
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStabilizing bubble and droplet interfaces using dipeptide hydrogels
Fernando
Aviño
 a,
Andrew B.
Matheson
a,
Dave J.
Adams
b and
Paul S.
Clegg
*a
a,
Andrew B.
Matheson
a,
Dave J.
Adams
b and
Paul S.
Clegg
*a
aSchool of Physics and Astronomy, University of Edinburgh, James Clerk Maxwell Building, Peter Guthrie Tait Road, Edinburgh, EH9 3FD, UK. E-mail: paul.clegg@ed.ac.uk
bSchool of Chemistry, College of Science and Engineering, University of Glasgow, Glasgow, G12 8QQ, UK
First published on 5th July 2017
Abstract
Hydrophobic dipeptide molecules can be used to create interfacial films covering bubbles and droplets made from a range of oils. At high pH, the dipeptide molecules form micelles which transform into a hydrogel of fibres in response to the addition of salt. We characterize the properties of the hydrogel for two different salt (MgSO4) concentrations and then we use these gels to stabilize interfaces. Under high shear, the hydrogel is disrupted and will reform around bubbles or droplets. Here, we reveal that at low dipeptide concentration, the gel is too weak to prevent ripening of the bubbles; this then reduces the long-term stability of the foam. Under the same conditions, emulsions prepared from some oils are highly stable. We examine the wetting properties of the oil droplets at a hydrogel surface as a guide to the resulting emulsions.
Introduction
Proteins have been used by cooks to stabilize foams and emulsions for many centuries.1 The proteins are typically trapped on the interface and can denature, especially at liquid/air interfaces. By comparison, small peptides can potentially improve performance in two diametrically opposed ways. (1) Isolated small peptides will be adsorbed to the interface more quickly which can aid bubble or droplet formation.2 This tends to be the more challenging route. (2) These small molecules may self-assemble into larger structures which retain their order at the interface;2–9 the trapping at the interface can be much stronger than for individual proteins. Proteins have also been used as subunits within robust composite particles which themselves are interfacially active.10–14 Nonetheless, short peptide sequences offer the distinct advantages of ease of synthesis and design as well as the tendency to self-assemble at very low concentration.Recently, the behaviour of very short peptide sequences at liquid/air interfaces has begun to be explored.6,7 Here naphthalene-protected diphenylalanine (2NapFF) molecules have been investigated most. One approach was to investigate the self-assembly of the dipeptide at the air/water interface prepared via drop-casting. A thin interfacial film formed from a dispersion of 2NapFF at high-pH on contact with a low-pH subphase. FTIR and pH variation tests show that this self-assembly behaviour is associated with the carboxylic acid group of the dipeptide becoming rapidly protonated at the interface. The film was made up of self-assembled fibres and behaved like an elastic sheet.6 The same dipeptide (2NapFF) also formed a hydrogel via the addition of metal ions. This hydrogel can be used to stabilise aqueous foams for weeks. The dipeptides at 1 wt% concentration self-assemble into fibre-like networks at both, the air/water interface and in the bulk. Raman spectroscopy hinted at a possible change in the balance between π-stacking and hydrogen bonding, with the latter appearing to drive self-assembly close to the interface. The dipeptide concentration as well as the type and concentration of metal ions, affects the foam stability.7 When the bulk hydrogel had a yield stress larger than the Laplace pressure of the bubbles, ripening was suppressed.
Separately, the stabilization of emulsions using di-, tri- and hepta- peptides has also been investigated.2,5,8,9 Initial experiments showed that it was possible to stabilize chloroform droplets for months using an interfacial network of dipeptide fibres. Here the dipeptide sequence was protected by N-(fluorenyl-9-methoxycarbonyl) (Fmoc) with self-assembly being driven by π-stacking and hydrogen bonding. The fibres were self-assembled nanostructures and, for a well-chosen dipeptide sequence, remained stable in the presence of added salt or when the temperature was raised. Curiously, using the tri-peptide sequence Fmoc-FFF it was possible to stabilize water droplets in chloroform, demonstrating the importance of the hydrophobicity of the fibres.5 Unprotected tripeptide emulsifiers divided into two classes: those that self-assembled into fibres prior to adsorption and those that adsorbed to droplets as a single-molecule layer. The former were more effective at stabilizing droplets of rapeseed oil.2 As an alternative demonstration of the key role played by self-assembly, an Fmoc protected dipeptide sequence, Fmoc-tyrosine-leucine (FmocYL) was used as a switchable emulsifier. An enzyme was employed to cleave a phosphate group which was initially attached to the tyrosine group. With the phosphate group attached chloroform droplets are stable for less than an hour; by contrast, when emulsification is carried out in the presence of the enzyme, the dipeptide self-assembles into nanofibres which then stabilize the emulsion for months.8 These studies strengthen the claim that it is the fibres which are most effective at stabilizing the liquid interfaces and not the separate molecules. Very recently a seven peptide molecule has been designed to self-assemble into fibres with excellent surfactant properties.9 At this length, the short peptide sequence has properties which are robust to changes in some parts of the sequence. Even more recently a low molecular weight gelator has been synthesized at a water–oil interface to create stable droplets which can be induced to coalescence by a trigger.15
The formation of bulk hydrogels by our dipeptide of choice, 2NapFF (Fig. 1a), has been studied in great detail.16–18 Gelation can be induced by initially dispersing the molecule at high pH and then adding salt or lowering the pH. Alternatively, the 2NapFF can be dispersed in dimethyl sulphoxide (DMSO) and then subsequently this can be diluted with water. While the gel strength does not change significantly for these different routes, the microstructure and yielding behaviour do alter.18 When gelation is triggered by the addition of salt (here Ca2+), the system exhibits a significant sensitivity to the initial high pH chosen which may be due to the differing degrees of lateral association between micelles. Further studies have shown that the use of divalent cations (Mg2+, Ca2+) leads to the formation of hydrogels with higher storage and loss moduli than monovalent cations (Li+, Na+, K+). The choice of anion has some additional effect on the moduli. These experiments also suggested that worm-like micelles always preceded salt-induced hydrogelation.16 More recently, a subtler relationship has been revealed: at high pH, 2NapFF self-assembles into spherical micelles above a first cmc of 0.01 wt%, with worm-like micelles are found above a second cmc of 0.07 wt%.17 At around 1 wt%, the worm-like micelles are found to aggregate. Typically addition of the calcium salt transforms worm-like micelles into hydrogels. However, above the first cmc, there appear to be structural transformations that occur on addition of the calcium salt, which allows gels to form in this intermediate regime. This is the concentration regime that we focus on here.
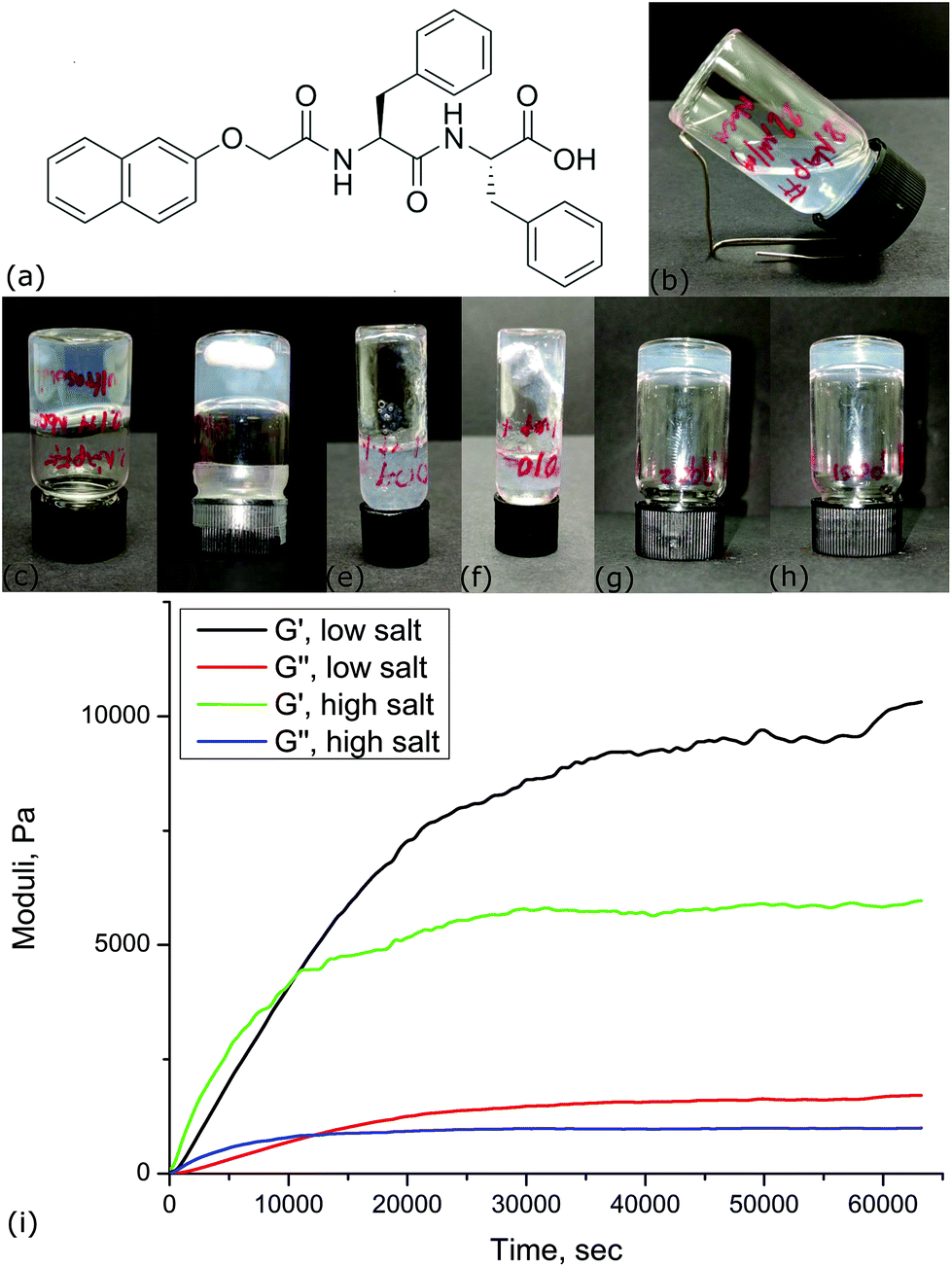 | ||
| Fig. 1 (a) Naphthalene dipeptide molecule used in this study. (b) Dispersion of 2 mM 2NapFF at pH 11 a few minutes after the ultrasonic bath; (c) dispersion 24 hours after the ultrasonic bath; (d) dispersion 24 hours after the magnetic bar; (e) hydrogel prepared at 18 mM MgSO4 dispersed at 25 °C, 2 minutes after addition of salt; (f) hydrogel prepared at 18 mM MgSO4 dispersed at 50 °C, 2 minutes after addition of salt. (g, h) Our preferred protocol for producing a hydrogel from 2 mM 2NapFF at <35 °C: (g) 18 mM MgSO4 and (h) 142 mM MgSO4. (i) Storage, G′, and loss, G′′, moduli measured using oscillating rheometry for hydrogels prepared at low and high salt concentrations. | ||
In this Paper, we outline the route to hydrogel formation induced by MgSO4 at low 2NapFF concentration (0.1 wt%). We then use this hydrogel to stabilize foams and emulsions. Our results show that using a low concentration of dipeptide and Mg2+ rather than Ca2+ undermines the stability of the foam compared to previous observations.7 By contrast, it is possible to create highly stable emulsions based on several different oils. We separately characterize the wetting behaviour of the oils and a bubble at a hydrogel interface. We consider the stability of our composites in the light of the wetting character of the hydrogel film at the fluid–fluid interface.
Results and discussion
In order to investigate foams and emulsions, the underlying hydrogels need to be repeatably produced. For this reason, we carefully studied the process of dispersing and gelling the dipeptide (see Experimental section). Whether and how long it takes for a gel to form and its ultimate properties are affected by factors such as pH, temperature, ionic strength and mechanical shear rate.16,17,19,20 A thorough study of the effect of temperature on dispersions of 2NapFF has been published very recently.21 Here, we compared the use of an ultrasound bath with a magnetic stirrer and found that the former resulted in more transparent dispersions. Dipeptide dispersion and gelation times initially differed between ostensibly identical hydrogels prepared using the ultrasonic bath presumably due to the ±8 °C variation in the bath temperature. For this reason, we established two temperatures (<35 °C and 50 °C) for dispersing the dipeptide. The rate of dipeptide dispersion (unsurprisingly) and the subsequent gelation time (unexpectedly) was found to be faster for the hydrogels prepared at 50 °C compared to those prepared at <35 °C. Our final protocol was to place the sample in an ultrasonic bath (at a controlled temperature) for 0.5 h until a translucent slightly viscous solution was formed, Fig. 1b.By leaving dispersions (prepared using either ultrasound or a magnetic stirrer) to stand for 24 hours at a room temperature (22 °C), we found that self-supported gels, able to pass the inversion vial test, sometimes formed (see Fig. 1c and d). Structural transitions, linked to increases in viscosity, have been observed before in 2NapFF dispersions which have been warmed in the temperature range of our bath (albeit at higher concentration). These changes in organization can then become trapped.22 We suggest that we might be seeing similar behaviour here. To avoid this, all of our hydrogels were prepared by the addition of salt within 5 minutes of removal from the ultrasound bath. Figures 1e and f demonstrate how the temperature of the ultrasound bath influences the speed of subsequent behaviour. Dipeptide dispersed at 50 °C has begun to gel within two minutes of the addition of salt. Typical hydrogels for two different concentrations of MgSO4 are shown in Fig. 1g and h.
The evolution of shear modulus was measured (see Experimental section) as a function of time to study the strength of hydrogels prepared at 18 mM and 142 mM MgSO4 (Fig. 1i). G′ and G′′ of hydrogels with high salt concentration rise faster than hydrogels with low salt concentration within the first 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 seconds. At this point, G′ and G′′ of hydrogels with high salt concentration hardly increase, reaching their maximum at 6 kPa for G′ after a few thousand seconds, while G′ and G′′ of low salt concentration hydrogels continue to increase even after 63000 seconds. Salt induced gelators which become weaker at higher salt concentration have been seen previously. The strength of a hydrogel prepared at 0.5 wt% of 2NapFF and Ca2+ salt was found to reach its maximum value at a concentration of 2:1 Ca2+ to dipeptide. Increasing further the concentration of Ca2+ resulted in weaker gels.16
000 seconds. At this point, G′ and G′′ of hydrogels with high salt concentration hardly increase, reaching their maximum at 6 kPa for G′ after a few thousand seconds, while G′ and G′′ of low salt concentration hydrogels continue to increase even after 63000 seconds. Salt induced gelators which become weaker at higher salt concentration have been seen previously. The strength of a hydrogel prepared at 0.5 wt% of 2NapFF and Ca2+ salt was found to reach its maximum value at a concentration of 2:1 Ca2+ to dipeptide. Increasing further the concentration of Ca2+ resulted in weaker gels.16
Fig. 2a and c shows foams formed from dispersions prepared at <35 °C which were gelled with 18 mM and 142 mM MgSO4, respectively. The formation method is described in the Experimental section. Foams with low salt concentration (Fig. 2a) resulted in higher-quality foams with small average bubble size and a higher volume of liquid entrained in the foam compared to those prepared at a high salt concentration (Fig. 2c). The bubble size is often related to foam stability due to the enhanced buoyancy of large bubbles; however, in this case we find that after 0.5 h it is the foam with smaller bubbles that has collapsed more, Fig. 2b and d. Evidently, while these hydrogels provide a good environment for creating bubbles the interfacial layer and sparse bulk gel are unable to prevent rapid coalescence. This is especially true with small bubbles which consume a greater proportion of fibres as the interfacial layer.
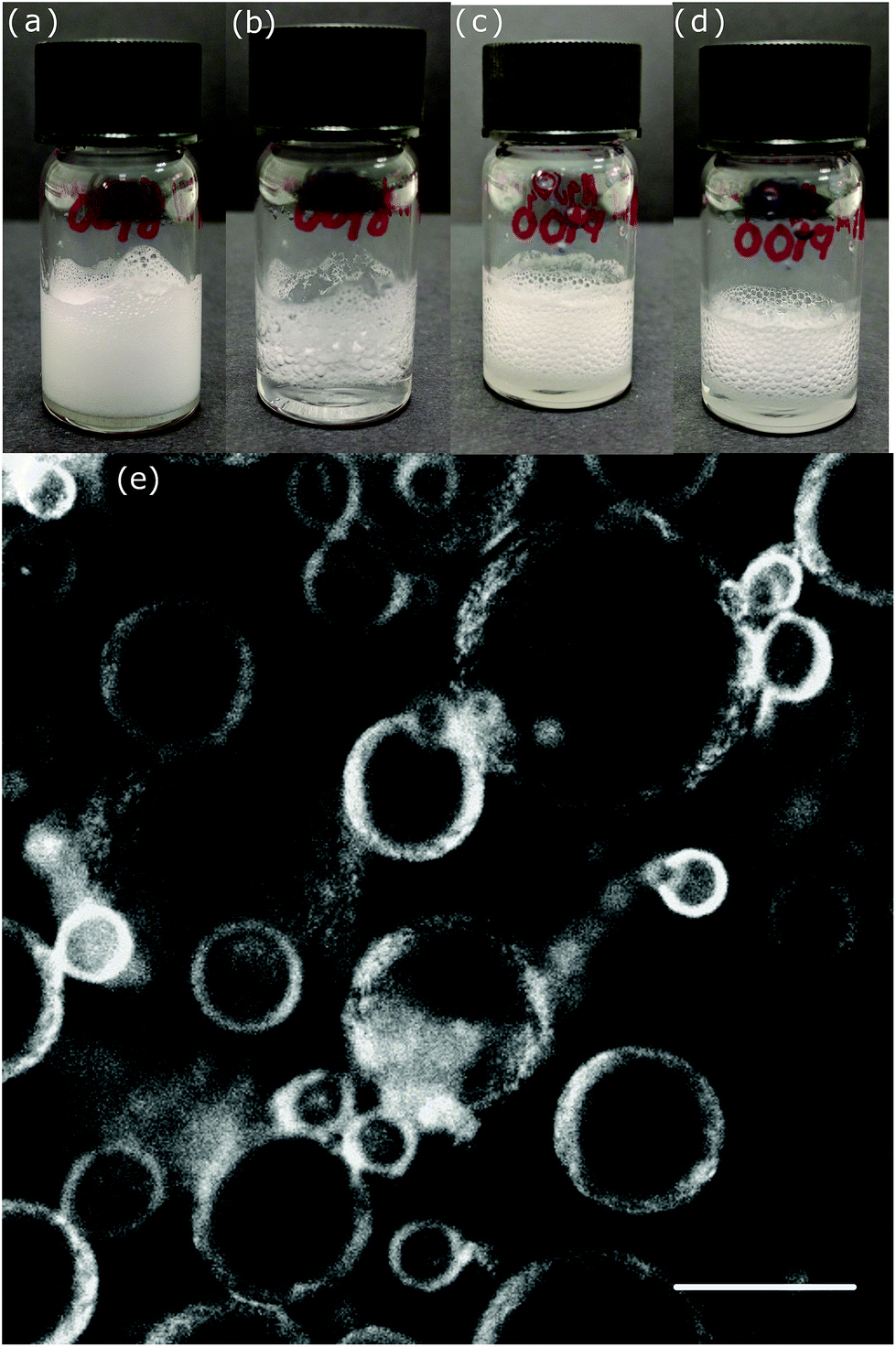 | ||
| Fig. 2 Illustrates fresh foams (a, c) and foams after 0.5 hours (b, d) prepared from 2 mM 2NapFF and dispersed at <35 °C; (a, b) 18 mM MgSO4 and (c, d) 142 mM MgSO4. (e) Shows a confocal micrograph of a fresh foam prepared at 2 mM 2NapFF, 142 mM, MgSO4 and dispersed at 50 °C. Dipeptides are dyed using Nile Blue; scale bar 100 μm. | ||
Fluorescence confocal microscopy was carried out to try to understand the foam performance. Fig. 2e shows the organization of a fresh foam prepared at 142 mM MgSO4 and 50 °C. The nanofibers prefer to adsorb at the air/aqueous interface (similar to ref. 23) rather than being dispersed in the continuous phase, Fig. 2e. However, over the next hour hydrogel begins to emerge between bubbles. This is very much reminiscent of the foams reported in ref. 7 which were created at significantly higher dipeptide concentration and gelled using CaCl2. In that work, the hydrogel initially formed on the bubble interfaces but then steadily appeared between the bubbles but now over a timescale of days rather than tens of minutes. At our very low concentrations of the dipeptide the foam lifetime is only two hours, indicating that the remnant hydrogel between the bubbles is now too weak to prevent coalescence and ripening.
As we have found changes in hydrogel properties depending on the ultrasound bath temperature, we repeated the same series of measurements starting from a dispersion made at both temperatures. No changes were observed. By contrast, using CaCl2 rather than MgSO4 to create the foam did lead to enhanced stability. With the calcium salt neither varying the salt concentration (15 mM and 120 mM) nor varying the initial ultrasound bath temperature, altered the foam properties noticeably. We conclude that any residual influence of the ultrasound bath temperature is negated once the rotor stator has been used to destroy the initial hydrogel. When the fibres re-form on the surface of the bubbles, the detailed properties of gelator system can remain influential.
In order to examine the organization of the molecules we have used FTIR, Fig. 3, for a dispersion of 2NapFF (black), a foam prepared at low salt concentration (red) and a foam prepared at high salt concentration (blue). The samples were allowed to dry prior to characterization. The original dispersion has a peak at 1630 cm−1, which may reflect the initial micellar ordering. No peak was observed at 1720 cm−1, indicating that the carboxylic acid group is deprotonated. A peak appears at 1650 cm−1 once salt has been added and grows with increasing salt concentration. This may well be related to the formation of a random coil structure, although we highlight that it is difficult to assign these dipeptides on the basis of conventional IR data.
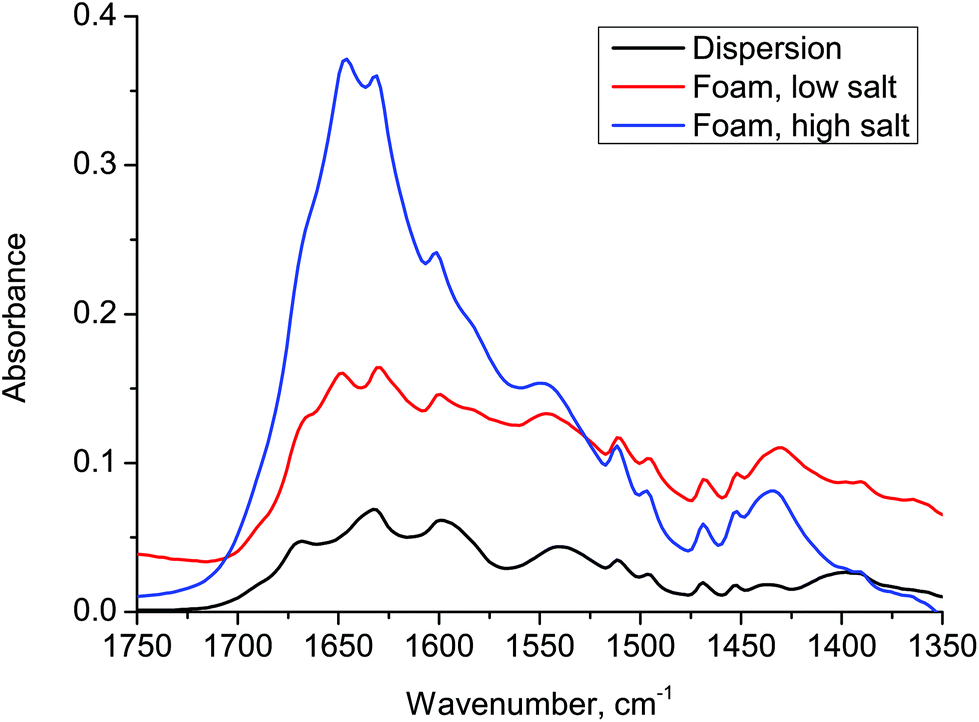 | ||
| Fig. 3 Fourier transform infra red absorbance data for a dispersion of 2NapFF and foams made at the two salt concentrations. There is a very significant growth in the peak around 1650 cm−1 corresponding to the formation of random coil structure. | ||
Emulsions with four different oils (isopropyl myristate, silicone oil, dodecane, octanol) were prepared in a similar manner to the foams described above. Three out of four emulsions remained stable for months (Fig. 4a,c and e). Isopropyl myristate emulsions (Fig. 4a) appeared the most ideal in that there was no creaming. At the other end of the range, octanol emulsions (Fig. 4g) could not be stabilised, phase separating immediately after preparation. Indeed it appears that the dipeptide material occupies the small volume of oil phase at the top of the sample and this plug floats above water, Fig. 4i. Less surprisingly, a small amount of creaming occurred within the first hour for silicone oil (Fig. 4c) and dodecane (Fig. 4e) emulsions. The volume of the clear aqueous phase in these emulsions barely increased after months. The same set of measurements were repeated at 50 °C and 142 mM MgSO4 and at <35 °C, 18 mM and 142 mM MgSO4 for each of the oils. The emulsification performance and stability are broadly similar.
 | ||
| Fig. 4 Shows the macroscopic and microscropic characteristics of four emulsions prepared at 2 mM 2NapFF, 18 mM MgSO4 at 50 °C; in the confocal micrographs the dipeptide has been dyed using Nile blue and the scale bar is 100 μm. The oil is (a, b) isopropyl myristate; (c, d) silicone oil; (e, f) dodecane; (g, h) octanol. (i) Shows the octanol sample tipped to one side, indicating that there is a light solid plug floating on a bath of aqueous phase. | ||
Confocal images of the emulsions are presented in Fig. 4b,d,f and h. Here, the dipeptides show a preference for the oil phase in the following sequence: octanol (3.4) > isopropyl myristate (3.3) > silicone oil (2.6) > dodecane (2). The numbers in brackets are the dielectric constants. Self-assembled dipeptides in isopropyl myristate emulsions (Fig. 4b) show a clear preference for the interfaces. The concentration of dipeptide in the aqueous phase increases slightly for silicone oil emulsions (Fig. 4d) and further still for the dodecane emulsions (Fig. 4f). The octanol samples are comprised of some sparse aqueous droplets within a background of dipeptide in the oil phase (Fig. 4h). This unexpected behaviour is consistent with our observations of the macroscopic emulsion, Fig. 4g and i.
It is tempting to consider the role of the interfacial dipeptide fibres to be similar to the interfacial particles in a Pickering emulsion.24 Under this scenario, whether the dipeptide fibres become trapped at the interface depends on their wettability by the two fluid phases. To explore this idea we have carried out an experiment to observe the wetting behaviour of the oils† at an interface between the hydrogel and an aqueous phase of the same salt concentration, Fig. 5a. If the hydrogel surface was precisely horizontal and reasonably flat we would be able to use the angle with which the droplet or bubble meets the hydrogel surface (measured through the aqueous phase) as a quantitative measurement of the wettability of the hydrogel. Unfortunately, since the surface is somewhat rough it is not possible to be quantitative. The air and the dodecane appear to make approximately similar (small) angles to the hydrogel surface (Fig. 5c and d). This suggests that in both cases the hydrogel greatly prefers contact with the aqueous phase over contact with the droplet/bubble. By contrast, isopropyl myristate makes a more gentle angle with the surface of the gel, Fig. 5b. The preference of the hydrogel for contact with the aqueous phase is significantly less marked, this is consistent with the formation of a stable emulsion. If the behaviour of octanol, described above, was because of the wetting characteristics of this oil at a hydrogel surface then we would expect to see the droplet spread out to maximize the contact area with the hydrogel. This would be reflected in a large three phase contact angle. What we see, Fig. 5e, is a quite standard contact angle as though the octanol only partially wets the hydrogel. Evidently, the behaviour of this system does not fit easily within the Pickering emulsion picture. We note that the droplet is becoming darker, which might point to more complex changes taking place.
 | ||
| Fig. 5 (a) The experimental apparatus used to evaluate the wetting characteristics of droplets and bubbles. The hydrogel is visible via the Nile blue dye; scale bar 18 mm. Droplets/bubbles of (b) isopropyl myristate (c) air (d) dodecane (e) octanol. | ||
Conclusions
Following previous studies stabilizing emulsions and foams using dipeptide hydrogels, we have investigated the behaviour of one dipeptide system at low concentration with the gelation induced by the addition of salt. We have presented a route to achieving repeatable gel formation based on the dipeptide 2NapFF and using the salt MgSO4. Using this gel system we have demonstrated that foams become less stable at low dipeptide concentration. The fibre behaviour is found to be closely similar to that at higher concentrations but evidently the gel structure is now too weak. By contrast, we have been able to form long lived emulsions using three out of the four oil phases that we have tested. Here the droplets could be stabilized by an interfacial film (isopropyl myristate) or by hydrogel both in the aqueous phase and on the interface (dodecane and silicone oil). The oil which could not be emulsified in this way (octanol) could not be described simply using the wetting characteristics of the hydrogel. This observation warns against a simple comparison between the behavior of these self-assembled fibres and the particulate emulsifiers used in Pickering emulsions. The new process, not observed in Pickering emulsions, is that some components partition into the oil phase but apparently not the intact fibres which are preferentially wet by the aqueous phase.Experimental
Hydrogel preparation
2NapFF was synthesised as described previously.25 All chemicals were purchased from Sigma-Aldrich and Fisher Chemicals and used as received. Vials and instruments were cleaned with hexane, followed by methanol and rinsed with ultra pure water before use. An equimolar quantity of NaOH (1 M, aq) was added to millipore water (resistivity = 18.2 MΩ cm) to give a basic solution (pH 11 ± 0.5). Then 2 mM 2NapFF was added and the mixture was dispersed. Ultimately this was carried out using an ultrasound bath (VWR model USC 300 T with power 80 W). The pH was 11 ± 0.5 measured with a Seven Easy pH probe (Mettler Toledo AG).Gelation was triggered by adding MgSO4·7H2O or CaCl2·2H2O aliquots on top of the translucent dispersion. The form of addition matters. By adding the salt already dissolved, hydrogels are more homogeneous compared to those prepared by adding the salt as a powder. It is possible that the latter favours the formation of two phase samples. In practice, all samples were prepared using 0.1 wt% dipeptide and were gelled using pre-dissolved salt to give a 1 mL sample. This was then left to stand for 12 h at room temperature before use in subsequent experiments.
Foam and emulsion preparation
Foams were prepared from the gels by using a rotor stator homogeniser (Polytron, PT-MR 3100) with a 12 mm diameter head, operating at 15000 rpm (shear rate ≈25700 s−1) for 50 seconds. Immediately following foam formation, the samples were sealed with parafilm to prevent liquid evaporation; the height of the foam was recorded as a function of time.
Emulsions with four different oils (isopropyl myristate, silicone oil, dodecane, octanol) were prepared in a similar manner to the foams described above. In each case 200 μL of the oil was placed on top of the hydrogel prior to high-shear mixing.
Rheology
Initial tests for gelation were performed by vial inversion. Oscillatory shear measurements, were conducted on a TA Instruments AR 2000 rheometer with cross-hatched parallel plates with 40 mm diameter and 1.0 mm gap distance. A 4 mL (0.1 wt%) dispersion solution of the dipeptide was previously prepared in a 7 mL glass vial, as described above. Gels were triggered on the bottom plate of the rheometer by loading the 4 mL dispersion and adding 18 mM MgSO4 for hydrogels with low salt concentrations and 142 mM MgSO4 for hydrogels with high salt concentration. The sides of the plates were covered with low viscosity mineral oil to avoid water evaporating from the hydrogel. Gelation process was observed by measuring the shear modulus (storage modulus G′ and loss modulus G′′) as a function of time at a frequency of 1 Hz and at a constant strain of 0.5% for a period of ∼18 hours. Experiments were carried out at 22 °C.Confocal microscopy
The measurements were carried out by using a Zeiss AxioObserver.Z1 inverted microscope and a Zeiss LSM 700 scanning system 9 with a 20× (0.40 NA) objective. Nile Blue at a concentration of 0.55 mM was used to label the dipeptides; this was excited using a 633 nm semiconductor laser.FTIR
IR spectra were measured using a Smiths Illuminat IR module coupled to a Renishaw inVia Raman microscope. Each spectrum was scanned between 4000 and 650 cm−1 at a resolution of 2 cm−1 and corrected for the influence of the optics and the substrate.Contact angle
The hydrogel (blue) was created, using our standard protocol, in the base of a sample vial in the presence of Nile blue dye. Following gelation, additional salt solution was added above the hydrogel. This two-phase cell was then inverted with the use of a petri dish. A droplet of oil or a bubble of air was added using a syringe with a U-shaped needle, Fig. 5a.Acknowledgements
We are grateful to the BBSRC for funding via grant BB/M027597/1. DA thanks the EPSRC for a Fellowship (EP/L021978/1).References
- S. Damodaran, J. Food Sci., 2005, 70, R54–R66 CrossRef CAS.
- G. G. Scott, P. J. McKnight, T. Tuttle and R. V. Ulijn, Adv. Mater., 2016, 28, 1381–1386 CrossRef CAS PubMed.
- A. F. Dexter, A. S. Malcolm and A. P. J. Middelberg, Nat. Mater., 2006, 5, 502–506 CrossRef CAS PubMed.
- A. F. Dexter and A. P. J. Middelberg, J. Phys. Chem. C, 2007, 111, 10484–10492 CAS.
- S. Bai, C. Pappas, S. Debnath, P. W. Frederix, J. Leckie, S. Fleming and R. V. Ulijn, ACS Nano, 2014, 8, 7005–7013 CrossRef CAS PubMed.
- T. Li, M. Kalloudis, A. Z. Cardoso, D. J. Adams and P. S. Clegg, Langmuir, 2014, 30, 13854–13860 CrossRef CAS PubMed.
- T. Li, F. Nudelman, J. W. Tavacoli, H. Vass, D. J. Adams, A. Lips and P. S. Clegg, Adv. Mater. Interfaces, 2016, 3, 1500601 CrossRef.
- I. P. Moreira, I. R. Sasselli, D. A. Cannon, M. Hughes, D. A. Lamprou, T. Tuttle and R. V. Ulijn, Soft Matter, 2016, 12, 2623–2631 RSC.
- S. Mondal, M. Varenik, D. N. Bloch, Y. Atsmon-Raz, G. Jacoby, L. Adler-Abramovich, L. J. Shimon, R. Beck, Y. Miller, O. Regev and E. Gazit, Nat. Commun., 2017, 8, 14018 CrossRef CAS PubMed.
- J. T. Russell, Y. Lin, A. Böker, L. Su, P. Carl, H. Zettl, J. He, K. Sill, R. Tangirala, T. Emrick, K. Littrell, P. Thiyagarajan, D. Cookson, A. Fery, Q. Wang and T. P. Russell, Angew. Chem., Int. Ed., 2005, 44, 2420–2426 CrossRef CAS PubMed.
- L. Isa, J.-M. Jung and R. Mezzenga, Soft Matter, 2011, 7, 8127 RSC.
- A. Ye, X. Zhu and H. Singh, Langmuir, 2013, 29, 14403–14410 CrossRef CAS PubMed.
- M. Sarker, N. Tomczak and S. Lim, ACS Appl. Mater. Interfaces, 2017, 9, 11193–11201 CAS.
- Z. Gao, J. Zhao, Y. Huang, X. Yao, K. Zhang, Y. Fang, K. Nishinari, G. O. Phillips, F. Jiang and H. Yang, LWT–Food Sci. Technol., 2017, 76, 1–8 CrossRef CAS.
- Y. Nishida, A. Tanaka, S. Yamamoto, Y. Tominaga, N. Kunikata, M. Mizuhata and T. Maruyama, Angew. Chem., Int. Ed., 2017 DOI:10.1002/anie.201704731.
- L. Chen, G. Pont, K. Morris, G. Lotze, A. Squires, L. C. Serpell and D. J. Adams, Chem. Commun., 2011, 47, 12071–12073 RSC.
- A. Z. Cardoso, L. L. E. Mears, B. N. Cattoz, P. C. Griffiths, R. Schweins and D. J. Adams, Soft Matter, 2016, 12, 3612–3621 RSC.
- C. Colquhoun, E. Draper, R. Schweins, M. Marcello, L. Serpell, D. Vadukul and D. Adams, Soft Matter, 2017, 13, 1914–1919 RSC.
- J. Shi, Y. Gao, Y. Zhang, Y. Pan and B. Xu, Langmuir, 2011, 27, 14425–14431 CrossRef CAS PubMed.
- L. Chen, T. O. McDonald and D. J. Adams, RSC Adv., 2013, 3, 8714 RSC.
- D. J. Adams, E. Draper, H. Su, C. Brasnett, R. Poole, S. Rogers, H. Cui and A. Seddon, Angew. Chem., Int. Ed., 2017 DOI:10.1002/anie.201705604.
- A. Z. Cardoso , Personal Communication.
- R. G. Alargova, D. S. Warhadpande, V. N. Paunov and O. D. Velev, Langmuir, 2004, 20, 10371–10374 CrossRef CAS PubMed.
- B. P. Binks, Curr. Opin. Colloid Interface Sci., 2002, 7, 21–41 CrossRef CAS.
- L. Chen, S. Revel, K. Morris, L. C. Serpell and D. J. Adams, Langmuir, 2010, 26, 13466–13471 CrossRef CAS PubMed.
Footnote |
| † We have not considered the behaviour of the silicone oil because it is density matched with the aqueous phase and hence does not rise to meet the hydrogel interface. |
| This journal is © The Royal Society of Chemistry 2017 |