
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis and biological evaluation of the ascidian blood-pigment halocyamine A†
Hugo K. H.
Fong
 a,
Jean Michel
Brunel
b,
Arlette
Longeon
c,
Marie-Lise
Bourguet-Kondracki
c,
David
Barker
a and
Brent R.
Copp
*a
a,
Jean Michel
Brunel
b,
Arlette
Longeon
c,
Marie-Lise
Bourguet-Kondracki
c,
David
Barker
a and
Brent R.
Copp
*a
aSchool of Chemical Sciences, University of Auckland, 23 Symonds St, Auckland 1142, New Zealand. E-mail: b.copp@auckland.ac.nz
bCentre de Recherche en Cancérologie de Marseille (CRCM), CNRS, UMR7258, Institut Paoli Calmettes, Aix-Marseille Université, UM 105, Inserm, U1068, F-13009 Marseille, France
cLaboratoire Molécules de Communication et Adaptation des Micro-organismes, UMR 7245 CNRS, Muséum National d'Histoire Naturelle, 57 rue Cuvier (C.P. 54), 75005 Paris, France
First published on 4th July 2017
Abstract
Synthesis of the antimicrobial marine natural product halocyamine A has been achieved utilizing a combination of Sonogashira coupling, ruthenium complex/ytterbium triflate catalyzed hydroamidation and solid-phase peptide synthesis (SPPS) chemistry. The synthetic natural product exhibited only modest levels of antibacterial activities but significant antioxidant activity.
Introduction
Investigation of the natural product chemistry of blood cells of marine organisms known as ascidians over the years has led to the identification of a number of modified peptides bearing C-terminus decarboxy-enamide moieties.1 These natural products, collectively known as tunichromes, typically incorporate either a decarboxy-(E)-α,β-dehydro-3,4-dihydroxyphenyl alanine (dcΔDOPA) or a decarboxy-(E)-α,β-dehydro-3,4,5-trihydroxyphenyl alanine (dcΔTOPA) residue at the C-terminus.2 While no role has been ascribed with any confidence, potential ecological roles proposed for the tunichromes include iron or vanadium sequestration, cross-linking/tunic formation or as primitive wound repair or clotting agents.3 Unusual members of the tunichrome-family are halocyamines A (1) and B, two DOPA-containing modified tetrapeptides, isolated from the ascidian Halocynthia roretzi (Fig. 1).4 | ||
| Fig. 1 Structure of halocyamine A (1). | ||
NMR and mass spectrometry data were used to characterize the natural products, with acidic hydrolysis, labelling and HPLC analysis used to establish the configuration of the L-His and L-DOPA residues. The structures of the halocyamines were unusual additions to the tunichrome family of modified peptides in that they contained the rare Z-configuration 6-bromoindolic enamide moiety5,6 at the C-terminus. Biological evaluation of halocyamine A revealed a wide range of activities, including growth inhibition of Gram-positive bacteria,4a Gram-negative marine bacteria and fish RNA viruses.7 As part of our ongoing investigation of the synthesis and biological investigation of tunichromes,8 we now report the synthesis and structural confirmation of halocyamine A (1) and present the results of preliminary biological evaluation.
Results and discussion
Prior to attempting the synthesis of halocyamine A, we chose to target the phenylenamide-containing model compound 2 (Fig. 2).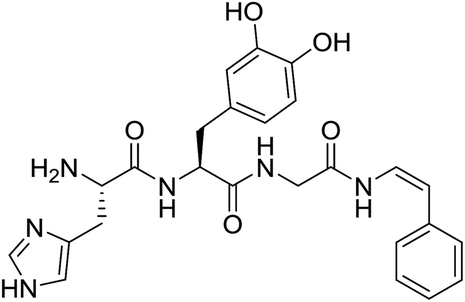 | ||
| Fig. 2 Target model compound 2. | ||
Established routes for the synthesis of enamides include dehydration9 or elimination10 methods, both of which favour the formation of the E-enamide product. In contrast, hydroamidation coupling of terminal alkynes with primary amides using a ruthenium complex/ytterbium triflate catalyst has been shown to give exclusively the Z-enamide product.11 Thus we envisaged that 2 could be prepared by reaction of the appropriately protected tripeptide L-His-L-DOPA-Gly-NH2 (3) with phenylacetylene (Scheme 1). Tripeptide 3 was prepared by standard Fmoc solid-phase peptide synthesis procedures using 2-chlorotrityl resin, protected amino acids Fmoc-His(Trt)-OH, Fmoc-DOPA(TBDMS)2-OH12 and Fmoc-Gly with HATU as the coupling agent. Cleavage from the resin using 2,2,2-trifluoroethanol afforded the protected tripeptide carboxylic acid 4 in 79% yield over seven steps. Subsequent reaction of 4 with HOBt·NH3 resulted in smooth conversion to the required tripeptide amide 3 (90% yield). Attempted hydroamidation of 3 and phenylacetylene using 5 mol% bis(2-methylallyl)(1,5-cyctooctadiene)ruthenium(II) heated at 60 °C for 6 h failed to afford the expected enamide-containing product. Unexpectedly, the only products detected were 1,4-disubstituted enynes 5 and 6, isolated as a mixture in a ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.8 in 80% yield. Comparison with literature NMR data previously reported for 5 and 6 confirmed their identities.13
:0.8 in 80% yield. Comparison with literature NMR data previously reported for 5 and 6 confirmed their identities.13
 | ||
| Scheme 1 Attempted synthesis of model enamide 2. | ||
Based on the outcome of this reaction, we speculated that the steric bulk of tripeptide-amide 3 resulted in crowding at the ruthenium catalytic centre, preventing the progress of the expected hydroamidation reaction.
We next explored an alternative route to model analogue 2: disconnection at the DOPA-Gly amide bond which would require dipeptide 7 and enamide 8 (Fig. 3). Protected dipeptide Fmoc-His(Trt)-DOPA(TBDMS)2-OH 7 was prepared by SPPS, in a similar manner to that described for 4, in 85% yield over five steps.
 | ||
| Fig. 3 Fragments 7 and 8. | ||
The synthesis of styryl enamide 8 was achieved in two steps, whereby hydroamidation of Fmoc-glycinamide (9) with phenylacetylene gave protected enamide 10 (96% yield, based upon stoichiometry of 9),14 which upon reaction with 20% piperidine/DMF gave 8 (84% yield) (Scheme 2). Alternatively, 8 could be prepared via hydroamidation of Boc-Gly-NH2 (11) with phenylacetylene to give 12 (78% yield) which was deprotected cleanly in TFA/TIS/H2O (95:2.5:2.5) to give 8 (78% yield).
 | ||
| Scheme 2 Synthesis of styryl enamide 8. | ||
With 7 and 8 in-hand, coupling using HBTU and HOBt in DMF gave 13 in 35% yield (Scheme 3). Stepwise deprotection of the N-terminus using piperidine/DMF (20 min) gave 14 (94% yield), followed by deprotection of the catechol group (triethylamine-trihydrogen fluoride, THF, 55 min) to give 15 (73% yield) and finally, removal of the trityl protecting group (0.01 N HCl/HFIP, 1 h) gave phenyl enamide model compound 2 as the dihydrochloride salt.
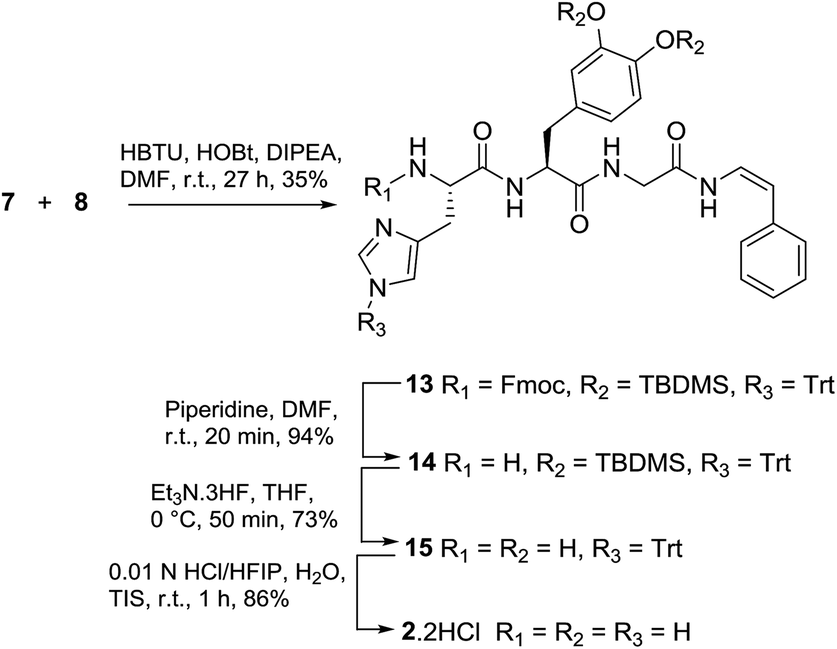 | ||
| Scheme 3 Synthesis of phenyl enamide model 2. | ||
With the successful synthesis of phenylenamide model 2, we chose to employ a similar disconnection methodology for the synthesis of halocyamine A (1), requiring protected dipeptide L-His-L-DOPA (7) and glycyl-indolic enamide 16 (Scheme 4). We have recently demonstrated that the ruthenium catalysed hydroamidation methodology allows rapid synthetic entry to indolic Z-enamides by coupling an appropriately substituted indole-3-alkyne (i.e.17) with a primary amide.15
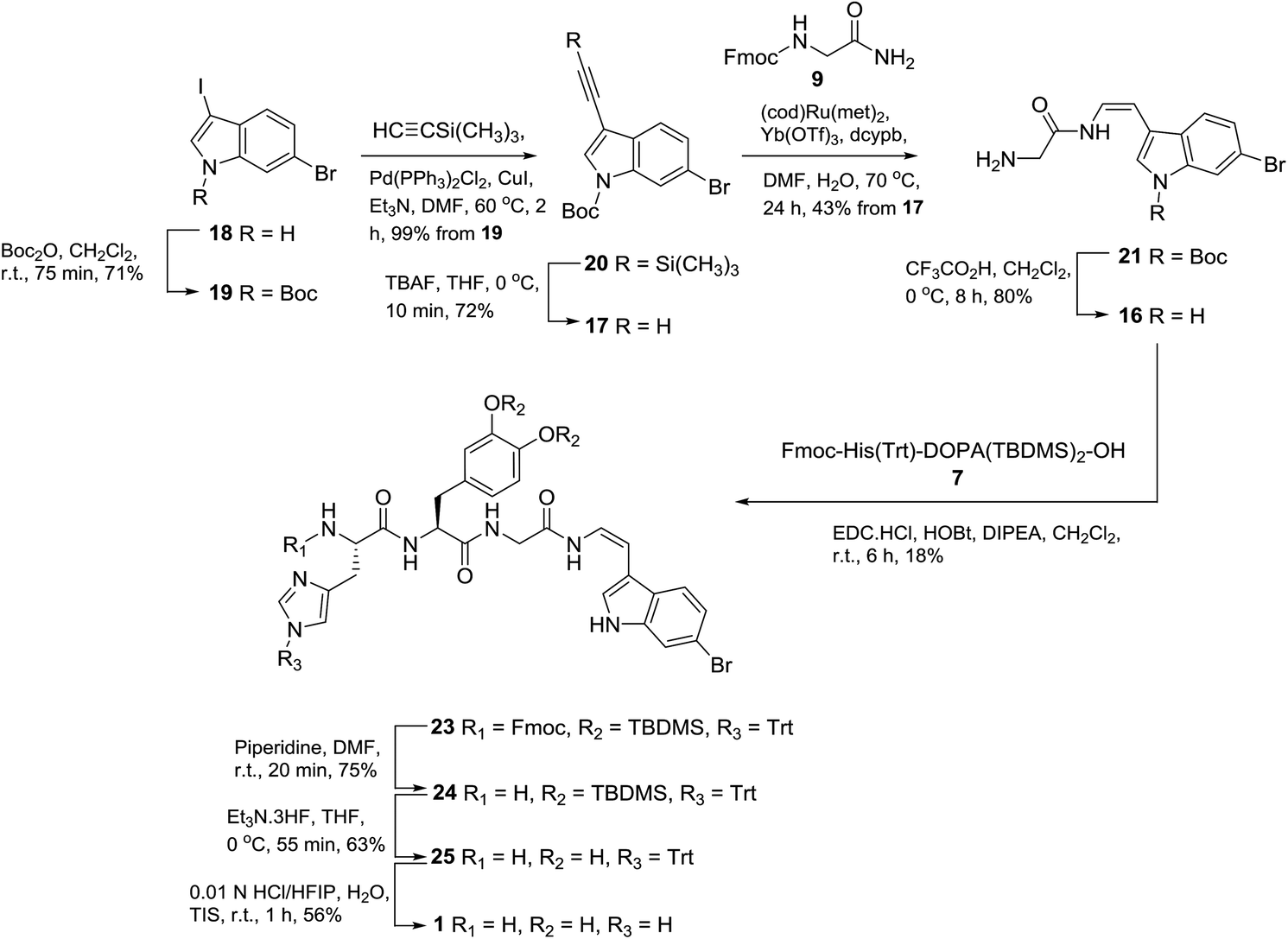 | ||
| Scheme 4 Synthesis of halocyamine A (1). | ||
Entry to 17 was achieved by Boc-protection of the known dihaloindole 1816,17 to give N-Boc-6-bromo-3-iodo indole (19), which, followed by Sonogashira alkynation, gave TMS-protected acetylene 20 in 99% yield (Scheme 4). Subsequent desilylation using TBAF in THF gave terminal acetylene 17 in 72% yield. Hydroamidation of 17 and Fmoc-glycinamide (9) using 5 mol% bis(2-methylallyl)(1,5-cyctooctadiene)ruthenium(II) heated at 70 °C for 24 h afforded glycyl enamide 21 (43%), exclusively as the Z-enamide. Of note was that the reaction product was deprotected at the N-terminus.
A minor (6%) by-product, E-enyne 22 (Fig. 4) was also purified from the product mixture. Detection of a sodiated molecular ion corresponding to C30H2879Br2N2O4Na, (observed [M + Na]+ 661.0302, calcd 661.0308) as well as two nearly identical sets of 1H NMR resonances attributable to a 3-substituted N-Boc-6-bromoindole fragment suggested 22 to be a dimer related to the starting material 17. Observation of E-alkene (δH 7.10, d, J = 16.4 Hz; δH 6.44, d, J = 16.4 Hz) and disubstituted alkyne resonances (δC 92.9, 83.0), combined with 2D NMR data analysis identified the minor product as the (E)-1,4-disubstituted enyne 22. Repeating the hydroamidation reaction in the absence of Fmoc-glycinamide afforded 22 in 28% yield. Of note, a number of transition metals are known to promote alkyne dimerization,18 including ruthenium,19 though with somewhat variable regio- (head-to-head vs. head-to-tail) and stereoselectivity.
 | ||
| Fig. 4 Structure of E-enyne 22. | ||
Removal of the Boc protecting group of enamide 21 using TFA/CH2Cl2 afforded 16 in 80% yield. Peptide coupling (EDC, HOBt, DIPEA, 6 h) of enamide 16 and dipeptide acid 7 gave protected halocyamine A 23 in a disappointing yield of 18%. Efforts to increase the yield of this reaction by altering the coupling agent (HATU or HBTU), reaction time (9 h or 24 h), and the ratio of reactants 7/16 (2:1, 1:1 or 1:2) met with no success (data not shown). Sequential Fmoc deprotection (piperidine, DMF, 20 min) gave 24 (75% yield), followed by desilylation (triethylamine trihydrofluoride, THF, 55 min) to give 25 (63% yield) and removal of the trityl group (HCl/HFIP, H2O, TIS, 1 h) gave the crude peptide that was purified by reversed-phase C8 column chromatography [H2O/MeOH] to afford halocyamine A (1) dihydrochloride salt in 56% yield. NMR (Table S1†) and optical rotation [+3.4 (c 1.07); lit.4a +5.2 (c 0.50)] data observed for synthetic 1 were in good agreement with those reported for the natural product.4a
The original reports of the halocyamines noted their abilities to inhibit the growth of Gram-positive bacteria, Gram-negative marine bacteria and fish RNA viruses.4a,7 Halocyamine A was evaluated against a panel of Gram-positive (Staphylococcus aureus ATCC 25923, Staphylococcus intermedius 1051997), Gram-negative (Pseudomonas aeruginosa ATCC 27853, Escherichia coli ATCC 25922, Enterococcus faecalis ATCC 29212), and marine Gram-negative bacteria (Vibrio harveyi ATCC 14126, Vibrio alginolyticus ATCC 17749 and Listonella anguillarum ATCC 19264) and for antioxidant activity in the DPPH radical scavenging and oxygen radical absorbance (ORAC) assays. Somewhat at odds with the original isolation report,4a,7 only modest antibacterial activity was observed for halocyamine A towards P. aeruginosa and E. faecalis, (both MIC 100 μM) and V. harveyi (IC50 129 μM); no antibacterial activity was observed against the other organisms (MIC > 200 μM). A significant antioxidant activity was observed in the DPPH assay (IC50 of 26.6 ± 2.9 μM; positive control ascorbic acid IC50 101 ± 8 μM) while in the ORAC assay, halocyamine A (1) was more active (relative ORAC value 1.29 ± 0.09) than Trolox, a water-soluble vitamin E analogue (ORAC value 1) and ascorbic acid (ORAC value 0.61 + 0.06).20
Conclusions
In summary, we have described a total synthesis of the marine natural product halocyamine A (1), making use of a ruthenium-catalysed hydroamidation of an indole acetylene with Fmoc-glycinamide to form the critical Z-enamide moiety. The natural product exhibits only mild levels of antibacterial activity. The relative ease of synthesis of halocyamine A now opens the door for future investigation of the potential ecological roles played by this unusual member of the tunichrome family of marine natural products.Experimental
General information and materials
Optical rotations were recorded using a 0.1 dm cell in methanol or dichloromethane. NMR spectra were recorded at either 500 or 400 MHz for 1H nuclei and 125 or 100 MHz for 13C nuclei. Residual solvent signals were used as reference (CD3OD: δH 3.31, δC 49.0; CDCl3: δH TMS 0, δC 77.16; DMSO-d6: δH 2.50, δC 39.52). 1H NMR data are reported as position (δ), relative integral, multiplicity (s = singlet, d = doublet, t = triplet, m = multiplet, br = broad, obs = obscured), coupling constant (J, Hz), and the assignment of the atom. 13C NMR data are reported as position (δ) and assignment of the atom. Assignments were based on 2D NMR data acquired using standard pulse sequences. (+)-ESI-MS data were acquired on a micrOTOF Q II mass spectrometer. Column chromatography was carried out with either C8 reversed-phase or silica gel. All solvents used were distilled analytical grade or better. Chemical reagents were purchased from a commercial supplier and used without purification. 6-Bromo-3-iodo-indole 18 was prepared by a literature method.16,17:15:5, 25 mL) was added and the mixture agitated for 20 min. The solution was drained and the procedure repeated. The Fmoc-glycine-loaded resin was rinsed with DMF before a solution of piperidine/DMF (10 mL, 1:4) was added to the resin and agitated for 10 min. The solution was drained and the procedure was repeated for a further 20 min. The solution was drained off while the remaining resin was washed with DMF (15 mL), isopropanol (15 mL), followed by n-hexane (15 mL). The resin was then extensively dried before storing in the desiccator for 16 h. CH2Cl2 (25 mL) was used to swell the resin, which was then drained, before a solution of Fmoc-DOPA(TBDMS)2-OH (2.59 g, 4.00 mmol), HOBt (1.01 g, 7.50 mmol), HATU (2.85 g, 7.50 mmol) and DIPEA (1.74 mL, 10.0 mmol) in DMF (7.50 mL) was added to the resin and shaken for 2 h. The solution was then drained and the resin was washed with DMF (10 mL). A solution of piperidine in DMF (10 mL, 1:4) was added to the Fmoc-DOPA(TBDMS)2-Gly-loaded resin and agitated. After 10 min, the solution was drained and the procedure was repeated for a further 20 min. The solution was drained and the DOPA(TBDMS)2-Gly-loaded resin was washed with DMF (10 mL) before a solution of Fmoc-His(Trt)-OH (3.1 g, 5.0 mmol), HOBt (1.01 g, 7.50 mmol), HATU (2.85 g, 7.50 mmol) and DIPEA (1.74 mL, 10.0 mmol) in DMF (7.50 mL) was added. The mixture was agitated for 90 min before the solution was drained. The Fmoc-His(Trt)-DOPA(TBDMS)2-Gly-loaded resin was washed with DMF (15 mL), isopropanol (15 mL), followed by n-hexane (15 mL) and was extensively dried before storing in the desiccator for 16 h. The protected peptide was cleaved from the resin using a solution of 2,2,2-trifluoroethanol in CH2Cl2 (25 mL, 1:4) to give the desired product 4 as a yellow solid (1.71 g, 79%). M.p. 130–131 °C; [α]22.7D −21.5 (c 0.14, CH2Cl2); Rf 0.52 (CH2Cl2/MeOH 9:1); IR (ATR) νmax 3278, 3036, 2930, 1662, 1508, 1446, 1251, 1128 cm−1; 1H NMR (CDCl3, 500 MHz) δ 7.75–7.69 (3H, m, NH-10, 2H-FmocAr), 7.54–7.44 (3H, m, H-6 and 2H-FmocAr), 7.32–7.24 (12H, m, NH-20, 2H-FmocAr, 9H-TrtAr), 7.20–7.18 (2H, m, 2H-FmocAr), 7.01–7.00 (6H, m, 6H-TrtAr), 6.71–6.64 (4H, m, H-8, H-14, H-17 and H-18), 6.40 (1H, br s, NH-1), 4.75 (1H, d, J = 4.4 Hz, H-11), 4.43 (1H, br s, H-2), 4.25–4.13 (3H, m, H2-21a and CO2CH2CH), 4.03 (1H, br s, CO2CH2CH), 3.69 (1H, br d, J = 17.4 Hz, H2-21b), 3.19 (1H, br d, J = 10.3 Hz, H2-12a), 3.12–3.10 (1H, m, H2-3a), 2.90–2.86 (1H, m, H2-12b), 2.80 (1H, br s, H2-3b), 0.93–0.91 (18H, m, 2SiC(CH3)3), 0.14–0.09 (12H, m, 2Si(CH3)2); 13C NMR (CDCl3, 125 MHz) δ 174.0 (C-22), 171.7 (C-19), 170.8 (C-9), 156.2 (CO2CH2CH), 146.8 (C-15), 145.7 (C-16), 144.0 (C-FmocAr), 143.9 (C-FmocAr), 141.5 (2C-FmocAr or 3C-TrtAr), 141.3 (2C-FmocAr or 3C-TrtAr), 137.5 (C-6), 134.8 (C-4), 129.9 (C-13), 129.7 (6C-TrtAr), 128.6 (3C-TrtAr), 128.4 (6C-TrtAr), 127.8 (2C-FmocAr), 127.2 (2C-FmocAr), 125.5 (C-FmocAr), 125.3 (C-FmocAr), 122.5 (C-14 or C-18), 122.0 (C-14 or C-18), 121.1 (C-17), 120.4 (C-8), 120.0 (2C-FmocAr), 76.5 (CAr3), 67.2 (COCH2CH), 55.5 (C-2), 55.1 (C-11), 47.2 (CO2CH2CH), 42.5 (C-21), 37.7 (C-12), 31.0 (C-3), 26.1 (2SiC(CH3)3), 18.5 (2SiC(CH3)3), −4.0 (2Si(CH3)2); (+)-HRESIMS [M + H]+ 1084.5052 (calcd for C63H74N5O8Si, 1084.5070).
:9 to EtOAc) afforded the desired product 3 as a white solid (0.90 g, 90%). M.p. 148–149 °C; [α]22.7D −8.1 (c 0.36, CH2Cl2); Rf 0.63 (CH2Cl2/MeOH 9:1); IR (ATR) νmax 3298, 1721, 1640, 1507, 1250 cm−1; 1H NMR (CDCl3, 500 MHz) δ 8.88 (1H, br s, NH-20), 7.76–7.73 (2H, m, 2H-FmocAr), 7.55–7.52 (2H, m, 2H-FmocAr), 7.38–7.36 (2H, m, 2H-FmocAr), 7.34–7.32 (10H, m, H-6 and 9H-TrtAr), 7.23–7.20 (2H, m, 2H-FmocAr), 7.09–7.07 (6H, m, 6H-TrtAr), 6.74 (1H, d, J = 7.6 Hz, H-17), 6.67 (1H, d, J = 1.7 Hz, H-14), 6.65 (1H, br s, H-8), 6.64 (1H, br s, NH2-23a), 6.56 (1H, d, J = 7.6 Hz, H-18), 6.44 (1H, d, J = 5.9 Hz, NH-10), 6.22 (1H, d, J = 5.0 Hz, NH-1), 5.16 (1H, br s, NH2-23b), 4.49–4.48 (1H, m, H-11), 4.33–4.28 (2H, m, CO2CH2CH), 4.23–4.22 (1H, m, H-2), 4.16–4.12 (2H, m, H2-21a and CO2CH2CH), 3.56 (1H, dd, J = 17.0, 5.1 Hz, H2-21b), 3.09 (1H, dd, J = 14.3, 5.2 Hz, H2-12a), 3.05–3.01 (2H, m, H2-3a and H2-12b), 2.82 (1H, dd, J = 15.4, 5.0 Hz, H2-3b), 0.95–0.93 (18H, m, 2SiC(CH3)3), 0.15–0.14 (12H, m, 2Si(CH3)2); 13C NMR (CDCl3, 125 MHz) δ 172.2 (C-9 and C-22), 171.3 (C-19), 156.3 (CO2CH2CH), 147.6 (C-15), 146.5 (C-16), 143.8 (2C-FmocAr), 142.2 (3C-TrtAr), 141.4 (2C-FmocAr), 138.8 (C-6), 135.5 (C-4), 129.8 (6C-TrtAr), 129.1 (C-13), 128.5 (3C-TrtAr), 128.4 (6C-TrtAr), 127.9 (2C-FmocAr), 127.2 (2C-FmocAr), 125.2 (2C-FmocAr), 121.9 (C-14 or C-18), 121.8 (C-14 or C-18), 121.5 (C-17), 120.5 (C-8), 120.2 (2C-FmocAr), 75.8 (CAr3), 67.4 (CO2CH2CH), 55.5 (C-2 and C-11), 47.2 (CO2CH2CH), 43.1 (C-21), 36.2 (C-12), 30.8 (C-3), 26.0 (2SiC(CH3)3), 18.6 (SiC(CH3)3), 18.5 (SiC(CH3)3), −3.9 (2Si(CH3)2); (+)-HRESIMS [M + H]+ 1083.5202 (calcd for C63H75N6O7Si, 1083.5230).
:1 to n-hexane/EtOAc 7:3) gave the desired product 10 as a yellow foam (0.38 g, 96%). M.p. 54–55 °C; Rf 0.68 (n-hexane/EtOAc 1:1); IR (ATR) νmax 3305, 1686, 1647, 1514, 1481, 1448, 1334, 1253 cm−1; 1H NMR (CDCl3, 500 MHz) δ 8.31 (1H, br d, J = 9.9 Hz, NH-4), 7.74 (2H, d, J = 7.2 Hz, 2H-FmocAr), 7.53 (2H, d, J = 7.2 Hz, 2H-FmocAr), 7.38 (2H, t, J = 7.2 Hz, 2H-FmocAr), 7.27 (4H, t, J = 7.2 Hz, 2H-FmocAr and 2H-9), 7.22 (2H, d, J = 7.2 Hz, 2H-8), 7.15 (1H, t, J = 7.2 Hz, H-10), 6.89 (1H, dd, J = 9.9, 9.7 Hz, H-5), 5.77 (1H, d, J = 9.7 Hz, H-6), 5.60 (1H, br s, NH-1), 4.36 (2H, d, J = 6.9 Hz, CO2CH2CH), 4.15 (1H, t, J = 6.9 Hz, CO2CH2CH), 3.87 (2H, br s, H2-2); 13C NMR (CDCl3, 125 MHz) δ 167.0 (C-3), 156.9 (CO2CH2CH), 143.7 (2C-FmocAr), 141.4 (2C-FmocAr), 135.3 (C-7), 129.1 (2C-9), 128.0 (2C-8), 127.9 (2C-FmocAr), 127.2 (2C-FmocAr and C-10), 125.1 (2C-FmocAr), 121.2 (C-5), 120.1 (2C-FmocAr), 111.5 (C-6), 67.5 (CO2CH2CH), 47.1 (CO2CH2CH), 44.9 (C-2); (+)-HRESIMS [M + Na]+ 421.1529 (calcd for C25H22N2NaO3, 421.1523).
:1 to n-hexane/EtOAc 7.5:2.5) gave the desired product 12 as a yellow oil (0.21 g, 78%). Rf 0.48 (n-hexane/EtOAc 7:3); IR (ATR) νmax 3333, 2977, 1674, 1512, 1453, 1368, 1252 cm−1; 1H NMR (CDCl3, 400 MHz) δ 8.42 (1H, d, J = 10.4 Hz, NH-4), 7.37 (2H, t, J = 7.7 Hz, 2H-9), 7.27 (2H, d, J = 7.7 Hz, 2H-8), 7.24–7.22 (1H, m, H-10), 6.93 (1H, dd, J = 10.4, 9.7 Hz, H-5), 5.79 (1H, d, J = 9.7 Hz, H-6), 5.08 (1H, br s, NH-1), 3.83 (2H, d, J = 6.0 Hz, H2-2), 1.42 (9H, s, CO2C(CH3)3); 13C NMR (CDCl3, 100 MHz) δ 167.6 (C-3), 156.3 (CO2C(CH3)3), 135.5 (C-7), 129.2 (2C-9), 128.0 (2C-8), 127.1 (C-10), 121.3 (C-5), 111.1 (C-6), 80.9 (CO2C(CH3)3), 44.9 (C-2), 28.4 (CO2C(CH3)3); (+)-HRESIMS [M + Na]+ 299.1370 (calcd for C15H20N2NaO3, 299.1366).
:1) gave the desired product 8 as a yellow oil (0.032 g, 84%).
Alternatively, a solution of TFA/TIS/H2O (0.5 mL, 95:2.5:2.5) was added to 12 (0.042 g, 0.15 mmol) and the solution was stirred at 0 °C under nitrogen for 20 min. The reaction was concentrated in vacuo. Purification using silica gel column chromatography (eluting with n-hexane/EtOAc 9:1 to EtOAc to CH2Cl2/MeOH 9:1) gave the desired product 8 as a yellow oil (0.021 g, 78%).
R
f 0.68 (n-hexane/EtOAc 1:1); IR (ATR) νmax 3305, 3023, 1677, 1645, 1503, 1477, 1442 cm−1; 1H NMR (CDCl3, 500 MHz) δ 9.85 (1H, br s, NH-4), 7.38 (2H, t, J = 7.3 Hz, 2H-9), 7.33 (2H, d, J = 7.3 Hz, 2H-8), 7.24 (1H, t, J = 7.3 Hz, H-10), 6.95 (1H, dd, J = 11.9, 9.6 Hz, H-5), 5.76 (1H, d, J = 9.6 Hz, H-6), 3.43 (2H, s, H2-2); 13C NMR (CDCl3, 125 MHz) δ 170.7 (C-3), 136.0 (C-7), 129.0 (2C-9), 128.0 (2C-8), 126.9 (C-10), 121.2 (C-5), 110.6 (C-6), 44.6 (C-2); (+)-HRESIMS [M + H]+ 177.1023 (calcd for C10H13N2O, 177.1022).
:15:5) was added to the mixture and shaken for 20 min. The solution was drained and the procedure was repeated. The resin was then washed with DMF (20 mL). Piperidine in DMF (15 mL, 1:4) was added to the resin mixture and shaken for 10 min. The liquid was drained off and the piperidine washing was repeated for another 20 min. The amino acid-loaded resin was thoroughly washed with DMF (20 mL), isopropanol (20 mL) and n-hexane (20 mL). The resin was dried under vacuum for 30 min and stored in a desiccator overnight. CH2Cl2 (40 mL) was added to the amino acid-loaded resin which was left to swell for 1 h. The solution was drained and a solution of HBTU (4.39 g, 11.6 mmol), HOBt (1.57 g, 11.6 mmol), Fmoc-His(Trt)-OH (4.79 g, 7.72 mmol) and DIPEA (2.69 mL, 15.4 mmol) in DMF (11.6 mL) was added. The amino acid resin mixture was agitated for 2 h. The solution was then drained and washed with DMF (20 mL), isopropanol (20 mL) and n-hexane (20 mL). The resin was extensively dried and stored in a desiccator overnight. 2,2,2-Trifluoroethanol in CH2Cl2 (11.7 mL, 1:4) was added to the amino acid-loaded resin and agitated for 1 h. The solution was drained and the organic solvent was removed in vacuo to afford 7 as a yellowish-brown foam (2.70 g, 85% yield). M.p. 129–130 °C; [α]21.9D −5.7 (c 0.71, CH2Cl2); Rf 0.61 (CH2Cl2/MeOH 9:1); IR (ATR) νmax 3320, 2930, 2857, 1723, 1655, 1509, 1446, 1251, 1128 cm−1; 1H NMR (CDCl3, 500 MHz) δ 7.94 (1H, br s, NH-10), 7.75 (2H, d, J = 7.3 Hz, 2H-FmocAr), 7.55–7.51 (3H, m, H-6 and 2H-FmocAr), 7.38 (2H, t, J = 7.3 Hz, 2H-FmocAr), 7.29–7.28 (2H, m, 2H-FmocAr), 7.26–7.23 (9H, m, 9H-TrtAr), 7.03–7.01 (6H, m, 6H-TrtAr), 6.75 (2H, br s, H-14 and H-17), 6.67–6.65 (2H, m, H-18 and H-8), 6.02 (1H, d, J = 7.8 Hz, NH-1), 4.72–4.66 (2H, m, H-2 and H-11), 4.29 (1H, dd, J = 12.9, 11.0 Hz, CO2CH2CH-a), 4.09–4.05 (2H, m, CO2CH2CH-b and CO2CH2CH), 3.33 (1H, d, J = 12.7 Hz, H2-3a), 3.07 (1H, dd, J = 13.6, 4.7 Hz, H2-12a), 3.01–2.97 (1H, m, H2-12b), 2.64 (1H, dd, J = 12.7, 12.7 Hz, H2-3b), 0.92–0.89 (18H, m, 2SiC(CH3)3), 0.11–0.08 (12H, m, 2Si(CH3)2); 13C NMR (CDCl3, 125 MHz) δ 175.6 (C-19), 170.7 (C-9), 155.7 (CO2CH2CH), 146.4 (C-16), 145.5 (C-15), 144.1 (C-FmocAr), 143.9 (C-FmocAr), 141.5 (3C-TrtAr), 141.3 (2C-FmocAr), 137.7 (C-6), 134.8 (C-4), 130.6 (C-13), 129.8 (6C-TrtAr), 128.5 (3C-TrtAr), 128.4 (6C-TrtAr), 127.8 (2C-FmocAr), 127.2 (2C-FmocAr), 125.6 (C-FmocAr), 125.3 (C-FmocAr), 122.7 (C-14 or C-18), 122.5 (C-14 or C-18), 120.9 (C-17), 120.3 (C-8), 120.0 (2C-FmocAr), 76.3 (CAr3), 67.1 (CO2CH2CH), 55.3 (C-2 or C-11), 55.0 (C-2 or C-11), 47.3 (CO2CH2CH), 38.4 (C-12), 32.4 (C-3), 26.1 (SiC(CH3)3), 26.0 (SiC(CH3)3), 18.5 (SiC(CH3)3), 18.4 (SiC(CH3)3), −3.9 (Si(CH3)2), −4.0 (Si(CH3)2); (+)-HRESIMS [M + H]+ 1027.4890 (calcd for C61H71N4O7Si2, 1027.4856).
:2 to n-hexane/EtOAc 6:4) gave the desired product 13 as a yellow foam (30.0 mg, 35%). [α]22.9D −1.2 (c 0.83, CH2Cl2). M.p. 94–96 °C; Rf 0.57 (n-hexane/EtOAc 1:1); IR (ATR) νmax 3301, 3025, 2929, 1652, 1509, 1493, 1252 cm−1; 1H NMR (CDCl3, 500 MHz) δ 9.26 (1H, br s, NH-20), 8.51 (1H, d, J = 10.7 Hz, NH-23), 7.75 (1H, d, J = 6.5 Hz, H-FmocAr), 7.73 (1H, d, J = 5.5 Hz, H-FmocAr), 7.54 (1H, d, J = 7.5 Hz, H-FmocAr), 7.51 (1H, d, J = 7.5 Hz, H-FmocAr), 7.38–7.34 (4H, m, 2H-28 and 2H-FmocAr), 7.31–7.30 (7H, m, 6H-TrtAr and H-6), 7.26–7.25 (4H, m, 2H-27 and 2H-FmocAr), 7.23–7.19 (3H, m, 3H-TrtAr), 7.07–7.05 (7H, m, H-29 and 6H-TrtAr), 6.75 (1H, dd, J = 10.7, 10.0 Hz, H-24), 6.72 (1H, d, J = 8.1 Hz, H-17), 6.66 (1H, s, H-14), 6.59 (1H, s, H-8), 6.52 (1H, dd, J = 8.1, 1.4 Hz, H-18), 6.15 (1H, d, J = 6.5 Hz, NH-10), 6.03 (1H, d, J = 5.4 Hz, NH-1), 5.68 (1H, d, J = 10.0 Hz, H-25), 4.69 (1H, ddd, J = 6.5, 6.5, 6.5 Hz, H-11) 4.31–4.23 (2H, m, CO2CH2CH), 4.18–4.17 (1H, m, H-2), 4.13 (1H, t, J = 7.2 Hz, CO2CH2CH), 3.99 (1H, dd, J = 16.3, 5.8 Hz, H2-21a), 3.84 (1H, dd, J = 16.3, 5.8 Hz, H2-21b), 3.07 (1H, dd, J = 14.7, 6.5 Hz, H2-12a), 3.00–2.95 (2H, m, H2-3a and H2-12b), 2.69 (1H, dd, J = 14.4, 3.6 Hz, H2-3b), 0.96–0.94 (18H, m, 2SiC(CH3)3), 0.16–0.15 (12H, m, 2Si(CH3)2); 13C NMR (CDCl3, 125 MHz) δ 172.3 (C-19), 170.7 (C-9), 167.5 (C-22), 156.1 (CO2CH2CH), 147.4 (C-15), 146.2 (C-16), 143.9 (C-FmocAr), 143.8 (C-FmocAr), 142.2 (3C-TrtAr), 141.4 (2C-FmocAr), 138.7 (C-6), 135.6 (C-26), 135.4 (C-4), 129.8 (6C-TrtAr), 129.3 (C-13), 129.1 (2C-28), 128.4 (3C-TrtAr), 128.3 (2C-27 and 6C-TrtAr), 127.9 (2C-FmocAr), 127.2 (2C-FmocAr), 127.1 (C-29), 125.3 (2C-FmocAr), 122.0 (C-18), 121.9 (C-14), 121.4 (C-17 and C-24), 120.7 (C-8), 120.1 (2C-FmocAr), 111.2 (C-25), 75.7 (CAr3), 67.2 (CO2CH2CH), 54.9 (C-2), 54.3 (C-11), 47.3 (CO2CH2CH), 44.0 (C-21), 36.3 (C-12), 31.0 (C-3), 26.1 (2SiC(CH3)3), 18.6 (2SiC(CH3)3), −3.9 (2Si(CH3)2); (+)-HRESIMS [M + H]+ 1185.5726 (calcd for C71H81N6O7Si2, 1185.5700).
:1), afforded the desired product 14 as a yellow oil (59.4 mg, 94%). [α]21.9D −24.2 (c 0.91, CH2Cl2); Rf 0.49 (CH2Cl2/MeOH 9:1); IR (ATR) νmax 3312, 2929, 2857, 1650, 1508, 1444, 1252, 1128 cm−1; 1H NMR (CDCl3, 400 MHz) δ 8.60 (1H, dd, J = 5.7, 5.6 Hz, NH-20), 8.51 (1H, d, J = 10.8 Hz, NH-23), 7.38–7.27 (15H, m, 9H-TrtAr, H-6, NH-10, 2H-27 and 2H-28), 7.20 (1H, t, J = 7.4 Hz, H-29), 7.09–7.06 (6H, m, 6H-TrtAr), 6.78 (1H, dd, J = 10.8, 9.8 Hz, H-24), 6.71 (1H, d, J = 8.1 Hz, H-17), 6.65 (1H, d, J = 2.0 Hz, H-14), 6.59 (1H, s, H-8), 6.54 (1H, dd, J = 8.1, 2.0 Hz, H-18), 5.71 (1H, d, J = 9.8 Hz, H-25), 4.45 (1H, ddd, J = 7.1, 7.1, 7.1 Hz, H-11), 3.93 (1H, dd, J = 16.3, 5.6 Hz, H2-21a), 3.87 (1H, dd, J = 16.3, 5.7 Hz, H2-21b), 3.40 (1H, dd, J = 5.5, 5.5 Hz, H-2), 3.09 (1H, dd, J = 14.1, 7.1 Hz, H2-12a), 2.98 (1H, dd, J = 14.1, 7.1 Hz, H2-12b), 2.82 (1H, dd, J = 14.9, 5.5 Hz, H2-3a), 2.71 (1H, dd, J = 14.9, 5.5 Hz, H2-3b), 0.97–0.96 (18H, m, 2SiC(CH3)3), 0.17–0.16 (12H, m, 2Si(CH3)2); 13C NMR (CDCl3, 125 MHz) δ 174.3 (C-9)a, 172.5 (C-19), 167.6 (C-22), 147.0 (C-15), 145.9 (C-16), 142.3 (3C-TrtAr), 138.8 (C-6), 136.4 (C-4), 135.5 (C-26), 130.2 (C-13), 129.8 (6C-TrtAr), 129.1 (2C-28), 128.3 (9C-TrtAr and 2C-27), 127.0 (C-29), 122.2 (C-18), 122.0 (C-14), 121.4 (C-24), 121.2 (C-17), 120.2 (C-8), 111.4 (C-25), 75.6 (CAr3), 55.2 (C-11), 54.7 (C-2), 44.0 (C-21), 36.1 (C-12), 32.3 (C-3), 26.1 (2SiC(CH3)3), 18.6 (SiC(CH3)3), 18.5 (SiC(CH3)3), −3.9 (2Si(CH3)2); (+)-HRESIMS [M + H]+ 963.5055 (calcd for C56H71N6O5Si2, 963.5019). a assignment by HMBC correlation.
:9), afforded 15 as a yellow oil (30.5 mg, 73%). [α]22.7D −21.9 (c 1.42, CH2Cl2); Rf 0.26 (CH2Cl2/MeOH 9:1); IR (ATR) νmax 3277, 3057, 2926, 1651, 1508, 1486, 1260 cm−1; 1H NMR (CD3OD, 500 MHz) δ 7.42 (1H, d, J = 1.0 Hz, H-6), 7.36–7.33 (13H, m, 2H-27, 2H-28 and 9H-TrtAr), 7.20–7.17 (1H, m, H-29) 7.14–7.12 (6H, m, 6H-TrtAr), 6.74 (1H, d, J = 9.7 Hz, H-24), 6.70 (1H, s, H-8), 6.64 (1H, d, J = 1.9 Hz, H-14), 6.58 (1H, d, J = 8.1 Hz, H-17), 6.44 (1H, dd, J = 8.1, 1.9 Hz, H-18), 5.77 (1H, d, J = 9.7 Hz, H-25), 4.49 (1H, dd, J = 9.0, 5.6 Hz, H-11), 3.98 (1H, d, J = 16.8 Hz, H2-21a), 3.84 (1H, d, J = 16.8 Hz, H2-21b), 3.72 (1H, t, J = 5.6 Hz, H-2), 2.98 (1H, dd, J = 13.8, 5.6 Hz, H2-12a), 2.78 (1H, dd, J = 14.8, 5.6 Hz, H2-3a), 2.73–2.66 (2H, m, H2-3b and H2-12b); 13C NMR (CD3OD, 125 MHz) δ 174.3 (C-9 or C-19), 174.2 (C-9 or C-19), 169.7 (C-22), 146.3 (C-15), 145.3 (C-16), 143.6 (3C-TrtAr), 140.0 (C-6), 136.9 (C-4), 136.7 (C-26), 130.9 (6C-TrtAr), 129.8 (3C-TrtAr), 129.5 (C-13), 129.4 (2C-27 or 2C-28), 129.34 (2C-27 or 2C-28), 129.28 (6C-TrtAr), 128.0 (C-29), 122.1 (C-24), 121.64 (C-8 or C-18), 121.57 (C-8 or C-18), 117.4 (C-14), 116.3 (C-17), 113.4 (C-25), 76.9 (CAr3), 56.3 (C-11), 55.2 (C-2), 43.7 (C-21), 38.1 (C-12), 32.7 (C-3); (+)-HRESIMS [M + H]+ 735.3292 (calcd for C44H43N6O5, 735.3289).
:2.5:2.5) was added to 15 (35.0 mg, 47.7 μmol) and the solution was stirred at r.t. for 1 h. The solution was then dried under nitrogen and the crude product was purified by C8 column chromatography (eluting with H2O to H2O/MeOH 6:4) to afford the desired product 2 as a white solid (18.0 mg, 77%). M.p. 240 °C (decomposed); [α]20.9D −5.7 (c 3.12, MeOH); Rf 0.66 (butan-1-ol/acetic acid/water 2:1:1); IR (ATR) νmax 3023, 2924, 1653, 1517, 1493, 1445, 1260, 1078, 1031 cm−1; 1H NMR (DMSO-d6, 500 MHz) δ 9.54 (1H, d, J = 10.2 Hz, NH-23), 8.65 (1H, br s, H-20), 8.08 (1H, br s, NH-10), 7.54 (1H, s, H-6), 7.39–7.34 (4H, m, 2H-27 and 2H-28), 7.22 (1H, tt, J = 6.8, 1.8 Hz, H-29), 6.81 (1H, br s, H-8), 6.76 (1H, dd, J = 10.2, 10.0 Hz, H-24), 6.60–6.59 (2H, m, H-14 and H-17), 6.41 (1H, dd, J = 8.2, 2.0 Hz, H-18), 5.69 (1H, d, J = 10.0 Hz, H-25), 4.44 (1H, br s, H-11), 3.96 (1H, dd, J = 16.7, 5.4 Hz, H2-21a), 3.89 (1H, dd, J = 16.7, 5.4 Hz, H2-21b), 3.34 (1H, dd, J = 8.2, 4.1 Hz, H-2), 2.90 (1H, dd, J = 13.9, 4.3 Hz, H2-12a), 2.78 (1H, dd, J = 14.4, 4.1 Hz, H2-3a), 2.67 (1H, dd, J = 13.9, 9.0 Hz, H2-12b), 2.48 (1H, d, J = 8.2 Hz, H2-3b); 13C NMR (DMSO-d6, 125 MHz) δ 174.1 (C-9), 171.9 (C-19), 168.1 (C-22), 144.8 (C-15), 143.7 (C-16), 135.4 (C-26), 134.9 (C-6), 128.6 (2C-28), 128.4 (C-13), 128.2 (2C-27), 126.5 (C-29), 121.7 (C-24), 120.0 (C-18), 116.6 (C-14), 115.2 (C-17), 110.2 (C-25), 54.9 (C-2), 53.7 (C-11), 42.4 (C-21), 37.1 (C-12), 32.1 (C-3); (+)-HRESIMS [M + H]+ 493.2182 (calcd for C25H29N6O5, 493.2194).
16,17 (0.66 g, 2.0 mmol), DMAP (0.02 g, 0.2 mmol) and di-tert-butyl dicarbonate (0.67 g, 3.1 mmol) were dissolved in CH2Cl2 (4 mL) and stirred for 75 min at r.t. under nitrogen. 10% aqueous HCl (20 mL) was added and extracted with CH2Cl2 (3 × 30 mL). The organic layers were combined, dried (MgSO4) and filtered. The solvent was then removed in vacuo. The crude product was subjected to silica gel column chromatography (n-hexane) to yield 19 as an orange solid (0.86 g, 71% yield). M.p. 148–149 °C; Rf 0.70 (n-hexane/EtOAc 9:1); IR (ATR) νmax 2986, 1732, 1602, 1427, 1365, 1244, 1115 cm−1; 1H NMR (CDCl3, 400 MHz) δ 8.35 (1H, br s, H-7), 7.68 (1H, s, H-2), 7.42 (1H, dd, J = 8.3, 1.8 Hz, H-5), 7.25 (1H, d, J = 8.3 Hz, H-4), 1.67 (9H, s, CO2C(CH3)3); 13C NMR (CDCl3, 100 MHz) δ 148.4 (CO2C(CH3)3), 135.5 (C-7a), 131.2 (C-3a), 130.7 (C-2), 126.8 (C-5), 122.8 (C-4), 119.5 (C-6), 118.4 (C-7), 85.1 (CO2C(CH3)3), 65.0 (C-3), 28.2 (CO2C(CH3)3); (+)-HRESIMS [M + Na]+ 443.9060 (calcd for C13H1379Br126INNaO2, 443.9067).
:1) to afford 20 as a brown oil (0.489 g, 99% yield). Rf 0.93 (n-hexane/EtOAc 9:1); IR (ATR) νmax 2980, 2162, 1734, 1432, 1364, 1247, 1154, 1092 cm−1; 1H NMR (CDCl3, 400 MHz) δ 8.35 (1H, br s, H-7), 7.72 (1H, s, H-2), 7.51 (1H, d, J = 8.4 Hz, H-4), 7.41 (1H, dd, J = 8.4, 1.9 Hz, H-5), 1.66 (9H, s, CO2C(CH3)3), 0.28 (9H, s, Si(CH3)3); 13C NMR (CDCl3, 100 MHz) δ 148.8 (CO2C(CH3)3), 135.3 (C-7a), 129.9 (C-2), 129.5 (C-3a), 126.6 (C-5), 121.4 (C-4), 119.1 (C-6), 118.6 (C-7), 103.6 (C-3), 98.8 (C-9), 96.2 (C-8), 85.0 (CO2C(CH3)3), 28.2 (CO2C(CH3)3), 0.2 (Si(CH3)3); (+)-HRESIMS [M + Na]+ 414.0495 (calcd for C18H2279BrNNaO2Si, 414.0495).
:1); IR (ATR) νmax 3292, 2987, 1735, 1456, 1364, 1312, 1251 cm−1; 1H NMR (CDCl3, 400 MHz) δ 8.31 (1H, br s, H-7), 7.73 (1H, s, H-2), 7.47 (1H, d, J = 8.3 Hz, H-4), 7.37 (1H, dd, J = 8.3, 1.8 Hz, H-5), 3.23 (1H, s, H-9), 1.66 (9H, s, CO2C(CH3)3); 13C NMR (CDCl3, 100 MHz) δ 148.6 (CO2C(CH3)3), 135.2 (C-7a), 130.2 (C-2), 129.3 (C-3a), 126.6 (C-5), 121.1 (C-4), 119.1 (C-6), 118.5 (C-7), 102.3 (C-3), 85.0 (CO2C(CH3)3), 81.2 (C-9), 75.2 (C-8), 28.1 (CO2C(CH3)3); (+)-HRESIMS [M + Na]+ 342.0099 (calcd for C15H1479BrNNaO2, 342.0100).
:1) gave enyne 22 as a brown oil (37.2 mg, 6% yield) and (eluting with n-hexane/EtOAc 9:1 to EtOAc) 21 as a yellow oil (0.164 g, 43% yield).
:1); IR (ATR) νmax 2980, 2934, 2860, 1736, 1602, 1431, 1360, 1249, 1081 cm−1; 1H NMR (CDCl3, 400 MHz) δ 8.35 (2H, d, J = 9.9 Hz, H-7 and H-15), 7.72 (1H, s, H-13), 7.65 (1H, s, H-2), 7.59 (1H, d, J = 8.4 Hz, H-4), 7.54 (1H, d, J = 8.3 Hz, H-18), 7.40 (1H, dd, J = 8.3, 1.7 Hz, H-17), 7.39 (1H, dd, J = 8.4, 1.7 Hz, H-5), 7.10 (1H, d, J = 16.4 Hz, H-8), 6.44 (1H, d, J = 16.4 Hz, H-9), 1.68 (18H, s, 3H3-21 and 3H3-24); 13C NMR (CDCl3, 100 MHz) δ 149.0 (C-19), 148.7 (C-22), 136.7 (C-7a), 135.4 (C-14a), 132.2 (C-8), 129.3 (C-18a), 128.9 (C-13), 127.0 (C-3a), 126.6 (C-5 and C-17), 126.5 (C-5 and C-17), 125.0 (C-2), 121.3 (C-18), 121.0 (C-4), 119.1 (C-6 or C-16), 118.8 (C-3 and C-7 or C-15), 118.6 (C-7 or C-15), 118.3 (C-6 or C-16), 108.3 (C-9), 103.7 (C-12), 92.9 (C-10), 85.0 (C-20 or C-23), 84.9 (C-20 or C-23), 83.0 (C-11), 28.2 (3C-21 and 3C-24); (+)-HRESIMS [M + Na]+ 661.0302 (calcd for C30H2879Br2N2O4Na, 661.0308).
:1) to give 16 as a black oil (0.10 g, 80% yield). Rf 0.90 (CH2Cl2/MeOH 9:1); IR (ATR) νmax 3209, 2931, 1652, 1532, 1455, 1227, 1002 cm−1; 1H NMR (DMSO-d6, 400 MHz) δ 11.73 (1H, br s, NH-9), 7.69 (1H, br, s, H-8), 7.62 (1H, d, J = 1.8 Hz, H-10), 7.56 (1H, d, J = 8.5 Hz, H-13), 7.17 (1H, dd, J = 8.5, 1.8 Hz, H-12), 6.73 (1H, d, J = 9.4 Hz, H-5), 5.95 (1H, d, J = 9.4 Hz, H-6), 3.53 (2H, br s, H2-2); 13C NMR (DMSO-d6, 100 MHz) δ 168.6 (C-3), 136.7 (C-9a), 125.7 (C-13a), 124.5 (C-8), 122.0 (C-12), 120.3 (C-13), 118.3 (C-5), 114.6 (C-11), 114.3 (C-10), 110.0 (C-7), 102.2 (C-6), 42.8 (C-2); (+)-HRESIMS [M + Na]+ 316.0060 (calcd for C12H1279BrN3NaO, 316.0056).
:2 to n-hexane/EtOAc 4:6) gave 23 as a yellow oil (61.2 mg, 18% yield). Rf 0.61 (CH2Cl2/MeOH 9:1); [α]23.0D +12.6 (c 1.03, CH2Cl2); IR (ATR) νmax 3317, 2930, 2857, 1657, 1506, 1446, 1251, 1229, 1158, 1129, 1041 cm−1; 1H NMR (CDCl3, 500 MHz) δ 9.06 (1H, br s, NH-28), 9.00 (1H, br s, NH-20), 8.21 (1H, d, J = 10.7 Hz, H-23), 7.74 (1H, d, J = 7.7 Hz, H-FmocAr), 7.73 (1H, d, J = 7.7 Hz, H-FmocAr), 7.52 (1H, d, J = 7.7 Hz, H-FmocAr), 7.50 (1H, d, J = 7.7 Hz, H-FmocAr), 7.49 (1H, br s, H-29), 7.41 (1H, d, J = 8.3 Hz, H-32), 7.37 (2H, t, J = 7.7 Hz, 2H-FmocAr), 7.32 (1H, s, H-6), 7.28–7.23 (11H, m, 2H-FmocAr, 9H-TrtAr), 7.21 (1H, d, J = 8.3 Hz, H-31), 7.16 (1H, br s, H-27), 6.99–6.98 (6H, m, 6H-TrtAr), 6.75–6.68 (3H, m, H-14, H-17 and H-24), 6.58 (1H, br s, H-8), 6.54 (1H, d, J = 7.6 Hz, H-18), 6.30 (1H, br s, NH-10), 5.99 (1H, br s, NH-1), 5.79 (1H, d, J = 9.1 Hz, H-25), 4.78–4.76 (1H, m, H-11), 4.33–4.30 (3H, m, H-2 and CO2CH2CH), 4.23–4.18 (1H, m, H2-21a), 4.15 (1H, t, J = 6.9 Hz, CO2CH2CH), 3.70 (1H, dd, J = 16.7, 4.2 Hz, H2-21b), 3.11 (1H, dd, J = 14.1, 6.1 Hz, H2-12a), 2.98 (1H, dd, J = 14.8, 4.2 Hz, H2-3a), 2.94 (1H, dd, J = 14.1, 6.1 Hz, H2-12b), 2.77 (1H, dd, J = 14.8, 4.2 Hz, H2-3b), 0.96–0.95 (18H, m, 2SiC(CH3)3), 0.16 (12H, s, 2Si(CH3)2); 13C NMR (CDCl3, 125 MHz) δ 172.0 (C-19), 170.8 (C-9), 166.6 (C-22), 156.3 (CO2CH2CH), 147.4 (C-15), 146.4 (C-16), 143.8 (C-FmocAr), 143.7 (C-FmocAr), 142.0 (3C-TrtAr), 141.4 (2C-FmocAr), 138.8 (C-6), 136.6 (C-28a), 135.2 (C-4), 129.7 (6C-TrtAr), 129.0 (C-13), 128.3 (9C-TrtAr), 128.0 (2C-FmocAr), 127.2 (2C-FmocAr), 126.0 (C-32a), 125.1 (2C-FmocAr), 123.4 (C-27), 123.2 (C-31), 122.3 (C-18), 122.0 (C-14), 121.4 (C-17), 120.8 (C-8), 120.2 (2C-FmocAr and C-32), 119.3 (C-24), 116.3 (C-30), 114.4 (C-29), 111.2 (C-26), 102.8 (C-25), 75.8 (CAr3), 67.3 (CO2CH2CH), 54.9 (C-2), 54.0 (C-11), 47.3 (CO2CH2CH), 43.9 (C-21), 36.6 (C-12), 30.9 (C-3), 26.0 (2SiC(CH3)3), 18.6 (2SiC(CH3)3), −3.9 (2Si(CH3)2); (+)-HRESIMS [M + H]+ 1302.4954 (calcd for C73H8179BrN7O7Si2, 1302.4914).
:1), afforded 24 as a yellow oil (15.6 mg, 75% yield). Rf 0.59 (CH2Cl2/MeOH 9:1); [α]20.4D −20.5 (c 1.07, CH2Cl2); IR (ATR) νmax 3257, 2929, 1663, 1509, 1445, 1252, 1202, 1129, 1023 cm−1; 1H NMR (CDCl3, 500 MHz) δ 9.58 (1H, br s, NH-28), 8.70 (1H, t, J = 5.9 Hz, NH-20), 8.22 (1H, d, J = 10.7 Hz, NH-23), 7.50 (1H, d, J = 1.6 Hz, H-29), 7.42 (1H, d, J = 8.5 Hz, H-32), 7.34 (1H, d, J = 0.9 Hz, H-6), 7.30–7.25 (9H, m, 9H-TrtAr), 7.22–7.19 (2H, m, H-27 and H-31), 7.01–6.99 (6H, m, 6H-TrtAr), 6.88 (1H, d, J = 7.5 Hz, NH-10), 6.73 (1H, d, J = 8.2 Hz, H-17), 6.71 (1H, dd, J = 10.7, 9.2 Hz, H-24), 6.67 (1H, d, J = 2.1 Hz, H-14), 6.60 (1H, d, J = 0.9 Hz, H-8), 6.56 (1H, dd, J = 8.2, 2.1 Hz, H-18), 5.80 (1H, d, J = 9.2 Hz, H-25), 4.68 (1H, ddd, J = 7.5, 7.5, 7.5 Hz, H-11), 4.14 (1H, dd, J = 16.9, 5.9 Hz, H2-21a), 3.81 (1H, dd, J = 16.9, 5.9 Hz, H2-21b), 3.54 (1H, dd, J = 5.2, 5.2 Hz, H-2), 3.08 (1H, dd, J = 14.1, 7.5 Hz, H2-12a), 2.98 (1H, dd, J = 14.1, 7.5 Hz, H2-12b), 2.88 (1H, dd, J = 14.8, 5.2 Hz, H2-3a), 2.74 (1H, dd, J = 14.8, 5.2 Hz, H2-3b), 0.97–0.96 (18H, m, 2SiC(CH3)3), 0.17–0.16 (12H, m, 2Si(CH3)2); 13C NMR (CDCl3, 125 MHz) δ 174.7 (C-9), 172.3 (C-19), 166.6 (C-22), 147.1 (C-15), 146.1 (C-16), 142.0 (3C-TrtAr), 138.9 (C-6), 136.4 (C-28a), 135.7 (C-4), 129.6 (9C-TrtAr), 129.4 (C-13), 128.2 (6C-TrtAr), 125.9 (C-32a), 123.6 (C-27), 123.0 (C-31), 122.1 (C-18), 121.9 (C-14), 121.2 (C-17), 120.6 (C-8), 120.1 (C-32), 119.0 (C-24), 116.0 (C-30), 114.2 (C-29), 110.9 (C-26), 102.9 (C-25), 75.5 (CAr3), 54.4 (C-11), 54.2 (C-2), 43.7 (C-21), 36.5 (C-12), 33.0 (C-3), 25.9 (2SiC(CH3)3), 18.5 (SiC(CH3)3), 18.4 (SiC(CH3)3), −4.1 (2Si(CH3)2); (+)-HRESIMS [M + H]+ 1080.4262 (calcd for C58H7179BrN7O5Si2, 1080.4233).
:9), afforded 25 as a yellow oil (56.5 mg, 63% yield). Rf 0.10 (CH2Cl2/MeOH 4:1); [α]27.3D −8.8 (c 1.96, CH2Cl2); IR (ATR) νmax 3282, 2929, 1655, 1532, 1446, 1338, 1041 cm−1; 1H NMR (CD3OD, 500 MHz) δ 7.52 (1H, d, J = 1.7 Hz, H-29), 7.43–7.41 (2H, m, H-27 and H-32), 7.36 (1H, br s, H-6), 7.32–7.30 (9H, m, 9H-TrtAr), 7.13 (1H, dd, J = 8.6 1.7 Hz, H-31), 7.08–7.06 (6H, m, 6H-TrtAr), 6.69 (1H, br s, H-8), 6.65–6.62 (3H, m, H-14, H-17 and H-24), 6.49 (1H, dd, J = 8.0, 1.9 Hz, H-18), 5.92 (1H, d, J = 9.9 Hz, H-25), 4.55 (1H, dd, J = 9.1, 5.2 Hz, H-11), 3.90 (1H, d, J = 16.7 Hz, H2-21a), 3.86 (1H, d, J = 16.7 Hz, H2-21b), 3.53 (1H, dd, J = 5.9, 5.9 Hz, H-2), 3.02 (1H, dd, J = 14.0, 5.2 Hz, H2-12a), 2.79–2.68 (3H, m, H2-3 and H2-12b); 13C NMR (CD3OD, 125 MHz) δ 175.9 (C-9), 174.6 (C-19), 169.3 (C-22), 146.3 (C-15), 145.2 (C-16), 143.6 (3C-TrtAr), 139.8 (C-6), 138.3 (C-28a), 137.6 (C-4), 130.8 (6C-TrtAr), 129.7 (C-13), 129.24 (3C-TrtAr), 129.22 (6C-TrtAr), 127.2 (C-32a), 125.5 (C-27), 123.6 (C-31), 121.7 (C-18 or C-8), 121.6 (C-18 or C-8), 121.0 (C-32), 119.5 (C-24), 117.3 (C-14), 116.4 (C-30), 116.3 (C-17), 115.3 (C-29), 111.3 (C-26), 105.4 (C-25), 76.8 (CAr3), 56.3 (C-11), 55.4 (C-2), 44.0 (C-21), 37.9 (C-12), 33.9 (C-3); (+)-HRESIMS [M + H]+ 852.2518 (calcd for C46H4379BrN7O5, 852.2504).
:2.5:2.5) was added to 25 (29.0 mg, 34.1 μmol). The reaction was stirred at r.t. for 1 h, after which, the solution was dried under a stream of nitrogen. Purification by C8 column chromatography (eluting with H2O to H2O/MeOH 60:40) afforded 1 as a white solid (13.09 mg, 56% yield). M.p. 280 °C (decomposed); Rf 0.72 (butan-1-ol/acetic acid/H2O 2:1:1); [α]22.3D +3.4 (c 1.07, MeOH) (lit.4a [α]22.3D +5.2 (c 0.5, MeOH)); 1H NMR (DMSO-d6, 500 MHz) δ 11.51 (1H, br s, NH-28), 9.08 (1H, d, J = 10.0 Hz, NH-23), 8.88 (1H, br s, NH-20), 8.40 (1H, br s, NH-10), 7.72 (1H, s, H-27), 7.60 (1H, s, H-6), 7.58 (1H, d, J = 1.6 Hz, H-29), 7.56 (1H, d, J = 8.3 Hz, H-32), 7.16 (1H, dd, J = 8.3, 1.6 Hz, H-31), 6.86 (1H, s, H-8) 6.67 (1H, dd, J = 10.0, 9.7 Hz, H-24), 6.64 (1H, d, J = 1.5 Hz, H-14), 6.59 (1H, d, J = 8.0 Hz, H-17), 6.45 (1H, dd, J = 8.0, 1.5 Hz, H-18), 5.92 (1H, d, J = 9.7 Hz, H-25), 4.44 (1H, br s, H-11), 3.99 (1H, dd, J = 16.6, 5.8 Hz, H2-21a), 3.93 (1H, dd, J = 16.6, 5.8 Hz, H2-21b), 3.61 (1H, br s, H-2), 2.95 (1H, dd, J = 14.0, 4.1 Hz, H2-12a), 2.91–2.89 (1H, m, H2-3a), 2.73 (1H, dd, J = 14.3, 7.5 Hz, H2-3b), 2.66 (1H, dd, J = 14.0, 9.6 Hz, H2-12b); 13C NMR (DMSO-d6, 125 MHz) δ 171.9 (C-19), 171.6 (C-9), 167.5 (C-22), 144.9 (C-15), 143.7 (C-16), 136.4 (C-28a), 135.1 (C-6), 133.5 (C-4), 128.5 (C-13), 125.7 (C-32a), 124.9 (C-27), 121.9 (C-31), 120.1 (C-32), 119.9 (C-18), 118.6 (C-24), 116.6 (C-14), 115.3 (C-17), 114.3 (C-30), 114.1 (C-29), 109.7 (C-26), 102.2 (C-25), 54.5 (C-11), 53.8 (C-2), 42.6 (C-21), 36.7 (C-12), 30.5 (C-3); (+)-HRESIMS [M + H]+ 610.1393 (calcd for C27H2979BrN7O5, 610.1408).
Antibiotic susceptibility testing
The susceptibility of bacterial strains to antibiotics and compounds was determined in microplates using the standard broth dilution method in accordance with the recommendations of the Comité de l'AntibioGramme de la Société Française de Microbiologie (CA-SFM).21 Briefly, the minimal inhibitory concentrations (MICs) were determined with an inoculum of 105 CFU in 200 μL of MH broth containing two-fold serial dilutions of each drug. The MIC was defined as the lowest concentration of drug that completely inhibited visible growth after incubation for 18 h at 37 °C. To determine all MICs, the measurements were independently repeated at least three times. Minimum inhibitory concentration of positive control: colistin [P. aeruginosa (1 μM), E. coli (2 μM)], streptomycin [P. aeruginosa (21.5 μM), E. coli (21.5 μM), S. aureus (21.5 μM), S. intermedius (10.7 μM) and E. faecalis (21.5 μM)] and chloramphenicol [S. aureus (1.5–3 μM), S. intermedius (3–6 μM) and E. faecalis (1.5–3 μM)].Marine bacteria susceptibility testing
The antibacterial activity assay was performed on the marine environmental bacterial strains Gram-negative V. harveyi ATCC 14126, V. alginolyticus ATCC 17749 and L. anguillarum ATCC 19264 by the liquid growth inhibition in 96-well microplates. A pre-culture of 5 mL marine broth (MB) was prepared by inoculating a colony of each bacterial strain and was incubated at 30 °C with stirring overnight. The concentration of the pre-culture was assessed by measuring the optical density (OD) at 620 nm and was adjusted by dilution in order to obtain a suspension of 0.03 OD. An aliquot of 200 μL of the bacterial suspension was distributed in each well and 10 μL of a serial dilution in DMSO of the pure compound were added in triplicate. The 96-well microplates were incubated at 30 °C overnight with shaking (450 rpm). The optical density of the wells was measured at 620 nm with a microplate reader and the inhibition (IC50) was calculated and plotted versus test concentrations.Antioxidant testing
Quantitative ORAC assay was run as previously described.22 The result is expressed as relative Trolox (6-hydroxy-2,5,7,8-tetramethyl chroman-2-carboxylic acid) equivalents.DPPH free radical scavenging assay
The potential antioxidant activity was evaluated in a 96-well microplate format assay whereby an aliquot of 10 μL of a serial dilution in MeOH of the pure compound and 190 μL of DPPH (200 μM, MeOH) were added in triplicate. After 1 h at room temperature in the dark, the absorbance was recorded at 510 nm in a microplate reader (DPPH radical has a characteristic absorption in MeOH at 510 nm, which disappears with acceptance of an electron from the antioxidant sample). Ascorbic acid was used as a positive control (IC50 101 ± 8 μM). The antioxidant activity of the tested compounds was evaluated by the IC50, which represents the sample concentration required to scavenge 50% of the DPPH free radical.Acknowledgements
We thank Dr M. Schmitz for assistance with NMR data acquisition, and Mr T. Chen for MS data. We gratefully acknowledge funding from the University of Auckland.Notes and references
- (a) R. C. Bruening, E. M. Oltz, J. Furukawa, K. Nakanishi and K. Kustin, J. Am. Chem. Soc., 1985, 107, 5298–5300 CrossRef CAS; (b) E. M. Oltz, R. C. Bruening, M. J. Smith, K. Kustin and K. Nakanishi, J. Am. Chem. Soc., 1988, 110, 6162–6172 CrossRef CAS PubMed; (c) E. Bayer, G. Schiefer, D. Waidelish, S. Scippa and M. de Vicentiis, Angew. Chem., Int. Ed. Engl., 1992, 31, 52–54 CrossRef; (d) J. A. Tincu, A. G. Craig and S. W. Taylor, Biochem. Biophys. Res. Commun., 2000, 270, 421–424 CrossRef CAS PubMed; (e) J. A. Tincu and S. W. Taylor, J. Nat. Prod., 2002, 65, 377–378 CrossRef CAS PubMed.
- M. Sugumaran and W. E. Robinson, Mar. Drugs, 2010, 8, 2906–2935 CrossRef CAS PubMed.
- M. Sugumaran and W. E. Robinson, Comp. Biochem. Physiol., Part B: Biochem. Mol. Biol., 2012, 163, 1–25 CrossRef CAS PubMed.
- (a) K. Azumi, H. Yokosawa and S. Ishii, Biochemistry, 1990, 29, 159–165 CrossRef CAS PubMed; (b) K. Azumi, H. Yokosawa and S. Ishii, Experientia, 1990, 46, 1020–1023 CrossRef CAS.
- E. Dumdei and R. J. Andersen, J. Nat. Prod., 1993, 56, 792–794 CrossRef CAS.
- D. R. Appleton, M. J. Page, G. Lambert, M. V. Berridge and B. R. Copp, J. Org. Chem., 2002, 67, 5402–5404 CrossRef CAS PubMed.
- K. Azumi, M. Yoshimizu, S. Suzuki, Y. Ezura and H. Yokosawa, Experientia, 1990, 46, 1066–1068 CrossRef CAS PubMed.
- M. A. Pullar, D. Barker and B. R. Copp, Tetrahedron Lett., 2015, 56, 5604–5606 CrossRef CAS.
- (a) Y. Ma, K. Yakushijin, F. Miyake and D. Horne, Tetrahedron Lett., 2009, 50, 4343–4345 CrossRef CAS; (b) L. Rivas, L. Quintero, J.-L. Fourrey and R. Benhida, Tetrahedron Lett., 2002, 43, 7639–7641 CrossRef CAS.
- (a) S. Su, H. Kakeya, H. Osada and J. A. Porco Jr., Tetrahedron, 2003, 59, 8931–8946 CrossRef CAS; (b) X. Wang and J. A. Porco Jr., J. Org. Chem., 2001, 66, 8215–8221 CrossRef CAS PubMed; (c) G. K. Min, D. Hernández, A. T. Lindhardt and T. Skrydstrup, Org. Lett., 2010, 12, 4716–4719 CrossRef CAS PubMed; (d) P. García-Reynaga, A. K. Carrillo and M. S. Van Nieuwenhze, Org. Lett., 2012, 14, 1030–1033 CrossRef PubMed; (e) A. Furstner, C. Brehm and Y. Cancho-Grande, Org. Lett., 2001, 3, 3955–3957 CrossRef CAS PubMed; (f) M. Chakrabarty, R. Basak and Y. Harigaya, Synthesis, 2003, 2011–2014 CrossRef CAS.
- (a) L. J. Gooßen, K. S. M. Salih and M. Blanchot, Angew. Chem., Int. Ed., 2008, 47, 8492–8495 CrossRef PubMed; (b) L. J. Gooßen, M. Blanchot, K. S. M. Salih and K. Gooßen, Synthesis, 2009, 2283–2288 CrossRef.
- M. J. Sever and J. J. Wilker, Tetrahedron, 2001, 57, 6139–6146 CrossRef CAS.
- M. Hoshi, H. Nakayabu and K. Shirakawa, Synthesis, 2005, 1991–2007 CrossRef CAS.
- Compounds 5 and 6 were also present as a 1.5:1 ratio in 47% yield (yield calculated based upon reaction stoichiometry of phenylacetylene).
- E. Dickson, B. R. Copp and D. Barker, Tetrahedron Lett., 2013, 54, 5239–5242 CrossRef CAS.
- Y. Tanoue, M. Hamada, N. Kai, K. Sakata, M. Hashimoto and T. Nagai, J. Heterocycl. Chem., 2005, 42, 1195–1199 CrossRef CAS.
- Y. Tanoue, A. Terada, K. Sakata, M. Hashimoto, S.-I. Morishita, M. Hamada, N. Kai and T. Nagai, Fish. Sci., 2001, 67, 726–729 CrossRef CAS.
- (a) B. M. Trost and J. T. Masters, Chem. Soc. Rev., 2016, 45, 2212–2238 RSC; (b) Y. Zhou, Y. Zhang and J. Wang, Org. Biomol. Chem., 2016, 14, 6638–6650 RSC.
- (a) C. S. Yi and N. Liu, Organometallics, 1996, 15, 3968–3971 CrossRef CAS; (b) X. Chen, P. Xue, H. H. Y. Sung, I. D. Williams, M. Peruzzini, C. Bianchini and G. Jia, Organometallics, 2005, 24, 4330–4332 CrossRef CAS; (c) A. Coniglio, M. Bassetti, S. E. García-Garrido and J. Gimeno, Adv. Synth. Catal., 2012, 354, 148–158 CrossRef CAS.
- Compounds 2, 13–15 displayed no activity (MIC > 200 μM) when tested against S. aureus ATCC 25923, S. intermedius, 1051997, P. aeruginosa ATCC 27853 and E. coli ATCC 25922.
- G. Carret, J. D. Cavallo, H. Chardon, C. Chidiac, P. Choutet, P. Courvalin, H. Dabernat, H. Drugeon, L. Dubreuil, F. Goldstein, V. Jarlier, R. Leclercq, M. H. Nicolas-Chanoine, A. Philippon, C. Quentin-Noury, B. Rouveix, J. Sirot and C. J. Soussy, Int. J. Antimicrob. Agents, 2003, 21, 364–391 CrossRef.
- A. Longeon, B. R. Copp, E. Quévrain, M. Roué, B. Kientz, T. Cresteil, S. Petek, C. Debitus and M.-L. Bourguet-Kondracki, Mar. Drugs, 2011, 9, 879–888 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: 1H and 13C spectral data. See DOI: 10.1039/c7ob01122a |
| This journal is © The Royal Society of Chemistry 2017 |