylene linker and characterize them chemically, electrochemically and biochemically in order to develop them as useful redox labels for DNA.
Results and discussion
Synthesis
From previous studies it is well known that dNTPs bearing modifications at position 5 of pyrimidines or at position 7 of 7 deazapurines are good substrates for DNA polymerases25,26 and some dNTPs bearing less bulky aryl or alkynyl groups can even be better substrates than natural dNTPs.27–29 Therefore, we selected 2′-deoxycytidine linked through position 5 and 2′-deoxy-7-deazaadenine linked through position 7 for attachment of PT either directly via s single bond or through a conjugate ethynyl tether. The syntheses started from commercial 2′-deoxy-5-iodocytidine (dCI) or from well-known 2′-deoxy-7-iodo-7-deazaadenosine (dAI),30 which were triphosphorylated to the corresponding dNTPs (dCITP and dAITP).5 The Suzuki–Miyaura cross-coupling of iodinated nucleosides (dCI or dAI) with PTZ-linked pinacolborane 1 under aqueous conditions31 in the presence of Pd(OAc)2 and tris(3-sulfonatophenyl)phosphine (TPPTS) gave the arylated nucleosides (dCPT and dAPT) in good yields of 75% or 96%, respectively (Scheme 1, Table 1). The Sonogashira cross-coupling reactions of dCI or dAI with ethynylphenothiazine 2 were performed in the presence of Pd(PPh3)2Cl2 and CuI in DMF to give labelled nucleosides dCEPT and dAEPT in similarly good yields (76% and 93%). The Suzuki–Miyaura reactions of iodinated dNITPs with PT-Bpin (1) under aqueous conditions gave the desired PT-linked dNPTTPs (dCPTTP in 53% and dAPTTP in 68%), whereas the aqueous Sonogashira reactions of dNITPs with EPT (2) afforded the PT-acetylene-linked dNEPTTPs (dCEPTTP in 48% and dAEPTTP in 49%). Taking into account partial hydrolysis of the dNTPs during the reaction and isolation, these yields are very good. Alternatively, the dNEPTTPs were prepared by triphosphorylation of the corresponding nucleosides dNEPT in moderate yields (43 and 45%). In all cases the PT-labelled dNPTTPs or dNEPTTPs were isolated in ca. 30–60 mg amounts by HPLC and fully characterized.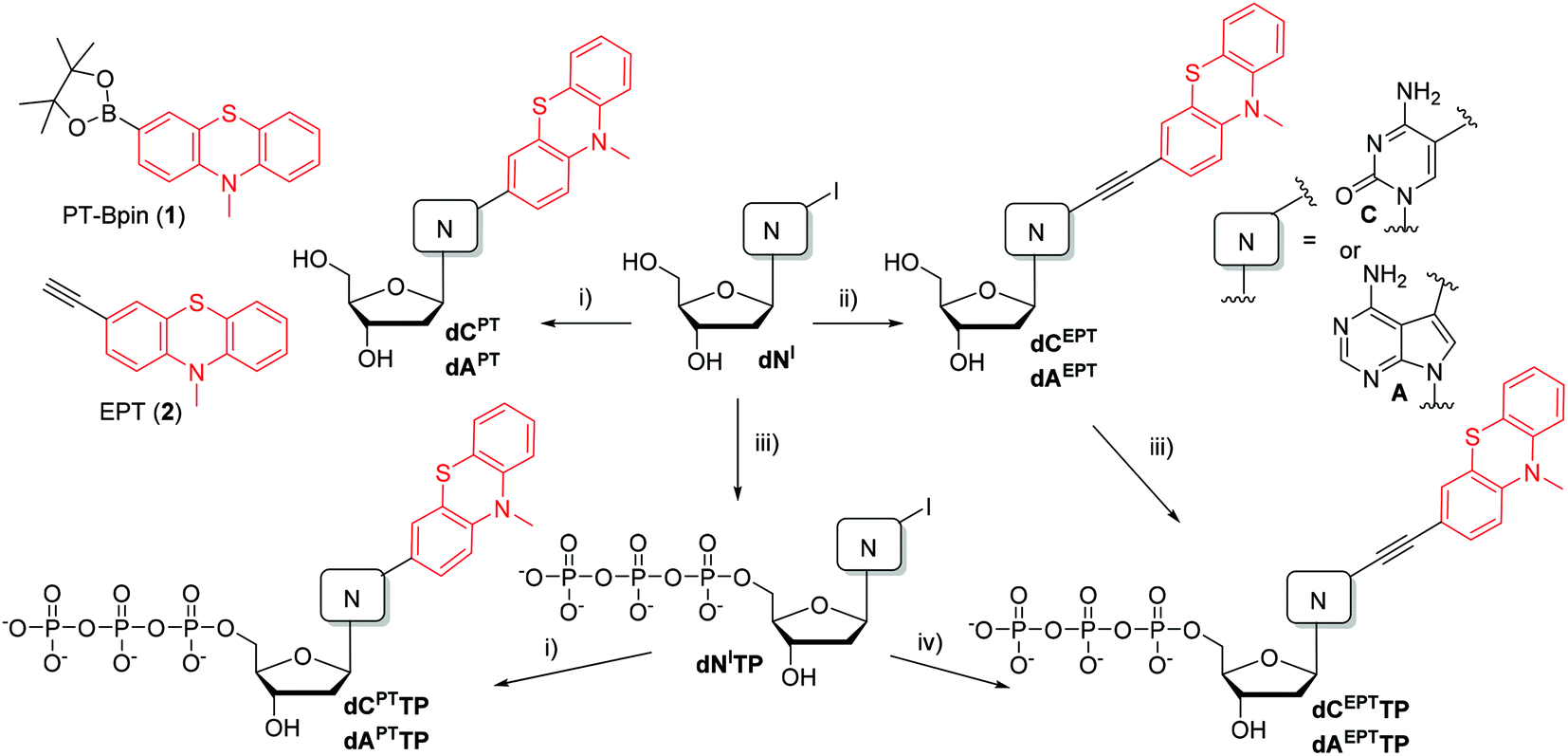 | ||
Scheme 1 Reagents and conditions: (i) PT-Bpin (1), Pd(OAc)2, TPPTS, Cs2CO3, AN/H2O (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), 50 °C, 40 min; (ii) EPT (2), Pd(PPh3)2Cl2, CuI, Et3N, DMF, 75 °C, 1 h; (iii) 1. POCl3, PO(OMe)3, 0 °C, 3 h; 2. (NHBu3)2H2P2O7, Bu3N, DMF, 0 °C, 1.5 h; 3. TEAB; (iv) EPT (2), Pd(OAc)2, CuI, TPPTS, Et3N, AN/H2O (1:1), 75 °C, 1 h. :1), 50 °C, 40 min; (ii) EPT (2), Pd(PPh3)2Cl2, CuI, Et3N, DMF, 75 °C, 1 h; (iii) 1. POCl3, PO(OMe)3, 0 °C, 3 h; 2. (NHBu3)2H2P2O7, Bu3N, DMF, 0 °C, 1.5 h; 3. TEAB; (iv) EPT (2), Pd(OAc)2, CuI, TPPTS, Et3N, AN/H2O (1:1), 75 °C, 1 h. | ||
| Starting compd. | Reagent | Catalyst | Solvent | Base | Product | Yield (%) |
|---|---|---|---|---|---|---|
| dAI | PT-Bpin (1) | Pd(OAc)2, TPPTS | AN/H2O (1/1) | Cs2CO3 | dAPT | 96 |
| dCI | dCPT | 75 | ||||
| dAITP | dA PTTP | 68 | ||||
| dCITP | dC PTTP | 53 | ||||
| dAI | EPT (2) | Pd(PPh3)2Cl2, CuI | DMF | Et3N | dAEPT | 93 |
| dCI | dCEPT | 76 | ||||
| dAITP | Pd(OAc)2, CuI, TPPTS | AN/H2O (1/1) | dAEPTTP | 49 | ||
| dCITP | dCEPTTP | 48 | ||||
| dAEPT | 1. PO(OMe)3, POCl3, 0 °C; 2. (NHBu3)2H2P2O7, Bu3N, DMF, 0 °C; 3) TEAB (2 M) | dAEPTTP | 43 | |||
| dCEPT | dCEPTTP | 45 | ||||
Biochemistry
The four new dNTPs (dAPTTP, dCPTTP, dAEPTTP and dCEPTTP) were then tested as substrates for DNA polymerases in primer extension experiments (PEX) using KOD XL, Vento(exo-) and Pwo polymerases (for sequences of primers, templates and products, see Table 2). The first experiment was a single nucleotide incorporation of each of the dNXTPs into a 15-nt primer followed by three natural dG using tempA or tempC templates (Fig. 1). In all cases, we obtained fully extended products as shown by the analysis by polyacrylamide gel electrophoresis (PAGE). All the products were also characterized by MALDI-TOF (see Table 3). The kinetic experiments (see Fig. S1–S4 in the ESI†) in the presence of KOD XL polymerase showed that the modified dNXTPs were incorporated at a slightly slower rate compared to dATP or dCTP but all of them were fully incorporated within max. 5–10 minutes. The slowest extension proceeded with dAEPTTP. | ||
| Fig. 1 Primer extension with tempA and tempC using KOD XL, Vent(exo-) and Pwo polymerases. Pr: primer; A+: product of PEX with natural dNTPs; A-: product of PEX with dGTP; APT: product of PEX with dAPTTP, dGTP; AEPT: product of PEX with dAEPTTP, dGTP; C+: product of PEX with dCTP, dGTP; C-: product of PEX with dGTP; CPT: product of PEX with dCPTTP, dGTP; CEPT: product of PEX with dCEPTTP, dGTP. | ||
| Name | Sequence |
|---|---|
| a For magnetoseparation of the extended primer strands, the templates were 5′- end biotinylated. b Primer sequences in the template are underlined. c 6-Carboxyfluorescein- (6-FAM-) labeled primerrnd was used for visualization in PEX experiments and TDT elongation. | |
| Primerrndc | 5′-CATGGGCGGCATGGG-3′ |
| Primer NICK | 5′-CCGATCTAGTGAGTCCTCG-3′ |
| Temprnd16 | 5′-CTAGCATGAGCTCAGT![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) ![[A with combining low line]](https://www.rsc.org/images/entities/char_0041_0332.gif) ![[T with combining low line]](https://www.rsc.org/images/entities/char_0054_0332.gif) ![[G with combining low line]](https://www.rsc.org/images/entities/char_0047_0332.gif) -3′ -3′ |
| Nick_1A | 5′-CACTCACGACcgag-3′ |
| Nick_1C | 5′-CACTCATGACcgag-3′ |
| Nick_2A(2C) | 5′-CAGTCATGAAcgag-3′ |
| Nick_4A(4C) | 5′-CATGATCAGTACGTACcgag-3′ |
| TempA | 5′-CCCT-3′ |
| TempC | 5′-CCCG-3′ |
| TemptermA | 5′-T-3′ |
| TemptermC | 5′-G-3′ |
| ONrnd16APT | 5′-CATGGGCGGCATGGGAPTCTGAPTGCTCAPTTGCTAPTG-3′ |
| ONrnd16AEPT | 5′-CATGGGCGGCATGGG AEPTCTG AEPTGCTC AEPTTGCTAEPTG-3′ |
| ONrnd16CPT | 5′-CATGGGCGGCATGGGA CPTTGAGCPTCPTATGCPTTAG-3′ |
| ONrnd16CEPT | 5′-CATGGGCGGCATGGGA CEPTTGAGCEPTTCEPTATGCEPTTAG-3′ |
| ONrnd16AEPT CEBF | 5′-CATGGGCGGCATGGGAEPTCEBFTGAEPTGCEBFTCEBFAEPTTGCEBFTAEPTG-3′ |
| ONrnd16AEPT UNO2 | 5′-CATGGGCGGCATGGGAEPTCUNO2GAEPTGCUNO2CAEPTUNO2GCUNO2AEPTG-3′ |
| ONNick_1AAPT | 5′-P-GTCGTGAPTGTG-3′ |
| ONNick_1AAEPT | 5′-P-GTCGTGAEPTGTG-3′ |
| ONNick_1CCPT | 5′-P-GTCPTATGAGTG -3′ |
| ONNick_1CCEPT | 5′-P-GTCEPTATGAGTG-3′ |
| ONNick_2AAPT | 5′-P-TTCAPTTGAPTCTG-3′ |
| ONNick_2AAEPT | 5′-P-TTCAEPTTGAEPTCTG-3′ |
| ONNick_2CCPT | 5′-P-TTCPTATGA CPTTG -3′ |
| ONNick_2CCEPT | 5′-P-TTCEPTATGA CEPTTG-3′ |
| ONNick_4AAPT | 5′-P-T APTGCAPTTGCTAPTCGTCAPTG-3′ |
| ONNick_4AAEPT | 5′-P-T AEPTGCAEPTTGCTAEPTCGTCAEPT G-3′ |
| ONNick_4ACPT | 5′-P-TAGCPTATGCPTTACPTGTCPTAG-3′ |
| ONNick_4ACEPT | 5′-P-TAGCEPTATGCEPTTACEPTGTCEPTAG-3′ |
| ONAAPT | 5′-CATGGGCGGCATGGGAPTGGG-3′ |
| ONAAEPT | 5′-CATGGGCGGCATGGGAEPTGGG-3′ |
| ONCCPT | 5′-CATGGGCGGCATGGGCPTGGG-3′ |
| ONCCEPT | 5′-CATGGGCGGCATGGGCEPTGGG-3′ |
| ONtermAAPT | 5′-CATGGGCGGCATGGGAPT-3′ |
| ONtermAAEPT | 5′-CATGGGCGGCATGGGAEPT-3′ |
| ONtermCCPT | 5′-CATGGGCGGCATGGGCPT-3′ |
| ONtermCCEPT | 5′-CATGGGCGGCATGGGCEPT-3′ |
| Oligonucleotide | M calcd (Da) | M found (Da) |
|---|---|---|
| ONrnd16APT | 10458.5 |
10459.0 |
| ONrnd16AEPT | 10554.5 |
10555.3 |
| ONrnd16CPT | 10462.5 |
10463.8 |
| ONrnd16CEPT | 10558.5 |
10559.3 |
| ONrnd16AEPTCEBF | 11031.1 |
11032.9 |
| ONrnd16AEPTTNO2 | 10886.9 |
10887.3 |
| ONNick_1AAPT | 3388.3 | 3389.4 |
| ONNick_1AAEPT | 3412.3 | 3413.4 |
| ONNick_1CCPT | 3373.7 | 3374.5 |
| ONNick_1CCEPT | 3397.4 | 3398.4 |
| ONNick_2AAPT | 3517.6 | 3519.3 |
| ONNick_2AAEPT | 3565.6 | 3566.8 |
| ONNick_2CCPT | 3519.6 | 3520.4 |
| ONNick_2CCEPT | 3567.6 | 3568.7 |
| ONNick_4AAPT | 5801.4 | 5802.8 |
| ONNick_4AAEPT | 5897.4 | 5898.7 |
| ONNick_4CCPT | 5805.4 | 5806.6 |
| ONNick_4CCEPT | 5901.4 | 5902.5 |
| ONAAPT | 6185.2 | 6186.5 |
| ONAAEPTZ | 6209.2 | 6210.6 |
| ONCCPT | 6162.2 | 6163.5 |
| ONCCEPT | 6186.2 | 6187.5 |
Then, each of the modified dNXTPs was incorporated into a longer 31-mer oligonucleotide (ON) using temprnd16 designed to encode for four incorporations at each of the four nucleotides and four modifications if one of the dNTPs is modified. Furthermore, the PEX experiments gave full-length products in the presence of all tested polymerases (Fig. 2). The PEX products have slightly different electrophoretic mobility but their correct length and sequence was verified by MALDI (Table 3).
 | ||
| Fig. 2 Primer extension with temprnd16 using KOD XL, Vent(exo-) and Pwo polymerases. Pr: primer; +: product of PEX with natural dNTPs; A-: product of PEX with dCTP, dTTP, dGTP; C-: product of PEX with dATP, dTTP, dGTP; APT: product of PEX with dAPTTP, dCTP, dTTP, dGTP; AEPT: product of PEX with dAEPTTP, dCTP, dTTP, dGTP; CPT: product of PEX with dCPTTP, dATP, dTTP, dGTP; CEPT: product of PEX with dCEPTTP, dATP, dTTP, dGTP. | ||
In order to test the possibility of use of the PT-labelled nucleotides for redox coding of DNA bases, we synthesized ON containing redox-labelled A and in combination with redox-labelled C or T (APT + CEBF or AMOP + UNO2). The PEX experiments combining the use of dAPTTP with previously reported dCEBFTP9 or dUNO2TP5 were also successful to give full-length ONs bearing four APT labels in combination with either four CEBF or four UNO2 modifications (Fig. 3 and 4). These ON products were also characterized by MALDI (Table 3).
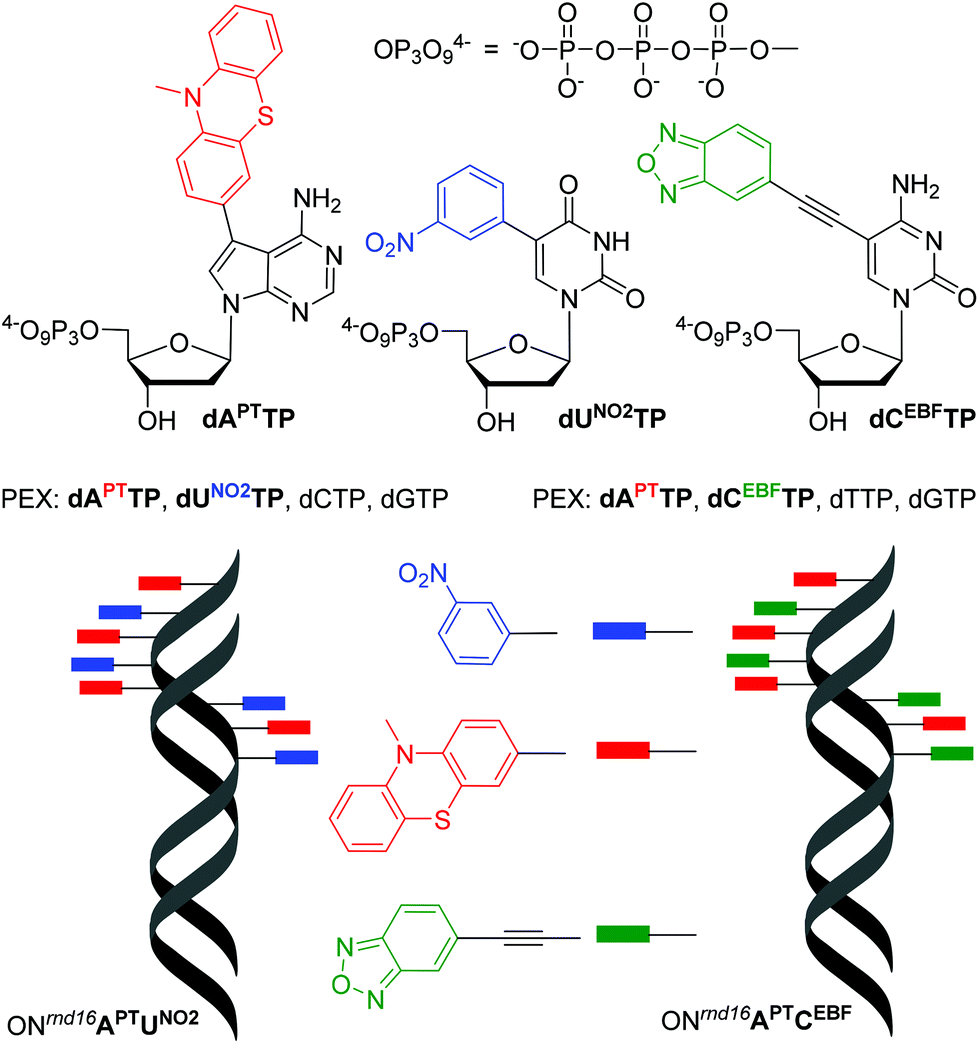 | ||
| Fig. 3 Redox coding of DNA bases: structures of redox-labelled dNTPs and DNA products of the mixed PEX. | ||
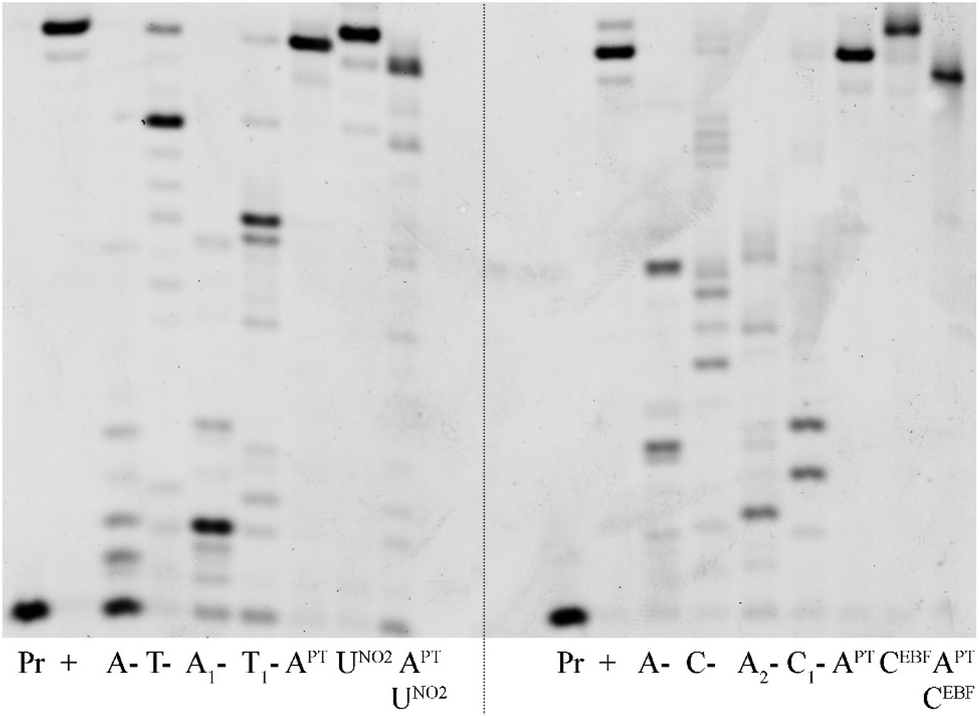 | ||
| Fig. 4 Primer extension with temprnd16 using KOD XL polymerase and combinations of redox-labelled dNTPs. Pr: primer; +: product of PEX with natural dNTPs; A-: product of PEX with dCTP, dTTP, dGTP; T-: product of PEX with dATP, dCTP, dGTP; A1-: product of PEX with dTNO2TP, dCTP, dGTP; T1-: product of PEX with dAPTTP, dCTP, dGTP; APT: product of PEX with dAPTTP, dCTP, dTTP, dGTP; UNO2: product of PEX with dUNO2TP, dATP, dCTP, dGTP; APTUNO2: product of PEX with dAPTTP, dUNO2TP, dCTP, dGTP; C-: product of PEX with dATP, dTTP, dGTP; A2-: product of PEX with dCEBFTP, dTTP, dGTP, C1-: product of PEX with dAPTTP, dTTP, dGTP; CEBF: product of PEX with dCEBFTP, dATP, dTTP, dGTP; APTCEBF: product of PEX with dAPTTP, dCEBFTP, dTTP, dGTP. | ||
We also tried the use of the dNXPTTPs in PCR amplifications, but the PCR reactions using KOD XL did not give a significant product (see Fig. S6 in ESI†). Since the dNXPTTPs are reasonably good substrates for these polymerases in the PEX experiments, the problem is probably in the limited ability of the enzymes to read through the PT-modified templates. We also tested PCR reactions with mixtures of natural dATP and modified dAPTTP or dAEPTTP, and only the experiment with 50% of dAPTTP in the presence of natural dATP gave a significant product of amplification (see Fig. S7 and S8†).
Next, we tested whether the new dNXPTTPs could be used in the nicking enzyme amplification reaction (NEAR), which is used for enzymatic synthesis of short single-stranded ONs.32 The principle of the method is that a PEX reaction is performed in the presence of Vent (exo-) polymerase and a nicking endonuclease (in our case Nt·BstNBI), which cleaves the extended strand to release the modified ssON.33 We tested the NEAR amplification using three different templates designed for the synthesis of ON containing either one, two or four modifications (10-mer ON containing either one or two modifications or 16-mer ON containing four modifications). Fig. 5 (and Fig. S5 in the ESI†) shows the outcome of these experiments. The ssONs containing one modification were obtained efficiently in case of each of the four dNXPTTPs, whereas the products containing two or four labels were still obtained (though in lower yield).
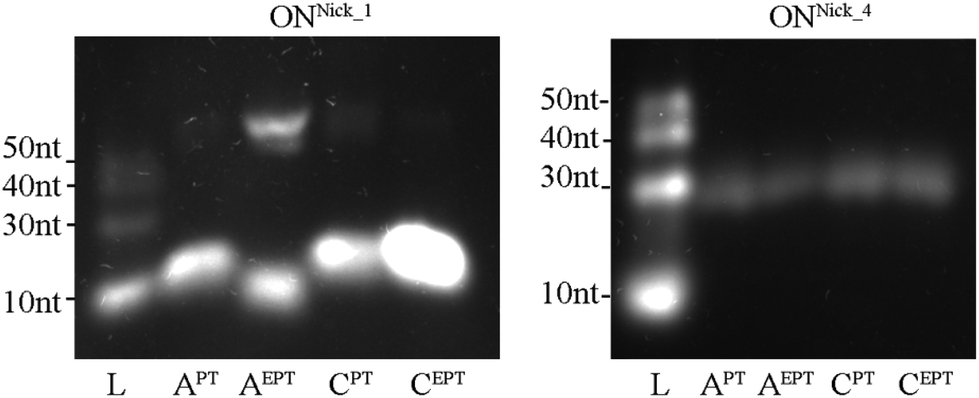 | ||
| Fig. 5 Incorporation of modified dNTPs in NEAR using Nick_1A, Nick_1C and Nick_4A(4C) templates. L: DNA ladder; APT: product of NEAR with dAPTTP, dCTP, dGTP, dTTP; AEPT: product of NEAR with dAEPTTP, dCTP, dGTP, dTTP, CPT: product of NEAR with dCPTTP, dATP, dGTP, dTTP, CEPT: product of NEAR with dCEPTTP, dATP, dGTP, dTTP. | ||
The last enzymatic method tested in this work was the non-templated 3′-tail labelling by terminal deoxynucleotidyl transferase (TdT).34,35Fig. 6 shows that the TdT-catalyzed elongation of the primer using dAPTTP gave almost the perfect product of single nucleotide extension, whereas the other dNXPTTPs were less efficient. The single nucleotide extension can advantageously be used for specific incorporation of one PT-redox label at the 3′-end of ON analytes.
 | ||
| Fig. 6 TdT-catalyzed DNA chain elongation. Pr: primerrnd; S: standard (PEX product of temptermA with dATP or temptermC with dCTP); A+, APT and AEPT: products of primerrnd elongation using terminal transferase and either dATP, dAPTTP or dAEPTTP respectively; C+, CPT and CEPT: products of primerrnd elongation using terminal transferase and either dCTP, dCPTTP or dCEPTTP respectively (for the full gel image, see Fig. S9 and S10†). | ||
Fluorescence
In order to verify the possible applications in fluorescence labelling of DNA, we measured the absorption and emission spectra of PT-modified nucleosides and triphosphates. Table 4 shows that the nucleosides in ethanol showed significant fluorescence with emission maxima at 462–476 nm, whereas the fluorescence of dNXPTTPs in water was negligible. The fluorescence of the PEX product ONrnd16AEPT containing four AEPT modifications was moderate (see Fig. S13 in ESI†).| Compound | Solvent |
λ
absa [nm] |
ε [103 M−1 cm−1]b |
λ
emc [nm] |
Φ
fd |
|---|---|---|---|---|---|
| a Position of the absorption maximum, ±1 nm. b Confidence interval did not exceed ±0.2 × 103 M−1 cm−1. c Position of the emission maximum, ±1 nm. d Quantum yield of fluorescence measured using quinine sulfate in 0.5 M H2SO4 (Φf = 0.546 at 25 °C) as a standard. | |||||
| dAEPT | EtOH | 344 | 16.4 | 462 | 0.1889 ± 0.0116 |
| 296 | 32.8 | ||||
| 267 | 35.9 | ||||
| dCEPT | EtOH | 346 | 6.2 | 476 | 0.0101 ± 0.0004 |
| 277 | 14.4 | ||||
| dAEPTTP | H2O | 336 | 11.4 | 486 | 0.014 ± 0.0003 |
| 270 | 24.9 | ||||
| dCEPTTP | H2O | 340 | 8.4 | 489 | 0.0014 ± 0.0001 |
| 275 | 19.8 | ||||
Electrochemistry
Electrochemical properties of PT- and EPT-deoxynucleoside conjugates (dNPT or dNEPT, respectively) and of PT-modified oligonucleotides were studied by means of cyclic (CV) and square-wave (SWV) voltammetry at the basal-plane pyrolytic graphite electrode (PGE). First, we compared electrochemical oxidation of free PT with individual modified nucleosides (Fig. 7, 8, S36 and S37†). PT alone yielded two oxidation signals in the potential range between +0.5 and +1.0 V. According to the literature,15 the first signal (denominated here as peak PTox1, around +0.6 V, Fig. 7a) corresponds to one-electron reversible oxidation to form a radical cation at the PT nitrogen atom. The second one-electron oxidation step (reflected in peak PTox2 around +0.9 V) is irreversible and results in the formation of the corresponding sulfoxide. Cyclic voltammograms of PT (Fig. 7a) accorded with the above mechanism: when the anodic CV scan was turned back at +0.8 V, i.e., without applying potentials sufficiently positive for the sulfoxide formation, a cathodic counter peak to the peak PTox1 was observed, giving evidence about the reversibility of the first oxidation step. On the other hand, when the CV scan was turned “after” the peak PTox2, the cathodic signal disappeared in agreement with the overall irreversibility of the two-step PT oxidation. Another irreversible anodic signal was observed around +1.3 V, suggesting further oxidation of the PT sulfoxide (Fig. 7a). Results of SWV measurements (Fig. 8), including inspection of the forward and backward components of the SWV current to evidence the (ir)reversibility of the processes (see Fig. S37†), accorded with CV data.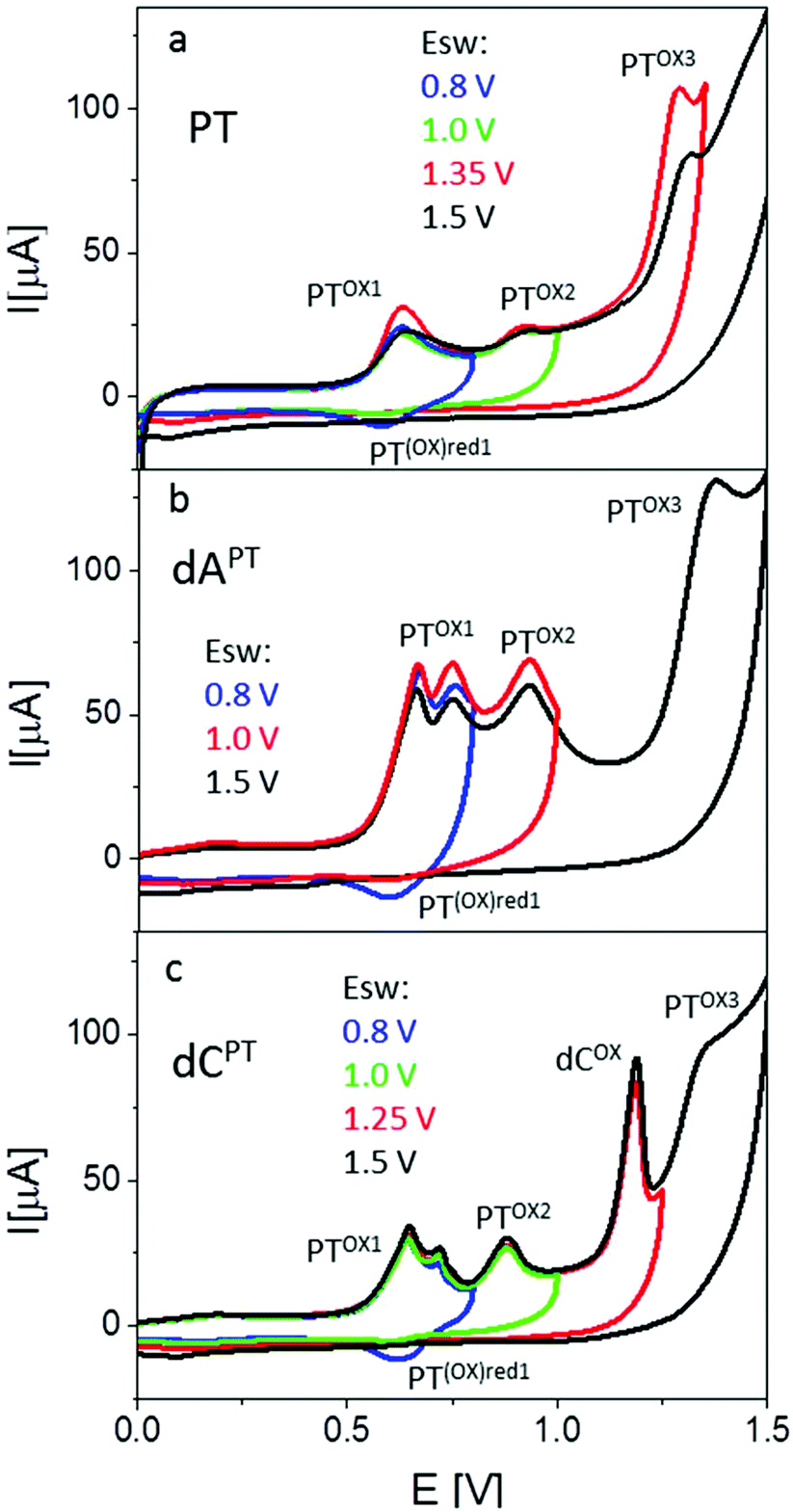 | ||
| Fig. 7 CV responses of phenothiazine (a), dAPT (b), and dCPT (c) at PGE. C = 40 μM, background electrolyte 0.2 M acetate buffer (pH 5.0). CV parameters: scan rate 1 V s−1, Ei = 0.0 V, Esw see legend in the figure. | ||
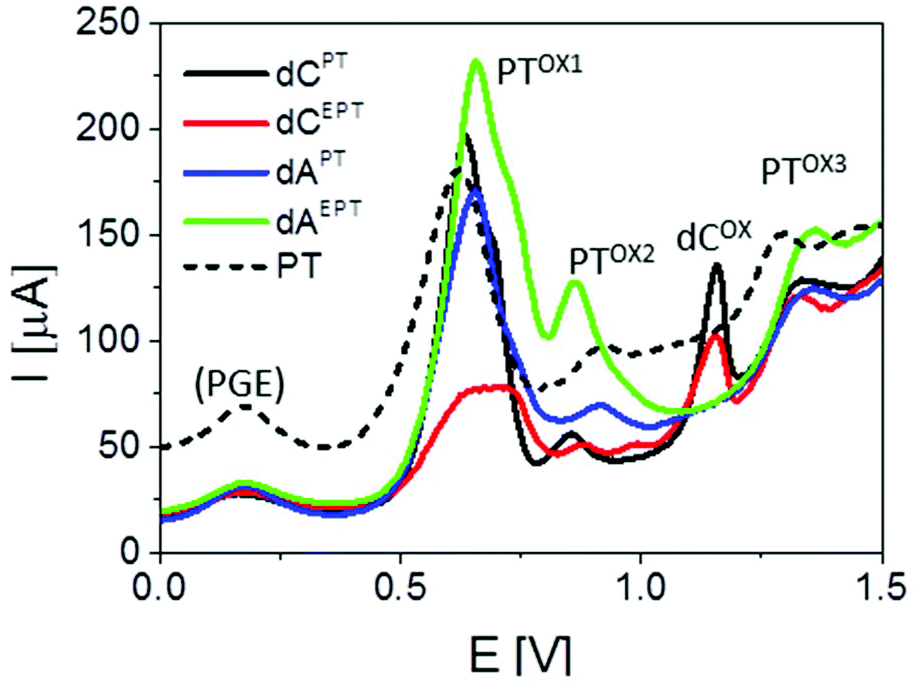 | ||
| Fig. 8 Comparison of SWV responses of PTZ and PT- or EPT-modified nucleosides. SWV parameters: frequency 200 Hz, amplitude 50 mV, Ei = 0.0 V. Peak denoted as (PGE) is produced by the electrode due to the presence of oxygenous functional groups at its surface. For other details, see Fig. 7. | ||
Basically, CVs and SWVs of dAPT and dCPT displayed signals characteristic for the PT moiety (Fig. 7b and c, S36†); in the case of the conjugates only the peak PTox1 was apparently split into two distinct signals. The reversibility of PT oxidation was retained when the CV scan was turned after the more positive one of them (at +0.80 V), but lost when the scan was turned at +1.0 V. The signals around +0.85–+0.90 V were thus identified as the peak PTox2 corresponding to the second irreversible oxidation step. In addition to the PT-specific signals, the dCPT conjugate yielded a well-developed, irreversible peak close to +1.2 V. An analogous peak was produced by dCEPT but not by either of the dAPT or dAEPT conjugates (see CVs in Fig. S36† and SWVs in Fig. 8), suggesting involvement of the cytosine moiety in the corresponding electrode process. The absence of a base-specific signal on the voltammograms of dAPT or dAEPT may be rather surprising as 7-deazaadenine was reported to undergo irreversible electrochemical oxidation at the PGE both in its underivatized form (around +1.1 V)36 and in conjugates with, e.g., trisbipyridine complexes of Os or Ru (between +0.9 and +1.0 V).6 An explanation of the absence of a distinct 7-deazaA peak on voltammograms of its PT conjugates may lie in its overlap with a PT signal, possibly peak PTox2. A more extensive electrochemical study, which is out of the scope of this report, will be required to understand the electrooxidation processes of (E)PT and nucleobase moieties in the dNPT conjugates in more detail.
In the next experiments, we used SWV to measure the electrochemical responses of ONs with incorporated PT- or EPT-labelled nucleotides. In the first series, we analyzed short 10- or 16-mer ON-products of NEAR bearing 1, 2 or 4 dAPT conjugates (for sequences of templates see Table 2). Control unmodified NEAR products (blue curves in Fig. 9a and b, shown for ONNick_1C) yielded well-developed signals of the oxidation of natural purine bases (peaks Gox and Aox), the intensities of which reflected relative contents of G and A in the given ON (see sequences in Table 2). For the dAPT-modified ONs, peaks corresponding to individual oxidation steps of PT were observed; even for the ONnick_1AAPT bearing a single PT moiety, small but distinct peaks PTox1, PTox2 and PTox3 were detected. Their potentials were in general shifted to less positive values, as compared to the corresponding peak potentials measured with the nucleosides (see Table S1†). The first two peaks occurred at potentials sufficiently less positive than the potentials of the purine oxidation signals, making it possible to measure them independently, while peak PTox3 was overlapping with peak Aox and its observation was possible only owing to the fact that the dAPT-modified NEAR products did not contain any natural adenine residues (unlike the PEX products that always contained adenines, see Fig. 10). Following the intensities of the measured signals in dependence on the number of PT moieties incorporated, one can see a significantly non-linear behavior, with a large difference between NEAR products containing 1 and 2 PT labels (black and green curve in Fig. 9a) and very similar peak heights obtained for 2 and 4 PT moieties (green and red curve in Fig. 9a). Nevertheless, it should be noted that the latter ONnick_4AAPT was 16-mer (while ONnick_1AAPT and ONnick_2AAPT were 10-mers), and thus the relative content of PT in the 16-mer was higher only by a factor of 1.25 (instead of 2), compared to ONnick_2AAPT. Thus, the small difference between 4 and 2 PT moieties can be ascribed primarily to this fact. On the other hand, the difference between the decanucleotides may be due to a strong effect of the PT moieties on interactions of the ON with the PGE surface, such as preferential adsorption of the PT tags.
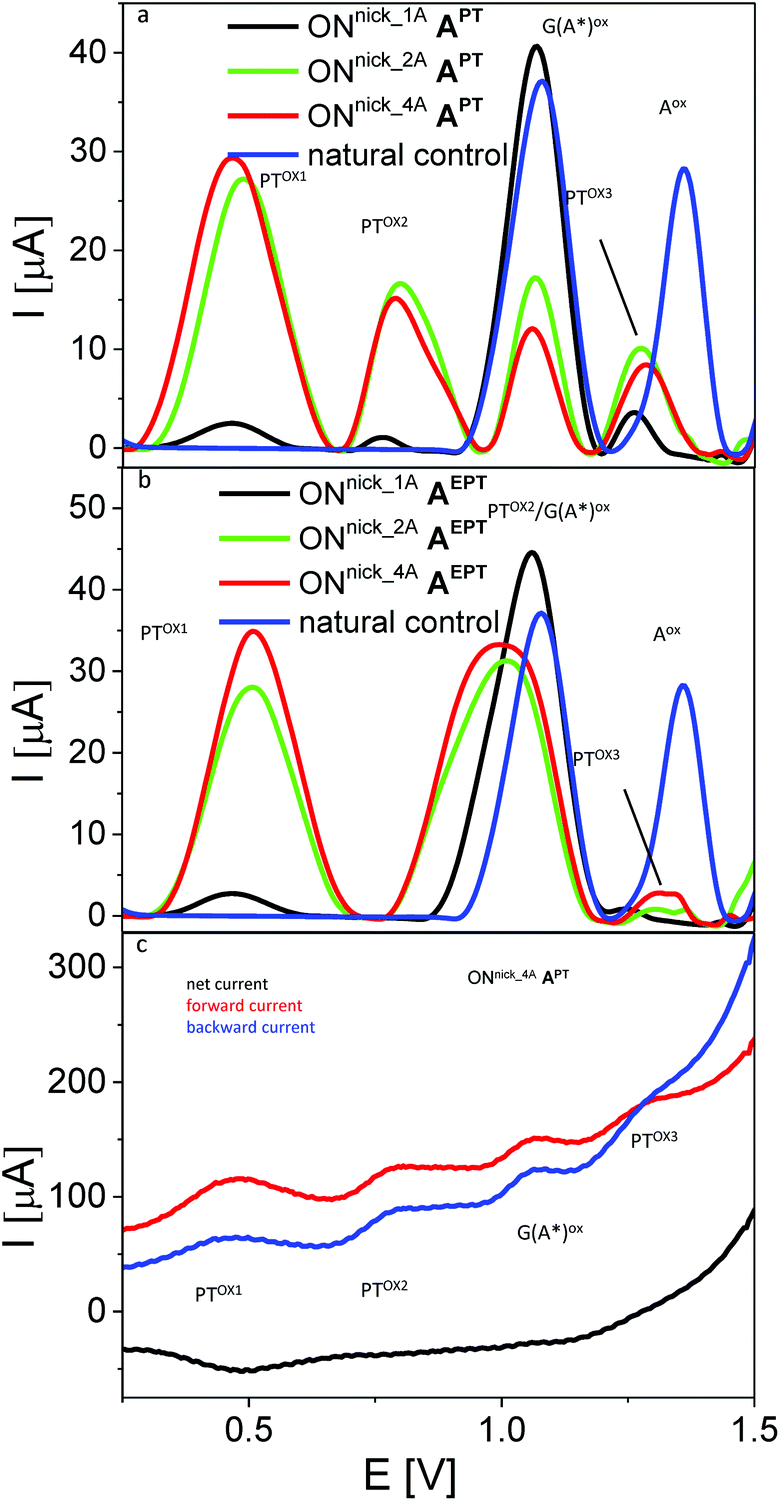 | ||
| Fig. 9 (a, b) Sections of baseline-corrected AdTS SWV responses of NEAR products involving different number of PT-modified nucleobases. (c) Components of the SWV current for sample ONnick_4AAPT. In all panels, ta = 1 min; for other details see Fig. 7 and 8. | ||
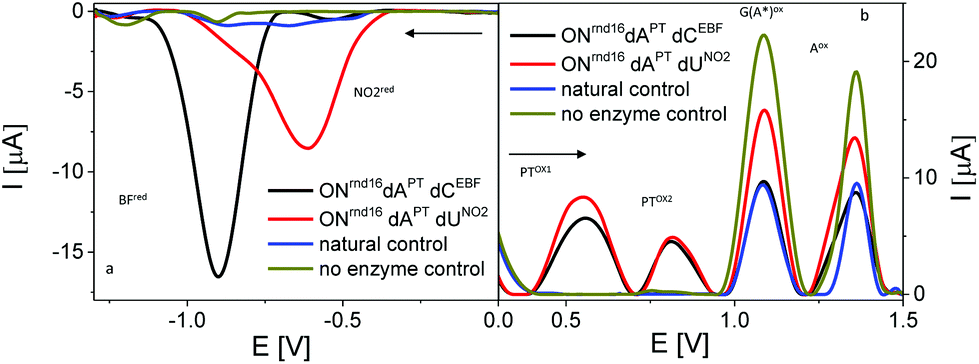 | ||
| Fig. 10 Sections of baseline-corrected AdTS SWV responses of PEX products bearing PT labels combined with EBF or PhNO2 labels. (a) Cathodic reduction of BF and NO2 and (b) anodic oxidation of PT and nucleobase moieties. In both cases Ei was set at 0.0 V; arrows indicate scan direction; for other details see Fig. 7 and 8. | ||
A different behavior was observed with analogous NEAR products labelled with dAEPT (Fig. 9b). While the peak PTox1 produced by these modified ONs was similar to that in the case of ONs bearing dAPT, well separated from other signals and exhibiting similar changes in peak heights depending on the number of EPT conjugates incorporated, peak PTox2 was shifted to a more positive potential, making it overlap with peak Gox. As a consequence, it was not possible to differentiate between these two signals and measure their intensities. Again, involvement of the ethynyl linker in the electrooxidation processes of EPT-modified DNA is a matter of specialized study and will be published elsewhere. Inspection of the SWV current components (Fig. 9c) confirmed the reversibility of the electrode process giving rise to the peak PTox1 for the (E)PT moiety incorporated into DNA, and the irreversibility of the more negative signals produced by either the PT or the nucleobases. Based on the above observations, for the next experiments we chose the dAPT conjugates producing distinct PT-specific and natural base-specific signals without mutual overlaps.
To test applicability of PT as a label in a “multicolor” redox coding system, we prepared PEX products modified with dAPT combined with either of the following reducible labels, ethynyl benzofurazane (as dCEBF) and nitrophenyl (as dUNO2). Again, the PT labels yielded characteristic oxidation peaks PTox1 and PTox2 on SWVs measured in the anodic direction, where the purine-specific peaks were also detected (Fig. 10). EBF and PhNO2 yielded, in agreement with previous literature,5,9 specific reduction signals around −0.95 V and −0.55 V, respectively. When the measurements were performed with ONs freshly adsorbed at the electrode and the initial potential (Ei) was set at 0.0 V, no interfering anodic signals of ONs modified with BF or PhNO2 were detected in the anodic scan. Similarly, no interfering reduction peak originating from PT labels was observed in the cathodic scans starting from 0.0 V (Fig. 10). Therefore, all three labels could easily be differentiated and determined independently by their peak potentials and/or by the direction of electron flow.
Conclusions
We developed the synthesis of 2′-deoxycytidine and 2′-deoxy-7-deazaadenosine nucleosides and dNTPs bearing phenothiazine linked directly or through an acetylene tether at position 5 of cytosine or at position 7 of 7-deazaadenine. The dNXPTTPs were good substrates for DNA polymerases and served as building blocks for enzymatic synthesis of modified ON and DNA through PEX and NEAR (but did not work well in PCR amplification). Interestingly, dAPTTP and dCPTTP gave efficient and selective non-templated single-nucleotide extension catalyzed by TdT. The nucleosides showed interesting fluorescence in EtOH but the nucleotides and EPT-modified DNA showed low fluorescence in water. The phenothiazine moiety is a useful redox label for nucleosides and DNA, giving two anodic peaks of PT oxidation, which are analytically useful. The directly linked APT is more useful than the ethynyl-linked AEPT base where the second oxidation peak overlaps with oxidation of guanine. The PT-redox label is orthogonal to previously reported benzofurazane9 and nitrophenyl labels5 and can be used for multipotential redox coding of DNA bases.12 The characteristic reversibility of the first PT signal and the irreversibility of the two-step oxidation process can be exploited for unambiguous identification of the PT label among other oxidizable moieties. Thus, although the presence of two oxidation peaks apparently limits the potential space for a combination of more oxidizable DNA labels, using proper measuring parameters can help one to attain satisfactory selectivity even in the case of overlapping primary oxidation signals. On the other hand, further studies of other alternative oxidizable labels are still needed to introduce the last (fourth) redox label in prospective four-label coding.Experimental
Chemicals, synthetic oligodeoxyribonucleotides, and enzymes were purchased from commercial suppliers and were used without further purification. Purification of nucleoside triphosphates was performed using HPLC on a column packed with 10 μm C18 reversed phase. NMR spectra were measured on a 500 MHz (1H at 500.0 MHz, 13C at 125.7 MHz and 31P at 202.3 MHz) or a 600 MHz (1H at 600.1 MHz and 13C at 150.9 MHz) NMR spectrometer in DMSO-d6 or D2O solutions at 25 °C. Chemical shifts (in ppm, δ scale) were referenced to the residual solvent signal in 1H spectra (δ((CHD2)SO(CD3)) = 2.5 ppm) or to the solvent signal in 13C spectra (δ((CD3)2SO) = 39.7 ppm). 1,4-Dioxane was used as an internal standard for D2O solutions (3.75 ppm for 1H and 69.3 ppm for 13C). Coupling constants (J) are given in Hz. The complete assignment of 1H and 13C signals was performed by an analysis of the correlated homonuclear H,H-COSY, and heteronuclear H,C-HSQC and H,C-HMBC spectra. Known starting compounds dAITP, dCITP, dUNO2TP,5dCEBFTP,9EPT,37 and PT-Bpin38 were prepared by published procedures.Synthesis of modified nucleosides – Suzuki–Miyaura cross-coupling
:1 mixture of H2O–CH3CN (2 mL) was added through a septum to an argon-purged flask containing a halogenated nucleoside dNI (1 equiv.), PT-Bpin (2 equiv.) and Cs2CO3 (3 equiv.). In a separate flask, Pd(OAc)2 (10 mol%), and TPPTS (2.5 equiv. with respect to Pd) were combined, the flask was evacuated and purged with argon, and then a 1:1 mixture of H2O–CH3CN (1 mL) was added. This catalyst solution was injected into the reaction mixture, which was then stirred at 50 °C for 40 min until complete consumption of the starting material, and then evaporated in vacuo. The products were purified by silica gel column chromatography using chloroform/methanol (100:0 to 90:10) as the eluent.
Synthesis of modified nucleosides – Sonogashira cross-coupling
:0 to 90:10) as the eluent.
Synthesis of modified nucleotides triphosphates – Suzuki–Miyaura cross-coupling
:1 mixture of H2O–CH3CN (1 mL) was added through a septum to an argon-purged flask containing a halogenated nucleotide dNITP (1 equiv.), PT-Bpin (2 equiv.) and Cs2CO3 (3 equiv.). In a separate flask, Pd(OAc)2 (10 mol%), and TPPTS (2.5 equiv. with respect to Pd) were combined, the flask was evacuated and purged with argon, and then a 1:1 mixture of H2O–CH3CN (0.5 mL) was added. This catalyst solution was injected into the reaction mixture, which was then stirred at 50 °C for 40 min until complete consumption of the starting material, and then evaporated in vacuo. The product was isolated from the crude reaction mixture by HPLC on a C18 column with the use of a linear gradient of 0.1 M TEAB (triethylammonium bicarbonate) in H2O to 0.1 M TEAB in H2O–MeOH (1:1) as the eluent. Several co-distillations with water and conversion to sodium salt form (Dowex 50WX8 in Na+ cycle) followed by freeze-drying from water gave the solid product.
Synthesis of modified nucleotide triphosphates – Sonogashira cross-coupling
:1 mixture of H2O–CH3CN (2 mL) was added through a septum to an argon-purged flask containing a halogenated nucleotide dNITP (1 equiv.), PTE (1.5 equiv.), CuI (10 mol%), and (iPr)2EtN (10 equiv.). In a separate flask, Pd(OAc)2 (5 mol%), and TPPTS (2.5 equiv. with respect to Pd) were combined, the flask was evacuated and purged with argon, and then a 1:1 mixture of H2O–CH3CN (0.5 mL) was added. This catalyst solution was injected into the reaction mixture, which was then stirred at 75 °C for 1 h until complete consumption of the starting material, and then evaporated in vacuo. The product was isolated from the crude reaction mixture by HPLC on a C18 column with the use of a linear gradient of 0.1 M TEAB (triethylammonium bicarbonate) in H2O to 0.1 M TEAB in H2O–MeOH (1:1) as the eluent. Several co-distillations with water and conversion to sodium salt form (Dowex 50WX8 in Na+ cycle) followed by freeze-drying from water gave solid product.
Synthesis of modified nucleosides triphosphates – triphosphorylation
:1) as the eluent. Several co-distillations with water and conversion to the sodium salt form (Dowex 50WX8 in Na+ cycle) followed by freeze-drying from water gave the solid product.
dCPT: Compound dCPT was prepared from dCI according to the general procedure (Method A). The product was isolated as a white solid (59 mg, 75%); m.p. 145 °C; 1H NMR (600.1 MHz, DMSO-d6): 2.06 (ddd, 1H, Jgem = 13.2, J2′b,1′ = 7.2, J2′b,3′ = 6.1, H-2′b); 2.13 (ddd, 1H, Jgem = 13.2, J2′a,1′ = 6.1, J2′a,3′ = 3.7, H-2′a); 3.33 (s, 3H, CH3N); 3.49, 3.54 (2 × ddd, 2 × 1H, Jgem = 11.8, J5′,OH = 5.1, J5′,4′ = 3.7, H-5′); 3.76 (q, 1H, J4′,3′ = J4′,5′ = 3.7, H-4′); 4.23 (ddt, 1H, J3′,2′ = 6.1, 3.7, J3′,OH = 4.3, J3′,4′ = 3.7, H-3′); 4.91 (t, 1H, JOH,5′ = 5.1, OH-5′); 5.17 (d, 1H, JOH,3′ = 4.3, OH-3′); 6.20 (dd, 1H, J1′,2′ = 7.2, 6.1, H-1′); 6.37 (bs, 1H, NHaHb); 6.94–7.01 (m, 3H, H-1,7,9-phenothiazine); 7.09 (d, 1H, J4,2 = 2.1, H-4-phenothiazine); 7.14 (dd, 1H, J2,1 = 8.3, J2,4 = 2.1, H-2-phenothiazine); 7.17 (m, 1H, H-6-phenothiazine); 7.23 (m, 1H, H-8-phenothiazine); 7.31 (bs, 1H, NHaHb); 7.77 (s, 1H, H-6). 13C NMR (150.9 MHz, DMSO-d6): 35.34 (CH3N); 40.66 (CH2-2′); 61.23 (CH2-5′); 70.36 (CH-3′); 85.18 (CH-1′); 87.39 (CH-4′); 106.93 (C-5); 114.77 (CH-9-phenothiazine); 114.96 (CH-1-phenothiazine); 122.17 (C-5a-phenothiazine); 122.72 (CH-7-phenothiazine); 122.77 (C-4a-phenothiazine); 127.00 (CH-6-phenothiazine); 127.46 (CH-4-phenothiazine); 127.97 (CH-8-phenothiazine); 128.26 (C-3-phenothiazine); 128.60 (CH-2-phenothiazine); 139.83 (CH-6); 144.95 (C-10a-phenothiazine); 145.29 (C-9a-phenothiazine); 154.60 (C-2); 163.66 (C-4). MS (ESI+): m/z (%): 461.1 (100) [M+ + Na]; HRMS (ESI+): calcd 439.14345 for C22H23N4O4S, found 439.14365; calcd 461.12540 for C22H22N4O4NaS, found 461.12549.
dAPT: Compound dAPT was prepared from dAI according to the general procedure (Method A). The product was isolated as a white solid (52 mg, 96%); m.p. 204 °C; 1H NMR (500.0 MHz, DMSO-d6): 2.18 (ddd, 1H, Jgem = 13.1, J2′b,1′ = 6.0, J2′b,3′ = 2.6, H-2′b); 2.55 (ddd, 1H, Jgem = 13.1, J2′a,1′ = 8.3, J2′a,3′ = 5.8, H-2′a); 3.35 (s, 3H, CH3N); 3.50 (dd, 1H, Jgem = 11.7, J5′b,4′ = 4.4, H-5′b); 3.57 (dd, 1H, Jgem = 11.7, J5′a,4′ = 4.7, H-5′a); 3.84 (ddd, 1H, J4′,5′ = 4.7, 4.4, J4′,3′ = 2.6, H-4′); 4.35 (dt, 1H, J3′,2′ = 5.8, 2.6, J3′,4′ = 2.6, H-3′); 5.05 (bs, 1H, OH-5′); 5.27 (bs, 1H, OH-3′); 6.17 (bs, 2H, NH2); 6.57 (dd, 1H, J1′,2′ = 8.3, 6.0, H-1′); 6.94–7.02 (m, 2H, H-7,9-phenothiazine); 7.06 (d, 1H, J1,2 = 8.3, H-1-phenothiazine); 7.18 (dd, 1H, J6,7 = 7.5, J6,8 = 1.5, H-6-phenothiazine); 7.24 (ddd, 1H, J8,9 = 8.2, J8,7 = 7.3, J8,6 = 1.5, H-8-phenothiazine); 7.26 (d, 1H, J4,2 = 2.0, H-4-phenothiazine); 7.28 (dd, 1H, J2,1 = 8.3, J2,4 = 2.0, H-2-phenothiazine); 7.50 (s, 1H, H-6); 8.14 (bs, 1H, H-2). 13C NMR (125.7 MHz, DMSO-d6): 35.39 (CH3N); 39.81 (CH2-2′); 62.20 (CH2-5′); 71.25 (CH-3′); 83.06 (CH-1′); 87.54 (CH-4′); 100.55 (C-4a); 114.84 (CH-9-phenothiazine); 115.11 (CH-1-phenothiazine); 115.56 (C-5); 120.59 (CH-6); 121.96 (C-5a-phenothiazine); 122.73 (CH-7-phenothiazine); 122.92 (C-4a-phenothiazine); 126.77 (CH-2-phenothiazine); 127.06 (CH-6-phenothiazine); 127.93 (CH-4-phenothiazine); 128.05 (CH-8-phenothiazine); 128.90 (C-3-phenothiazine); 144.32 (C-10a-phenothiazine); 145.42 (C-9a-phenothiazine); 150.57 (C-7a); 151.80 (CH-2); 157.49 (C-4). MS (ESI+): m/z (%): 462.1 (40) [M+ + H]; 484.1 (100) [M+ + Na]; HRMS (ESI+): calcd 462.15944 for C24H24N5O3S, found 462.15934; calcd 484.14138 for C24H23O3N5NaS, found 484.14141.
dCEPT: Compound dCEPT was prepared from dCI according to the general procedure (Method B). The product was isolated as a yellow solid (54 mg, 76%); m.p. 143 °C; 1H NMR (600.1 MHz, DMSO-d6): 2.02 (ddd, 1H, Jgem = 13.1, J2′b,1′ = 7.0, J2′b,3′ = 6.2, H-2′b); 2.17 (ddd, 1H, Jgem = 13.1, J2′a,1′ = 6.0, J2′a,3′ = 3.6, H-2′a); 3.33 (s, 3H, CH3N); 3.58, 3.65 (2 × ddd, 2 × 1H, Jgem = 11.8, J5′,OH = 5.0, J5′,4′ = 3.6, H-5′); 3.80 (q, 1H, J4′,3′ = J4′,5′ = 3.6, H-4′); 4.23 (ddt, 1H, J3′,2′ = 6.2, 3.6, J3′,OH = 4.3, J3′,4′ = 3.6, H-3′); 5.11 (t, 1H, JOH,5′ = 5.0, OH-5′); 5.21 (d, 1H, JOH,3′ = 4.3, OH-3′); 6.13 (dd, 1H, J1′,2′ = 7.0, 6.0, H-1′); 6.95 (d, 1H, J1,2 = 8.5, H-1-phenothiazine); 6.97–7.00 (m, 2H, H-7,9-phenothiazine); 7.01 (bs, 1H, NHaHb); 7.17 (dd, 1H, J6,7 = 7.8, J6,8 = 1.5, H-6-phenothiazine); 7.23 (ddd, 1H, J8,9 = 8.2, J8,7 = 7.4, J8,6 = 1.5, H-8-phenothiazine); 7.41 (dd, 1H, J2,1 = 8.5, J2,4 = 2.0, H-2-phenothiazine); 7.44 (d, 1H, J4,2 = 2.0, H-4-phenothiazine); 7.76 (bs, 1H, NHaHb); 8.27 (s, 1H, H-6). 13C NMR (150.9 MHz, DMSO-d6): 35.44 (CH3N); 41.00 (CH2-2′); 61.08 (CH2-5′); 70.13 (CH-3′); 81.64 (C5-C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C-phenothiazine); 85.55 (CH-1′); 87.58 (CH-4′); 89.90 (C-5); 93.30 (C5-CC-phenothiazine); 114.63 (CH-1-phenothiazine); 115.08 (CH-9-phenothiazine); 116.50 (C-3-phenothiazine); 121.54 (C-5a-phenothiazine); 122.28 (C-4a-phenothiazine); 123.04 (CH-7-phenothiazine); 127.02 (CH-6-phenothiazine); 128.11 (CH-8-phenothiazine); 129.32 (CH-4-phenothiazine); 130.99 (CH-2-phenothiazine); 144.59 (CH-6); 144.82 (C-9a-phenothiazine); 145.46 (C-10a-phenothiazine); 153.53 (C-2); 163.91 (C-4). MS (ESI-): m/z (%):461.1 (100) [M − H]; HRMS (ESI−): calcd 461.12890 for C24H21N4O4S, found 461.12835.
C-phenothiazine); 85.55 (CH-1′); 87.58 (CH-4′); 89.90 (C-5); 93.30 (C5-CC-phenothiazine); 114.63 (CH-1-phenothiazine); 115.08 (CH-9-phenothiazine); 116.50 (C-3-phenothiazine); 121.54 (C-5a-phenothiazine); 122.28 (C-4a-phenothiazine); 123.04 (CH-7-phenothiazine); 127.02 (CH-6-phenothiazine); 128.11 (CH-8-phenothiazine); 129.32 (CH-4-phenothiazine); 130.99 (CH-2-phenothiazine); 144.59 (CH-6); 144.82 (C-9a-phenothiazine); 145.46 (C-10a-phenothiazine); 153.53 (C-2); 163.91 (C-4). MS (ESI-): m/z (%):461.1 (100) [M − H]; HRMS (ESI−): calcd 461.12890 for C24H21N4O4S, found 461.12835.
dAEPT: Compound dAEPT was prepared from dAI according to the general procedure (Method B). The product was isolated as a yellow solid (50 mg, 93%); m.p. 208 °C; 1H NMR (600.1 MHz, DMSO-d6): 2.20 (ddd, 1H, Jgem = 13.1, J2′b,1′ = 6.0, J2′b,3′ = 2.8, H-2′b); 2.49 (ddd, 1H, Jgem = 13.1, J2′a,1′ = 8.0, J2′a,3′ = 5.7, H-2′a); 3.34 (s, 3H, CH3N); 3.53 (ddd, 1H, Jgem = 11.8, J5′b,OH = 5.9, J5′b,4′ = 4.4, H-5′b); 3.59 (ddd, 1H, Jgem = 11.8, J5′a,OH = 5.3, J5′a,4′ = 4.4, H-5′a); 3.84 (td, 1H, J4′,5′ = 4.4, J4′,3′ = 2.5, H-4′); 4.35 (dddd, 1H, J3′,2′ = 5.7, 2.8, J3′,OH = 4.1, J3′,4′ = 2.5, H-3′); 5.06 (dd, 1H, JOH,5′ = 5.9, 5.3, OH-5′); 5.27 (d, 1H, JOH,3′ = 4.1, OH-3′); 6.51 (dd, 1H, J1′,2′ = 8.0, 6.0, H-1′); 6.69 (bs, 2H, NH2); 6.95–7.01 (m, 3H, H-1,7,9-phenothiazine); 7.17 (dd, 1H, J6,7 = 7.8, J6,8 = 1.5, H-6-phenothiazine); 7.24 (ddd, 1H, J8,9 = 8.2, J8,7 = 7.3, J8,6 = 1.5, H-8-phenothiazine); 7.40 (d, 1H, J4,2 = 2.0, H-4-phenothiazine); 7.42 (dd, 1H, J2,1 = 8.4, J2,4 = 2.0, H-2-phenothiazine); 7.83 (s, 1H, H-6); 8.20 (bs, 1H, H-2). 13C NMR (150.9 MHz, DMSO-d6): 35.43 (CH3N); 40.04 (CH2-2′); 62.05 (CH2-5′); 71.12 (CH-3′); 82.97 (C5-CC-phenothiazine); 83.35 (CH-1′); 87.71 (CH-4′); 90.57 (C5-CC-phenothiazine); 95.18 (C-5); 102.50 (C-4a); 114.79 (CH-1-phenothiazine); 115.07 (CH-9-phenothiazine); 116.50 (C-3-phenothiazine); 121.56 (C-5a-phenothiazine); 122.54 (C-4a-phenothiazine); 123.05 (CH-7-phenothiazine); 126.64 (CH-6); 127.03 (CH-6-phenothiazine); 128.11 (CH-8-phenothiazine); 129.16 (CH-4-phenothiazine); 131.06 (CH-2-phenothiazine); 144.82 (C-9a-phenothiazine); 145.53 (C-10a-phenothiazine); 149.54 (C-7a); 152.83 (CH-2); 157.81 (C-4). MS (ESI−): m/z (%): 484.1 (30) [M − H]; HRMS (ESI−): calcd 484.14488 for C26H22N5O3S, found 484.14429.
dCPTTP: Compound dCPT was prepared from dCITP according to the general procedure (Method C). The product was isolated as a white solid (30 mg, 53%); 1H NMR (500.0 MHz, D2O, ref(dioxane) = 3.75 ppm): 2.35 (ddd, 1H, Jgem = 14.0, J2′b,1′ = 7.5, J2′b,3′ = 6.6, H-2′b); 2.42 (ddd, 1H, Jgem = 14.0, J2′a,1′ = 6.3, J2′a,3′ = 3.6, H-2′a); 3.38 (s, 3H, CH3N); 4.11–4.19 (m, 2H, H-5′); 4.22 (tdd, 1H, J4′,5′ = 4.2, J4′,3′ = 3.6, JH,P = 1.2, H-4′); 4.60 (dt, 1H, J3′,2′ = 6.6, 3.6, J3′,4′ = 3.6, H-3′); 6.31 (dd, 1H, J1′,2′ = 7.5, 6.3, H-1′); 7.01 (dd, 1H, J9,8 = 8.2, J9,7 = 1.2, H-9-phenothiazine); 7.02–7.09 (m, 2H, H-1,7-phenothiazine); 7.11 (d, 1H, J4,2 = 2.1, H-4-phenothiazine); 7.23 (dd, 1H, J2,1 = 8.4, J2,4 = 2.1, H-2-phenothiazine); 7.24–7.33 (m, 2H, H-6,8-phenothiazine); 7.61 (s, 1H, H-6). 13C NMR (125.7 MHz, D2O, ref(dioxane) = 69.3 ppm): 37.47 (CH3N); 41.52 (CH2-2′); 68.02 (d, JC,P = 5.5, CH2-5′); 73.31 (CH-3′); 88.20 (d, JC,P = 8.5, CH-4′); 88.77 (CH-1′); 112.32 (C-5); 117.48 (CH-9-phenothiazine); 117.95 (CH-1-phenothiazine); 124.97 (C-5a-phenothiazine); 125.85 (CH-7-phenothiazine); 126.16 (C-4a-phenothiazine); 129.16 (C-3-phenothiazine); 129.96 (CH-6-phenothiazine); 130.09 (CH-4-phenothiazine); 130.88 (CH-8-phenothiazine); 131.73 (CH-2-phenothiazine); 142.14 (CH-6); 148.12 (C-9a-phenothiazine); 148.65 (C-10a-phenothiazine); 159.73 (C-2); 167.20 (C-4). 31P{1H} NMR (202.3 MHz, D2O): −22.04 (dd, J = 20.0, 19.6, Pβ); −11.22 (d, J = 19.6, Pα); −6.39 (d, J = 20.0, Pγ). MS (ESI-): m/z (%): 597.1 (100) [M − H − H2PO3]; HRMS (ESI−): calcd 677.02789 for C22H24O13N4P3S, found 677.02661.
dAPTTP: Compound dAPT was prepared from dAITP according to the general procedure (Method C). The product was isolated as a white solid (38 mg, 68%); 1H NMR (500.0 MHz, D2O): 2.55 (ddd, 1H, Jgem = 13.9, J2′b,1′ = 6.2, J2′b,3′ = 3.4, H-2′b); 2.81 (ddd, 1H, Jgem = 13.9, J2′a,1′ = 7.8, J2′a,3′ = 7.0, H-2′a); 3.18 (bs, 3H, CH3N); 4.11–4.24 (m, 2H, H-5′); 4.19 (td, 1H, J4′,5′ = 4.4, J4′,3′ = 3.5, H-4′); 4.79 (m, 1H, H-3′); 6.59 (dd, 1H, J1′,2′ = 7.8, 6.2, H-1′); 6.74–6.83 (m, 3H, H-1,4,9-phenothiazine); 7.00 (t, 1H, J7,6 = J7,8 = 7.5, H-7-phenothiazine); 7.18–7.27 (m, 3H, H-2,6,8-phenothiazine); 7.41 (s, 1H, H-6); 8.06 (bs, 1H, H-2). 13C NMR (150.9 MHz, D2O): 37.27 (CH3N); 40.72 (CH2-2′); 68.30 (d, JC,P = 6.0, CH2-5′); 73.75 (CH-3′); 85.27 (CH-1′); 87.74 (d, JC,P = 8.6, CH-4′); 103.39 (C-4a); 117.22 (CH-9-phenothiazine); 117.57 (CH-1-phenothiazine); 119.88 (C-5); 122.21 (CH-6); 124.72 (C-5a-phenothiazine); 125.55 (CH-7-phenothiazine); 125.66 (C-4a-phenothiazine); 129.27 (CH-4-phenothiazine); 129.84 (CH-6-phenothiazine); 130.45 (CH-2-phenothiazine); 130.49 (C-3-phenothiazine); 130.64 (CH-8-phenothiazine); 147.29 (C-10a-phenothiazine); 147.94 (C-9a-phenothiazine); 152.48 (C-7a); 153.94 (CH-2); 159.68 (C-4). 31P{1H} NMR (202.3 MHz, D2O): −21.75 (dd, J = 20.1, 19.2, Pβ); −10.88 (d, J = 19.2, Pα); −5.83 (d, J = 20.1, Pγ). MS (ESI−): m/z (%): 620.1 (100) [M − H − H2PO3]; HRMS (ESI−): calcd 700.04387 for C24H25O12N5P3S, found 700.04255.
dCEPTTP: Compound dCEPT was prepared from dCITP according to Method D in 48% yield or from dCEPT according to Method E in 45% yield. The product was isolated as a yellow solid. 1H NMR (500.0 MHz, D2O, ref(dioxane) = 3.75 ppm): 2.23 (bm, 1H, H-2′b); 2.36 (bm, 1H, H-2′a); 3.28 (s, 3H, CH3N); 4.12–4.25 (m, 3H, H-4′,5′); 4.54 (bm, 1H, H-3′); 6.11 (bt, 1H, J1′,2′ = 6.7, H-1′); 6.85 (bd, 1H, J1,2 = 8.0, H-1-phenothiazine); 6.91 (bd, 1H, J9,8 = 7.9, H-9-phenothiazine); 6.99 (bt, 1H, J7,6 = J7,8 = 7.9, H-7-phenothiazine); 7.16 (bd, 1H, J6,7 = 7.9, H-6-phenothiazine); 7.21–7.26 (bm, 2H, H-4,8-phenothiazine); 7.37 (bd, 1H, J2,1 = 8.0, H-2-phenothiazine); 7.96 (s, 1H, H-6). 13C NMR (125.7 MHz, D2O, ref(dioxane) = 69.3 ppm): 37.50 (CH3N); 41.62 (CH2-2′); 67.91 (d, JC,P = 5.0, CH2-5′); 72.88 (CH-3′); 81.98 (C5-CC-phenothiazine); 88.09 (d, JC,P = 8.4, CH-4′); 89.10 (CH-1′); 95.59 (C-5); 97.96 (C5-CC-phenothiazine); 117.14 (CH-1-phenothiazine); 117.50 (CH-9-phenothiazine); 118.32 (C-3-phenothiazine); 124.36 (C-5a-phenothiazine); 125.18 (C-4a-phenothiazine); 125.86 (CH-7-phenothiazine); 129.80 (CH-6-phenothiazine); 130.82 (CH-8-phenothiazine); 132.15 (CH-4-phenothiazine); 134.24 (CH-2-phenothiazine); 146.45 (CH-6); 147.55 (C-9a-phenothiazine); 148.77 (C-10a-phenothiazine); 158.49 (C-2); 167.22 (C-4). 31P{1H} NMR (202.3 MHz, D2O): −21.55 (bs, Pβ); −10.91 (bs, Pα); −5.70 (bs, Pγ). MS (ESI−): m/z (%): 621.1 (100) [M − H − H2PO3]; HRMS (ESI−): calcd 701.02789 for C24H24O13N4P3S, found 701.02649.
dAEPTTP: Compound dAEPT was prepared from dAITP according to Method D in 49% yield or from dAEPT according to Method E in 43% yield. The product was isolated as a yellow solid. 1H NMR (500.0 MHz, D2O): 2.35, 2.50 (2 × bm, 2 × 1H, H-2′); 3.02 (bs, 3H, CH3N); 4.03–4.17 (bm, 2H, H-5′); 4.19 (btd, 1H, J4′,5′ = 4.4, J4′,3′ = 3.5, H-4′); 4.61 (bm, 1H, H-3′); 6.20 (bt, 1H, J1′,2′ = 6.2, H-1′); 6.38 (bm, 1H, H-1-phenothiazine); 6.68 (bm, 1H, H-9-phenothiazine); 6.82–6.92 (bm, 2H, H-4,7-phenothiazine); 6.98 (bm, 1H, H-6-phenothiazine); 7.00 (bm, 1H, H-2-phenothiazine); 7.12 (bm, 1H, H-8-phenothiazine); 7.35 (s, 1H, H-6); 7.69 (bs, 1H, H-2). 13C NMR (150.9 MHz, D2O): 37.38 (CH3N); 41.11 (CH2-2′); 68.40 (d, JC,P = 4.8, CH2-5′); 73.70 (CH-3′); 84.32 (C5-CC-phenothiazine); 85.49 (CH-1′); 87.61 (d, JC,P = 8.2, CH-4′); 94.95 (C5-CC-phenothiazine); 100.04 (C-5); 105.32 (C-4a); 116.45 (CH-1-phenothiazine); 117.29 (CH-9-phenothiazine); 118.22 (C-3-phenothiazine); 124.09 (C-5a-phenothiazine); 124.62 (C-4a-phenothiazine); 125.52 (CH-7-phenothiazine); 127.64 (CH-6); 129.56 (CH-6-phenothiazine); 130.57 (CH-8-phenothiazine); 131.22 (CH-4-phenothiazine); 133.32 (CH-2-phenothiazine); 147.15 (C-9a-phenothiazine); 147.65 (C-10a-phenothiazine); 150.49 (C-7a); 153.51 (CH-2); 159.05 (C-4). 31P{1H} NMR (202.3 MHz, D2O): −22.21 (t, J = 19.2, Pβ); −11.00 (d, J = 19.2, Pα); −8.41 (d, J = 19.2, Pγ). MS (ESI−): m/z (%): 644.1 (100) [M − H − H2PO3]; HRMS (ESI−): calcd 724.04387 for C26H25O12N5P3S, found 724.04273.
Biochemistry
Single incorporation. Method A: Reaction mixture (20 μL) contained temptempA (3 μM, 1 μl), 5′-(FAM)-labeled primerrnd (3 μM, 1.5 μl), dGTP (4 mM, 0.1 μl), either dATP or dAXTPTP (4 mM, 1 μl) DNA polymerase (0.125 U KOD XL or 0.2 U Vent(exo-)) and reaction buffer (2 μl) supplied by the manufacturer. In the case of Pwo polymerase, 1.25 U of the enzyme, 0.25 μl of dGTP (4 mM) and 2 μl of either dATP or dAXTPTP (4 mM) were used. The reaction mixture was incubated for 40 minutes at 60 °C. The PEX reaction was stopped by the addition of PAGE stop solution [20 μL, formamide (80%, v/v), EDTA (20 mM), bromophenol blue (0.025%, w/v), xylene cyanol (0.025%, w/v) and Milli-Q water] and heated for 2 minutes at 95 °C. Samples were analyzed by use of 12.5% denaturing polyacrylamide gel electrophoresis (1 h, 50 °C) and visualized by fluorescence imaging. Method B: PEX reactions with temptempC were performed in the same way as described for temptempA except either dCTP or dCXTPTP (4 mM, 1 μl for KOD XL and VENT(exo-) polymerases and 2 μl for Pwo polymerase) were used.
Multiple incorporation. The reaction mixture (20 μL) contained templaternd16 (3 μM, 1 μl), 5′-(FAM)-labeled primerrnd (3 μM, 1.5 μl), dNTP (either natural or modified, 4 mM, 1 μl for KOD XL and VENT(exo-) polymerases and 2 μl for Pwo polymerase), DNA polymerase (0.25 U KOD XL, 0.5 U Vent(exo-) or 12.5 U PWO) and reaction buffer (2 μl) supplied by the manufacturer. The reaction mixture was incubated for 40 minutes at 60 °C. The PEX reaction was stopped by the addition of PAGE stop solution [20 μL, formamide (80%, v/v), EDTA (20 mM), bromophenol blue (0.025%, w/v), xylene cyanol (0.025%, w/v) and Milli-Q water] and heated for 2 minutes at 95 °C. Samples were analyzed by use of 12.5% denaturing polyacrylamide gel electrophoresis (1 h, 50 °C) and visualized by fluorescence imaging.
(B) The PCR reaction mixture (20 μL) contained KOD XL (3 U), natural dNTPs (4 mM, 0.15 μl), primers (10 μM, 4 μL, LT25TH: 5′-CAAGGACAAAATACCTGTATTCCTT-3′ and 10 μM, 4 μL, L20-: 5′-GACATCATGAGAGACATCGC-3′), and a 98-mer template (1 μM, 0.5 μL, FVL-A: 5′-GACATCATGAGAGACATCGCCTCTGGGCTAATAGGACTACTTCTAATCTGTAAGAGCAGATCCCTGGACAGGCAAGGAATACAGGTATTTTGTCCTTG-3′), in reaction buffer (2 μL) supplied by the manufacturer. Modified dNTPs (either dAPTTP or dAEPTTP) were added in combination with natural dATP (100%: dAXPTTP (4 mM, 2 μl); 90%: dAXPTTP (4 mM, 1.8 μl), dATP (4 mM, 0.2 μl); 80%: dAXPTTP (4 mM, 1.6 μl), dATP (4 mM, 0.4 μl); 50%: dAXPTTP (4 mM, 1.0 μl), dATP (4 mM, 1.0 μl)) and 30 PCR cycles were run under the following conditions: denaturation for 1 min at 95 °C, annealing for 1 min at 53 °C, extension for 1.5 min at 72 °C, followed by the final extension step of 2 min at 72 °C. Reaction mixtures were than separated by use of a 2% agarose gel with GelRed as an intercalator. Visualization was performed using an electronic dual wave transilluminator equipped with a GBox iChemi XRQ Bio imaging system (Syngene).
(C) PCR reactions were performed in the same way as described above except 5′-(FAM)-labeled primers (10 μM, 4 μL, 5′-(FAM)-LT25TH: 5′ (FAM)-CAAGGACAAAATACCTGTATTCCTT-3′ and 10 μM, 4 μL, 5′-(FAM)-L20-: 5′ (FAM)-GACATCATGAGAGACATCGC-3′) were used and visualization was performed by fluorescence imaging.
NEAR general procedure. The reaction mixture contained the template (0.125 μM), primer (0.125 μM), modified dNXTP (160 μM), natural dNTPs (125 μM), 1 × ThermoPol buffer (10 mM KCl, 10 mM (NH4)2SO4, 20 mM Tris-HCl/pH 8.8/, 0.1% Triton-X-100, and 2 mM MgSO4; 5 μL for analytical scale and 50 μL for preparative scale), and 0.5 × NEBuffer 3 (50 mM NaCl, 25 mM Tris-HCl/pH 7.9, 5 mM MgCl2, and 0.5 mM DTT; 2.5 μL for analytical scale and 25 μL for preparative scale), Vent(exo-) (5 U for analytical scale and 80 U for preparative scale), Nt·BstNBI (30 U for analytical scale and 150 U for preparative scale). The reaction mixture was incubated at 55 °C for 3 h. The reaction was stopped by cooling to 4 °C.
NEAR on analytical scale. The analytical reactions were performed according to the general procedure in a volume of 50 μL. The products were analyzed by agarose gel electrophoresis using 4% agarose gels stained with GelRed (Lab Mark). Samples were prepared by mixing 1.6 μL of 6 × DNA Loading Dye (Thermo Scientific) and 8 μL of the reaction mixture or ss DNA ladder. The gel was run for 70 min at 120 V and imaged using an electronic dual wave transilluminator equipped with a GBox iChemi XRQ Bio imaging system (Syngene).
NEAR on preparative scale. The preparative reactions were performed according to the general procedure in a volume of 500 μL. After the reaction was stopped, the solution was concentrated on a vacuum concentrator to approximately 100 μL. The viscous concentrate was injected on an HPLC XBridge OST C18 Column (waters; 2.5 μm particle size, 4.6 mm × 50 mm) and separated using a gradient of triethylammonium acetate (TEAA) and acetonitrile at a flow rate of 1 mL min−1. Mobile phase A corresponded to 0.1 M TEAA in HPLC-grade water and mobile phase B to acetonitrile/0.1 M TEAA in HPLC grade water 20/80 (v/v). The fractions containing the product were evaporated on a vacuum concentrator. The products were analyzed by MALDI-TOF mass spectrometry.
Streptavidin magnetic particles (Roche, 50 μL) were washed with binding buffer TEN100 (10 mM Tris, 1 mM EDTA, 100 mM NaCl, pH 7.5; 3 × 200 μL). The reaction mixture after PEX was diluted with binding buffer TEN100 (50 μL), and then the solution was added to the prewashed magnetic beads and incubated for 30 min at 15 °C and 1400 rpm. After the incubation, the magnetic beads were collected on a magnet (DynaMag-2, Invitrogen) and the solution was discarded. The beads were washed successively with wash buffer TEN 500 (10 mM Tris, 1 mM EDTA, 500 mM NaCl, pH 7.5; 3 × 200 μL), and water (3 × 200 μL). Then water (50 μL) was added and the sample was denatured for 2 min at 55 °C and 900 rpm. The beads were collected on a magnet and the solution was transferred into a clean vial. The product was analyzed by MALDI-TOF mass spectrometry.
Electrochemistry
Nucleosides and free PT were analyzed by conventional in situ cyclic or square wave voltammetry (CV or SWV). Purified NEAR and PEX products were analyzed by ex situ (adsorptive transfer stripping) SWV. The NEAR and PEX products were accumulated at the surface of the working basal-plane pyrolytic graphite electrode for 60 s from 5 μL aliquots containing 0.2 M NaCl. The electrode was then rinsed with deionized water and placed in the electrochemical cell. CV settings: initial potential (Ei) 0.0 V, for switching potential (Esw) see legends in Fig. 7, scan rate 1 V s−1, SWV settings: initial potential 0.0 V, end potential 1.5 V, frequency 200 Hz, amplitude 50 mV. Background electrolyte: 0.2 M sodium acetate pH 5.0. All measurements were performed at room temperature using an Autolab analyzer (Eco Chemie, The Netherlands) in connection with a VA-stand 663 (Metrohm, Herisau, Switzerland). The three-electrode system was used with an Ag|AgCl|3 M KCl electrode as the reference and platinum wire as the auxiliary electrode.Acknowledgements
This work was supported by the Academy of Sciences of the Czech Republic (RVO: 61388963, Praemium Academiae award for M. H.), by the Czech Science Foundation (P206-12-G151 to A. S., L. H., M. F. and M. H.).References
- E. Paleček and M. Bartošík, Chem. Rev., 2012, 112, 3427–3481 CrossRef PubMed.
- M. Hocek and M. Fojta, Chem. Soc. Rev., 2011, 40, 5802–5814 RSC.
- D. A. Di Giusto, W. A. Wlassoff, S. Giesebrecht, J. J. Gooding and G. C. King, J. Am. Chem. Soc., 2004, 126, 4120–4121 CrossRef CAS PubMed.
- P. Brázdilová, M. Vrábel, R. Pohl, H. Pivonková, L. Havran, M. Hocek and M. Fojta, Chem. – Eur. J., 2007, 13, 9527–9533 CrossRef PubMed.
- H. Cahová, L. Havran, P. Brázdilová, H. Pivonková, R. Pohl, M. Fojta and M. Hocek, Angew. Chem., Int. Ed., 2008, 47, 2059–2062 CrossRef PubMed.
- M. Vrábel, P. Horáková, H. Pivonková, L. Kalachova, H. Cernocká, H. Cahová, R. Pohl, P. Sebest, L. Havran, M. Hocek and M. Fojta, Chem. – Eur. J., 2009, 15, 1144–1154 CrossRef PubMed.
- J. Balintová, R. Pohl, P. Horáková, P. Vidláková, L. Havran, M. Fojta and M. Hocek, Chem. – Eur. J., 2011, 17, 14063–14073 CrossRef PubMed.
- A. A. Gorodetsky, O. Green, E. Yavin and J. K. Barton, Bioconjugate Chem., 2007, 18, 1434–1441 CrossRef CAS PubMed.
- J. Balintová, M. Plucnara, P. Vidláková, R. Pohl, L. Havran, M. Fojta and M. Hocek, Chem. – Eur. J., 2013, 19, 12720–12731 CrossRef PubMed.
- J. Balintová, J. Špaček, R. Pohl, M. Brázdová, L. Havran, M. Fojta and M. Hocek, Chem. Sci., 2015, 6, 575–587 RSC.
- P. Dauphin-Ducharme and K. W. Plaxco, Anal. Chem., 2016, 88, 11654–11662 CrossRef CAS PubMed.
- A. M. Debela, S. Thorimbert, B. Hasenknopf, C. K. O'Sullivan and M. Ortiz, Chem. Commun., 2016, 52, 757–759 RSC.
- A. M. Debela, M. Ortiz, V. Beni, S. Thorimbert, D. Lesage, R. B. Cole, C. K. O'Sullivan and B. Hasenknopf, Chem. – Eur. J., 2015, 21, 17721–17727 CrossRef CAS PubMed.
- A. Simonova, J. Balintová, R. Pohl, L. Havran, M. Fojta and M. Hocek, ChemPlusChem, 2014, 79, 1703–1712 CAS.
- L. A. Tinker and A. J. Bard, J. Am. Chem. Soc., 1979, 101, 2316–2319 CrossRef CAS.
- B. Paduszek and M. K. Kalinowski, Electrochim. Acta, 1983, 28, 639–642 CrossRef CAS.
- A. M. M. Rawashdeh, Basic Sci. Eng., 2005, 14, 195–208 Search PubMed.
- A. A. Golriz, T. Kaule, M. B. Untch, K. Kolman, R. Berger and J. S. Gutmann, ACS Appl. Mater. Interfaces, 2013, 5, 2485–2494 CAS.
- N. Nakadan, S. Imabayashi and M. Watanabe, J. Electroanal. Chem., 2009, 632, 59–63 CrossRef CAS.
- G. Viola, L. Latterini, D. Vedaldi, G. G. Aloisi, F. Dall'Acqua, N. Gabellini, F. Elisei and A. Barbafina, Chem. Res. Toxicol., 2003, 16, 644–651 CrossRef CAS PubMed.
- M. T. Tierney and M. W. Grinstaff, Org. Lett., 2000, 2, 3413–3416 CrossRef CAS PubMed.
- M. T. Tierney and M. W. Grinstaff, J. Org. Chem., 2000, 65, 5355–5359 CrossRef CAS PubMed.
- X. Hu, M. T. Tierney and M. W. Grinstaff, Bioconjugate Chem., 2002, 13, 83–89 CrossRef CAS PubMed.
- S. A. N. Hashmi, X. Hu, C. E. Immoos, S. J. Lee and M. W. Grinstaff, Org. Lett., 2002, 4, 4571–4574 CrossRef CAS PubMed.
- M. Hocek, J. Org. Chem., 2014, 79, 9914–9921 CrossRef CAS PubMed.
- A. Hottin and A. Marx, Acc. Chem. Res., 2016, 49, 418–427 CrossRef CAS PubMed.
- P. Kielkowski, J. Fanfrlík and M. Hocek, Angew. Chem., Int. Ed., 2014, 53, 7552–7555 CrossRef CAS PubMed.
- H. Cahová, A. Panattoni, P. Kielkowski, J. Fanfrlík and M. Hocek, ACS Chem. Biol., 2016, 11, 3165–3171 CrossRef PubMed.
- A. Hottin, K. Betz, K. Diederichs and A. Marx, Chem. – Eur. J., 2017, 23, 2109–2118 CrossRef CAS PubMed.
- F. Seela, A. Zulauf, H. Rosemeyer and H. Reuter, J. Chem. Soc., Perkin Trans. 2, 1996, 2373–2376 RSC.
- K. H. Shaughnessy, Molecules, 2015, 20, 9419–9454 CrossRef CAS PubMed.
- P. Ménová, V. Raindlová and M. Hocek, Bioconjugate Chem., 2013, 24, 1081–1093 CrossRef PubMed.
- J. Van Ness, L. K. Van Ness and D. J. Galas, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 4504–4509 CrossRef CAS PubMed.
- A. M. Michelson and S. H. Orkin, J. Biol. Chem., 1982, 257, 14773–14782 CAS.
- P. Horáková, H. Macíčková-Cahová, H. Pivoňková, J. Spaček, L. Havran, M. Hocek and M. Fojta, Org. Biomol. Chem., 2011, 9, 1366–1371 Search PubMed.
- H. Pivonkova, P. Horakova, M. Fojtova and M. Fojta, Anal. Chem., 2010, 82, 6807–6813 CrossRef CAS PubMed.
- C. S. Kramer and T. J. J. Muller, Eur. J. Org. Chem., 2003, 3534–3548 CrossRef.
- C. S. Kramer, T. J. Zimmermann, M. Sailer and T. J. J. Muller, Synthesis, 2002, 1163–1170 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Additional gels and copies of spectra. See DOI: 10.1039/c7ob01439b |
| This journal is © The Royal Society of Chemistry 2017 |