
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Urotensin-II peptidomimetic incorporating a non-reducible 1,5-triazole disulfide bond reveals a pseudo-irreversible covalent binding mechanism to the urotensin G-protein coupled receptor†
Salvatore
Pacifico
a,
Aidan
Kerckhoffs
b,
Andrew J.
Fallow
c,
Rachel E.
Foreman
c,
Remo
Guerrini
a,
John
McDonald
d,
David G.
Lambert
*d and
Andrew G.
Jamieson
 *b
*b
aDepartment of Chemical and Pharmaceutical Sciences and LTTA, University of Ferrara, Ferrara, Italy
bSchool of Chemistry, Joseph Black Building. University Avenue, Glasgow, G12 8QQ, UK. E-mail: andrew.jamieson.2@glasgow.ac.uk
cDepartment of Chemistry, University of Leicester, University Road, LE1 9HN, UK
dDepartment of Cardiovascular Sciences, Division of Anaesthesia Critical Care & Pain Management, Robert Kilpatrick Clinical Sciences Building, Leicester Royal Infirmary, Leicester, LE2 7LX, UK. E-mail: dgl3@leicester.ac.uk
First published on 12th May 2017
Abstract
The urotensin-II receptor (UTR) is a class A GPCR that predominantly binds to the pleiotropic cyclic peptide urotensin-II (U-II). U-II is constrained by a disulfide bridge that induces a β-turn structure and binds pseudo-irreversibly to UTR and is believed to result in a structural rearrangement of the receptor. However, it is not well understood how U-II binds pseudo-irreversibly and the nature of the reorganization of the receptor that results in G-protein activation. Here we describe a series of U-II peptidomimetics incorporating a non-reducible disulfide bond structural surrogate to investigate the feasibility that native U-II binds to the G protein-coupled receptor through disulfide bond shuffling as a mechanism of covalent interaction. Disubstituted 1,2,3-triazoles were designed with the aid of computational modeling as a non-reducible mimic of the disulfide bridge (Cys5–Cys10) in U-II. Solid phase synthesis using CuAAC or RuAAC as the key macrocyclisation step provided four analogues of U-II(4–11) incorporating either a 1,5-triazole bridge (5, 6) or 1,4-triazole bridge (9, 10). Biological evaluation of compounds 5, 6, 9 and 10 was achieved using in vitro [125I]UII binding and [Ca2+]i assays at recombinant human UTR. Compounds 5 and 6 demonstrated high affinity (KD ∼ 10 nM) for the UTR and were also shown to bind reversibly as predicted and activate the UTR to increase [Ca2+]i. Importantly, our results provide new insight into the mechanism of covalent binding of U-II with the UTR.
Introduction
G-Protein Coupled Receptors (GPCRs) are important transmembrane proteins responsible for numerous cell-signaling processes. They play a fundamental role in normal physiology and their dysfunction is implicated in diseases including cardiovascular, neurological and reproductive among others.1 In view of this, GPCRs have become a major focus of the scientific community and are valuable drug targets. The G-protein cycle is initiated through the interaction of ligand molecules including hormones and neurotransmitters with the receptor. A conformation change of the receptor then causes G-protein activation and leads to intracellular signaling cascades.2 However, knowledge of the precise mechanism of receptor–ligand binding and structural rearrangement of GPCRs is lacking in many examples. There has been a recent explosion in GPCR structural biology research facilitated by the lipidic cubic phase method, mutation stabilized structures and advances in cryo-EM.3 This has highlighted commonalities in GPCR structural rearrangement resulting in activation, however this research has also demonstrated the many differences in ligand binding mechanisms. Despite these advances, full understanding of the scope and complexity of GPCR ligand interactions and their role in cell signaling is still to be determined.The urotensin-II receptor (UTR) is a subclass 1A GPCR with both central and peripheral expression. Recent evidence suggests that UTR isoforms are found in rat heart nuclear extract and can also play a role in the regulation of gene expression.4 UTR has two endogenous cyclic peptide ligands: urotensin-II (U-II) and urotensin-II related peptide (URP) (Fig. 1A and B).5,6 Together U-II and URP regulate the urotensinergic system and thus play a key role in the regulation of a number of human pathologies; the most well studied being cardiovascular. For example U-II is elevated in patients with heart failure.7 Despite this recent research, there is still a lot to learn to fully understand the nature of ligand recognition and the complexity of UTR involvement in cell signaling.
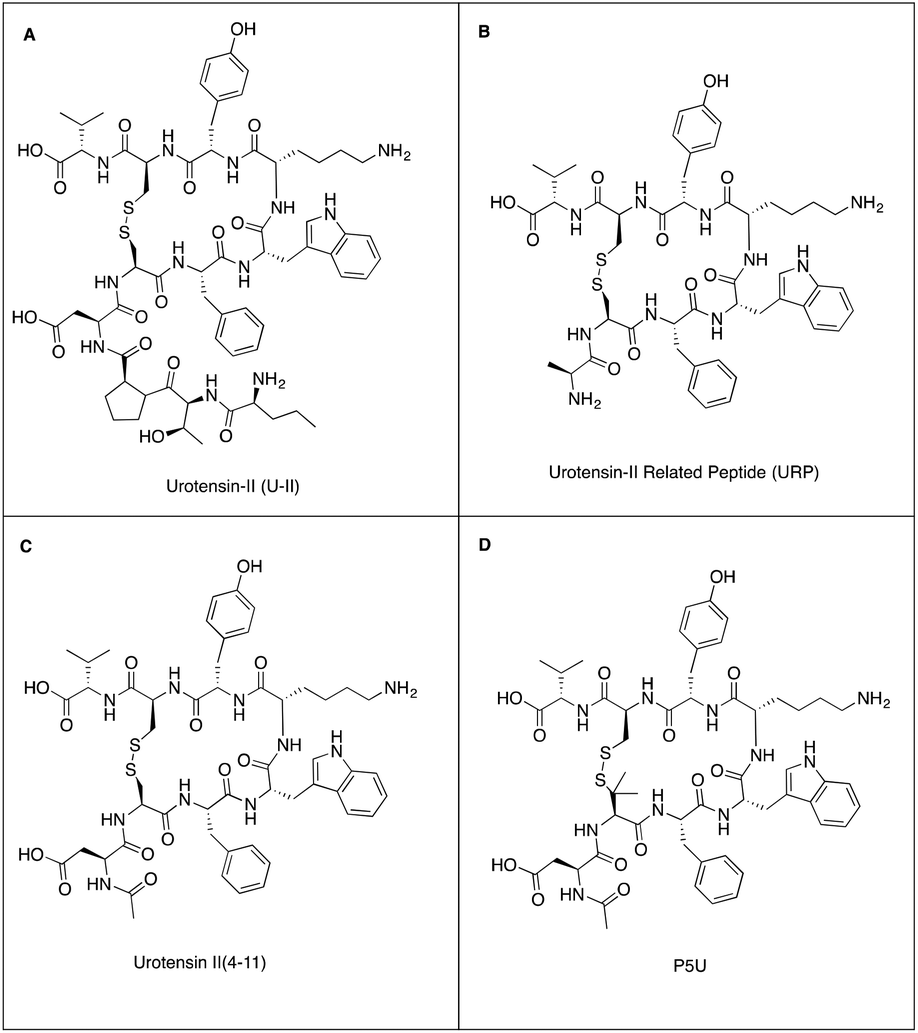 | ||
| Fig. 1 Structures of (A) urotensin-II, (B) urotensin II related peptide, (C) urotensin-II (4–11) and (D) synthetic agonist P5U. | ||
Consequently, the development of peptidomimetics based on U-II that act as agonists or antagonists of the UTR would be extremely useful tools to investigate the physiological role of the U-II/UTR system.
Structure–activity-relationship studies have identified the functionality and conformational preferences of U-II and URP that are responsible for mediating the molecular recognition event with the UTR. All U-II and URP analogues incorporate a conserved C-terminal cyclic hexapeptide that is structurally constrained by a disulfide bridge. This cyclic hexapeptide sequence adopts a β-turn in SDS micelle solution as determined by NMR spectroscopy and is proposed as the bioactive conformation.8
Several SAR studies have identified the amino acid residues that are essential for biological activity.9–12 An investigation of truncated analogues of U-II provided an octapeptide U-II(4–11) as the minimum fragment that retained full biological activity (Fig. 1C). The length and sequence of the U-II N-terminal residues vary across animal species. The role of the N-terminal tail is not well understood but may be involved in facilitating biased agonism.8 The intracyclic Trp7-Lys8-Tyr9 triad has been identified as the pharmacophore and is associated with biological activity.9,10 Most medicinal chemistry programs have thus focused on this amino acid triad within the cyclic hexapeptide sequence and have produced potent UTR peptidomimetic agonists: P5U and UPG84 (Fig. 1D);13 and also antagonists such as urantide.14,15
The U-II-UTR system is particularly intriguing as peptide binding is irreversible such that under normal physiological conditions (circulating U-II) the receptor is probably functionally silent.15 As a consequence, receptor function may be driven, not by increased peptide production, rather by modulation of receptor expression. To an extent UTR in primary ex vivo tissue will represent a partially desensitised state. Current peptide ligands are based on U-II and as such all display the unwanted effect of irreversibility; production of reversible U-II-like molecules is a current unmet need in the field.
Most irreversible agonists permanently bind to their receptor either by forming a covalent bond at the binding site or by binding with a dissociation rate of zero, relative to the limit of detection. U-II is a very tight binder of the UTR (KD 170 pM), however several analogues of U-II have been developed with lower binding affinity yet presumably are also pseudo-irreversible ligands.15 This intriguing effect led us to speculate whether the U-II disulfide bond that constrains the bioactive amino acid triad could undergo disulfide bond shuffling at the binding site and subsequently covalently cross-link to cysteine residues on the receptor.
A two-state acid (–SH)/base (–S–S–) model for GPCRs has previously been reported.16 Evidence for this model was obtained from the observation that the reducing agent DTT causes functional activation of the 5-HT2A GPCR in the presence or absence of agonist, and that the binding of agonists, but not antagonist, is pH dependent.
No X-ray crystal structure is available of the UTR, however the high-resolution crystal structure of human δ-opioid receptor (h-DOR, PDB ID: 4n6h)17 has previously been shown to be a high quality template to model the UTR.8 The h-DOR structure cannot provide the exact position of the inter-loop disulfide bond, however given the structural homology between h-DOR and UTR, an assumption of the general location of this structural motif may be gained. We were therefore extremely gratified to observe that UTR incorporates a disulfide bond between extracellular loop 1 (Cys123) and loop 2 (Cys199) (ESI Fig. S1†).18,19
Computation docking experiments have previously suggested that the U-II and URP binding site is between extracellular loops 2 and 3 occupying the hydrophobic cavity at the end of the helix bundle. Upon binding, disulfide bond shuffling is therefore possible and may result in a conformational change in the receptor and facilitate the observed cellular internalization. The reducing environment within the cytosol provides a mechanism for ligand release and recycling of the receptor at the nuclear membrane. To investigate this possibility, we designed a U-II peptidomimetic incorporating a non-reducible disulfide bond surrogate with the aim of developing a reversible UTR agonist.
The development of synthetic disulfide bond mimics is a challenge in peptide chemical biology. Technologies that effectively mimic disulfide bonds open the possibility of developing chemical probes that are resistant to reduction by glutathione and are thus more stable under physiological conditions. Designing synthetic functionality that accurately mimics the Cβ–Sγ–Sγ–Cβ atoms in combination with the correct spacing of the Cα has proven difficult to achieve.
The most successful strategies to mimic a disulfide bond to date include cystathionine (CH2–S, Ctt), diselenide (Se–Se), selenylsulfide (Se–S) and ditelluride (Te–Te) bonds.20 However, multiple steps are required for their preparation and so their use has been limited.21 Other synthetic disulfide bond surrogates that have been developed include thioether and olefin-based isosteres.22–25 However thioethers require multistep synthesis utilizing complex orthogonal protecting group strategies. Olefin-based isosteres rely on ring-closing metathesis (RCM) reactions that give cis/trans isomers, and required a subsequent palladium-catalyzed hydrogenation step to access a suboptimal, conformationally flexible alkane bridge.
Analogues of U-II incorporating a macrocyclic lactam in place of the disulfide bridge have been reported, although with reduced bioactivity compared to U-II indicating that the lactam functionality is not an accurate mimic of the disulfide bridge.26,27 Conformationally constraining the Cys5–Cys10 disulfide bridge within U-II(4–11), by replacing Cys5 with penicillamine (β,β-dimethyl cysteine) has given the most potent UTR agonists reported to date (Fig. 1D).13
Disubstitiuted 1,2,3-triazoles have been used extensively in peptides as surrogates for trans-amide bonds.28 1,2,3-Triazoles can be prepared as either the 1,4-isomer using a Copper-Catalyzed Azide–Alkyne Cycloaddition (CuAAC); or as the 1,5-isomer using a Ruthenium-Catalyzed Azide–Alkyne Cycloaddition (RuAAC). Several examples of bioactive peptides incorporating a 1,4-disubstituted 1,2,3-triazole as a disulfide bond mimic have been reported.29 However, in some instances it has been ineffective and caused structural unfolding leading to reduced biological activity.30 Kolmer was first to describe the use of a 1,5-disubstitiuted 1,2,3-triazole as a disulfide bond mimic. In this seminal work the disulfide bond within a macrocyclic sunflower trypsin inhibitor-I was replaced by a 1,5-triazole and the resulting peptidomimetic retained biological activity.31
Herein, we describe the development of a series of U-II(4–11) peptidomimetics that incorporate a triazole bridge to investigate whether the disulfide bridge of U-II could be responsible for the pseudo-irreversible binding mechanism. The most potent of these peptidomimetics is compound 6, which we refer to as Urotriazole because it incorporates a 1,5-triazole bridge that accurately mimics the disulfide bridge in U-II. Urotriazole 6 is a potent UTR agonist and was found to be a reversible binder in a radioligand U-II displacement assay with a full agonist profile at UTR.
Results and discussion
The NMR structure of U-II(4–11) in SDS micelle solution was used to computationally model the peptidomimetics and inform the design process (Fig. 2). Rather than mimicking the Cβ–Sγ–Sγ–Cβ disulfide bond we chose to design a bridge that would accurately mimic the orientation of the two Cα–Cβ bonds. To assess the effectiveness of the triazole bridges corresponding to compounds 5, 6, 9 and 10, computational overlays with the U-II NMR structure were created and the root mean square deviations (RMSD) calculated. In initial designs it was apparent that a symmetrical triazole bridge incorporating one methylene was too short to act as an effective surrogate. RMSD for structures corresponding to compounds 9 and 10 were a disappointing 1.173 Å and 0.783 Å, respectively. Addition of a second methylene group to the azide derived side of the bridge proved more effective. Overlay of compound 5, incorporating a 1,5-triazole provided a RMSD of 0.607 Å. The optimal design was Urotriazole 6, incorporating a 1,5-triazole that gave RMSD of 0.182 Å. | ||
| Fig. 2 Structure and overlays of energy minimized 3D models of U-II(4–11) variants 5, 6, 9 and 10 (blue nitrogen, green carbon of U-II(4–11); grey carbon atoms of 9–10 and 5–6; red oxygen, yellow sulfur atoms). Models were aligned at the respective Cα and Cβ on residues 5 and 10. RMSD calculated for the respective Cα and Cβ of residues 5 and 10 at the compared structures are given in Å. Measured distances (Å) between the Cα atoms of residues 5 and 10 are shown as pink dashed lines (U-II(4–11) is 3.95 Å for reference). | ||
Chemistry
Linear peptides 1 and 2 were synthesized by microwave-assisted Fmoc solid phase peptide synthesis (SPPS) on ChemMatrix™ 2-chlorotrityl resin using commercially available building blocks Fmoc-L-propargylglycine (Fmoc-Pra-OH) and Fmoc-L-azidoalanine (Fmoc-Aza-OH) or Fmoc-L-azido-homoalanine (Fmoc-Aha-OH) (Scheme 1). CuAAC is incompatible with trityl type resins due to the acidic nature of the reaction medium.32 Linear precursors 1 and 2 were therefore cleaved from the solid support under acidic conditions and then 1,4-disubstituted 1,2,3-triazoles 9 and 10 were prepared by CuAAC-mediated macrocyclisation from the unprotected precursors 7 and 8 in dilute solution. RuAAC reaction conditions are compatible with SPPS using acid sensitive resins. Macrocyclisation of linear peptides 1 and 2 was achieved on solid support using Cp*RuCl(cod) as the catalyst and microwave irradiation (60 °C) to give peptides 5 and 6 in quantitative conversion as determined by LCMS analysis of a cleaved peptide sample. Anhydrous reaction conditions were required to prevent decomposition of the ruthenium catalyst. Conversion of starting materials 1 and 2 into macrocyclic peptides 5, 6, 9 and 10 proceeds without a change in molecular weight, however an significant shift in LC retention time is observed and was used to determine the reaction progress. Disappearance of the azide IR absorption band at 2100 cm−1 was also used to confirm conversion of the starting materials to products. Triazole bridged peptides 5, 6, 9 and 10 were then purified by reverse-phase HPLC and characterized by analytical RP-HPLC and ESI-MS. | ||
Scheme 1 Synthesis of triazole bridged U-II(4–11) peptidomimetics. Reagents and conditions: (a) (i) 20% piperidine/DMF, 75 °C (MW); (ii) Fmoc-AA-OH, HCTU, DIEA, DMF, 75 °C (MW); (iii) repeat (i) & (ii); (iv) Ac2O, DIEA, DMF, RT; (b) Cp*RuCl(cod) (20 mol%), DMF, 60 °C (MW), 5 h; (c) 95![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2.5:2.5 TFA/TIS/H2O, RT, 3 h; (d) CuSO4·5H2O, NaAsc, DIEA, H2O, RT, 12 h. :2.5:2.5 TFA/TIS/H2O, RT, 3 h; (d) CuSO4·5H2O, NaAsc, DIEA, H2O, RT, 12 h. | ||
Biological data
The capacity of U-II(4–11) peptidomimetics 5, 6, 9 and 10, to act as orthosteric ligands and displace [125I]U-II from recombinant human UTR expressed in CHO cells (CHOhUTR) was assessed. [125I]U-II was displaced by native U-II in a concentration dependent and saturable manner (Fig. 3). Analysis of this data set yielded a pIC50 = KD of 9.36 nM for U-II (Table 1). Peptidomimetics 5 and 6 also displaced [125I]U-II binding, (Fig. 3) and had essentially identical IC50 = KD values of around 8 nM. Whilst chemical modification of U-II was tolerated there was a loss of affinity of ∼20 fold. Compound 10 displaced [125I]U-II by ∼40% at the highest concentration tested (1 μM). Compound 9 was inactive (Fig. 3 and Table 1). | ||
| Fig. 3 [125I]U-II binding data; isotope dilution. U-II, compounds 5 and 6 bound to UTR in CHOhUT cells with high (10 nM) affinity. Data are mean ± SEM (n = 5). In these experiments the concentration of [125I]U-II used was ultra low compared to its KD such that in this paradigm displacer IC50 = KD. | ||
| Compound | pKi | [125I]U-II binding reversibility | Intracellular Ca2+ | |
|---|---|---|---|---|
| pEC50 | E max (nM) | |||
| Data are mean SEM (n = 5 for binding and 3–4 for [Ca2+]i). | ||||
| U-II | 9.36 ± 0.19 | Irreversible | 9.65 ± 0.21 | 813 ± 200 |
| 5 | 7.98 ± 0.04 | Reversible | 8.49 ± 0.23 | 729 ± 66 |
| 6 | 7.96 ± 0.05 | Reversible | 8.47 ± 0.25 | 1036 ± 203 |
| 9 | Inactive | Inactive | Inactive | |
| 10 | <6 | Not tested | <6 | |
Having established that the peptidomimetics bind to the UTR, we next wanted to determine if this binding caused functional activation of the receptor and resulted in intracellular calcium release. To achieve this, [Ca2+]i was measured in Fura2 loaded CHOhUTR cells. At 1 μM 5, 6 and 10 produced monophasic increase in [Ca2+]i. Compound 9 was inactive (Fig. 4). The increase in [Ca2+]i observed with 5 and 6 was clearly concentration dependent and saturable; 10 did not saturate at 1 μM and 9 was inactive. Both 5 and 6 displayed a potency of ∼3 nM and this was ∼15 fold weaker than the native peptide U-II (Fig. 4 and Table 1). Similar [Ca2+]i data were obtained at endogenous UTR expressed at low levels in SJCRH30 rhabdomyosarcoma cells (ESI Fig. S2†).
 | ||
| Fig. 4 Intracellular Ca2+ data. U-II, compounds 5 and 6 and, at high concentrations 10, increased intracellular Ca2+ in CHOhUT cells. The increase for U-II and compounds 5 and 6 was concentration related and saturable. Data are mean ± SEM (n = 3–4). | ||
The purpose of the chemical modification was to favour reversibility of binding so we tested this in a wash on-wash off protocol. CHOhUTR cell membranes were labelled with a fixed concentration of U-II, 5 or 6. The membranes were then washed extensively (as in methods), incubated with a fixed concentration of [125I]U-II and increasing concentrations of unlabeled U-II added; essentially isotope dilution. Low binding of [125I]U-II indicated the original U-II challenge has bound irreversibly leaving few free receptors to which [125I]U-II could bind. In contrast, the binding of [125I]U-II to membranes that had been treated with 5 or 6 was indistinguishable from control untreated membranes; 5 and 6 binding was therefore reversible (Fig. 5).
 | ||
| Fig. 5 [125I]U-II binding data; reversibility. Binding of compounds 5 and 6 but not U-II to UTR in CHOhUT cells was reversible. Data are from a typical experiment (n = 5). Mean data are shown in Table 1. | ||
Conclusions
In conclusion, novel triazole bridged analogues of U-II have been computationally designed and prepared using a ruthenium or copper catalyzed macrocyclisation reaction. Biological evaluation using in vitro [Ca2+]i and [125I]UII binding assays demonstrated that compounds 5 and 6 are high affinity UTR ligands and bind reversibly. These peptidomimetics have provided new insight into the mechanism of pseudo-irreversible binding of U-II with the UTR. These data provide evidence that U-II binds covalently to the UTR. Future investigations in our laboratories will focus on structural studies to further understand the mechanism of U-II/UTR binding interactions and the nature of receptor conformation change.Experimental section
Materials and general methods
Solvents and reagents used in the experiments were purchased from Novabiochem or Sigma Aldrich and were used without purification. Fmoc-L-propargylglycine (Fmoc-Pra-OH) and Fmoc-L-azidoalanine (Fmoc-Aza-OH) were purchased from TCI and Fmoc-L-azido-homoalanine (Fmoc-Aha-OH) was purchased from Chiralix. Chloro(pentamethylcyclopentadienyl)(cyclooctadiene)ruthenium(II) (Cp*RuCl(cod)) catalyst was purchased from Sigma-Aldrich.All amino acids used in peptides synthesis were of L-configuration and were Fmoc protected. All amino acids had their side chains protected. DMF was dried over 4 Å molecular sieves and stored under nitrogen.
Solid Phase Peptide Synthesis was performed on a Biotage Alstra microwave-assisted, automated peptide synthesizer on 0.1 mmol scale. Solvents were evaporated under reduced pressure on Büchi vacuum rotary evaporators. Peptides were freeze-dried on a Labconco FreeZone 2.5 lyophiliser.
Analytical RP-HPLC analyses were preformed on a Gemini™ 5 μm C18 110 Å column (Phenomenex® Inc., Torrance, California, 150 mm × 4.6 mm, 5 μm) with a flow rate of 0.5 mL min−1 using a linear gradient of acetonitrile (0.1% trifluoroacetic acid (TFA)) in water (0.1% TFA). Retention times (tR) from analytical RP-HPLC are reported in minutes. Peptides were purified with a Gemini™ 5 μm C18 110 Å column (Phenomenex® Inc., Torrance, California, 250 mm × 21 mm, 5 μm, C18) using a specified linear gradient of acetonitrile (0.1% TFA) in water (0.1% TFA), with a flow rate of 10.6 mL min−1. UV detection wavelengths in analytical HPLC were 214 nm and 260 nm. UV detection wavelength in semi-preparative HPLC was 214 nm.
LCMS analysis was performed on a Waters Xevo Q ToF mass spectrometer coupled to a Water Acquity LC system with Waters Acquity UPLC BEH C18 column (2.1 × 50 mm). Solvent system was 0.1% formic acid in MeCN and 0.1% formic acid in H2O (deionised). Gradient was 5–100% of 0.1% formic acid in MeCN at 0.6 mL min−1. Mass accuracy was accomplished by using a reference lock mass scan once per second. ES cone voltage was 30 V. Collision energy was 4 eV. MS acquisition rate was 10 spectra per seconds with m/z range 50–2000 Da.
Peptide synthesis
Peptides were synthesized on a Biotage Alstra automated microwave-assisted solid-phase peptide synthesizer on 0.1 mmol scale using ChemMatrix 2-chlorotrityl resin (substitution: 0.66 mmol g−1, 100–200 mesh).Washing procedure: DMF was used for all of the wash cycles (4 washes between deprotection, coupling and acetylation reactions).
Analytical RP-HPLC indicated that all peptides had a purity of >97%, and the correct molecular weights were confirmed by LCMS (see ESI†).
Biological data
Acknowledgements
We thank the Universities of Leicester and Glasgow and the Leverhulme Trust (Research Project Grant RPG-2014-372) for financial support of this research. The authors thank Mick Lee (mass spectrometry) for technical assistance. S. P. would like to acknowledge the University of Ferrara for the award of a mobility grant (founded with 5 × 1000 to UniFe year 2013) to work in the lab of A. G. J. We also thank Dr Alfonso Carotenuto (University of Naples) for kindly providing the NMR structure of U-II.Notes and references
- M. C. Lagerström and H. B. Schiöth, Nat. Rev. Drug Discovery, 2008, 7, 339–357 CrossRef PubMed.
- T. H. Ji, M. Grossmann and I. Ji, J. Biol. Chem., 1998, 273, 17299–17302 CrossRef CAS PubMed.
- R. C. Stevens, V. Cherezov, V. Katritch, R. Abagyan, P. Kuhn, H. Rosen and K. Wüthrich, Nat. Rev. Drug Discovery, 2013, 12, 25–34 CrossRef CAS PubMed.
- N. D. Doan, T. T. M. Nguyen, M. Létourneau, K. Turcotte, A. Fournier and D. Chatenet, Br. J. Pharmacol., 2012, 166, 243–257 CrossRef CAS PubMed.
- S. A. Douglas and E. H. Ohlstein, Trends Cardiovasc. Med., 2000, 10, 229–237 CrossRef CAS PubMed.
- T. Sugo, Y. Murakami, Y. Shimomura, M. Harada, M. Abe, Y. Ishibashi, C. Kitada, N. Miyajima, N. Suzuki, M. Mori and M. Fujino, Biochem. Biophys. Res. Commun., 2003, 310, 860–868 CrossRef CAS PubMed.
- Y.-C. Zhu, Y.-Z. Zhu and P. K. Moore, Br. J. Pharmacol., 2006, 148, 884–901 CrossRef CAS PubMed.
- D. Brancaccio, F. Merlino, A. Limatola, A. M. Yousif, I. Gomez-Monterrey, P. Campiglia, E. Novellino, P. Grieco and A. Carotenuto, J. Pept. Sci., 2015, 21, 392–399 CrossRef CAS PubMed.
- S. Flohr, M. Kurz, E. Kostenis, A. Brkovich, A. Fournier and T. Klabunde, J. Med. Chem., 2002, 45, 1799–1805 CrossRef CAS PubMed.
- W. A. Kinney, H. R. Almond, J. Qi, C. E. Smith, R. J. Santulli, L. de Garavilla, P. Andrade-Gordon, D. S. Cho, A. M. Everson, M. A. Feinstein, P. A. Leung and B. E. Maryanoff, Angew. Chem., Int. Ed., 2002, 41, 2940–2944 CrossRef CAS PubMed.
- A. Brkovic, A. Hattenberger, E. Kostenis, T. Klabunde, S. Flohr, M. Kurz, S. Bourgault and A. Fournier, J. Pharmacol. Exp. Ther., 2003, 306, 1200–1209 CrossRef CAS PubMed.
- P. Labarrére, D. Chatenet, J. Leprince, C. Marionneau, G. Loirand, M. C. Tonon, C. Dubessy, E. Scalbert, B. Pfeiffer, P. Renard, B. Calas, P. Pacaud and H. Vaudry, J. Enzyme Inhib. Med. Chem., 2003, 18, 77–88 CrossRef PubMed.
- Rd. di Villa Bianca, E. Mitidieri, E. Donnarumma, F. Fusco, N. Longo, G. D. Rosa, E. Novellino, P. Grieco, V. Mirone, G. Cirino and R. Sorrentino, Asian J Androl., 2015, 17, 81–85 CrossRef CAS PubMed.
- R. Patacchini, P. Santicioli, S. Giuliani, P. Grieco, E. Novellino, P. Rovero and C. A. Maggi, Br. J. Pharmacol., 2003, 140, 1155–1158 CrossRef CAS PubMed.
- P. Tsoukas, É. Kane and A. Giaid, Front. Pharmacol., 2011, 2, 38 CAS.
- L. A. Rubenstein and R. G. Lanzara, J. Mol. Struct., 1998, 430, 57–71 CrossRef CAS.
- G. Fenalti, P. M. Giguere, V. Katritch, X. P. Huang, A. A. Thompson, V. Cherezov, B. L. Roth and R. C. Stevens, Nature, 2014, 506, 191–196 CrossRef CAS PubMed.
- H. Déméné, S. Granier, D. Muller, G. Guillon, M.-N. Dufour, M.-A. Delsuc, M. Hibert, R. Pascal and C. Mendre, Biochemistry, 2003, 42, 8204–8213 CrossRef PubMed.
- S. Boivin, I. Ségalas-Milazzo, L. Guilhaudis, H. Oulyadi, A. Fournier and D. Davoust, Peptides, 2008, 29, 700–710 CrossRef CAS PubMed.
- M. Muttenthaler, A. Andersson, A. D. de Araujo, Z. Dekan, R. J. Lewis and P. F. Alewood, J. Med. Chem., 2010, 53, 8585–8596 CrossRef CAS PubMed.
- T. S. Han, M.-M. Zhang, K. H. Gowd, A. Walewska, D. Yoshikami, B. M. Olivera and G. Bulaj, ACS Med. Chem. Lett., 2010, 1, 140–144 CrossRef CAS PubMed.
- L. Yu, Y. Lai, J. Wade and S. M. Coutts, Tetrahedron Lett., 1998, 39, 6633–6636 CrossRef CAS.
- J. Elaridi, J. Patel, W. R. Jackson and A. J. Robinson, J. Org. Chem., 2006, 71, 7538–7545 CrossRef CAS PubMed.
- S. Jiang, P. Li, S. L. Lee, C. Y. Lin, Y. Q. Long, M. D. Johnson, R. B. Dickson and P. P. Roller, Org. Lett., 2007, 9, 9–12 CrossRef CAS PubMed.
- M. A. Hossain, K. J. Rosengren, S. Zhang, R. A. Bathgate, G. W. Tregear, B. J. van Lierop, A. J. Robinson and J. D. Wade, Org. Biomol. Chem., 2009, 7, 1547–1553 CAS.
- P. Grieco, A. Carotenuto, R. Patacchini, C. A. Maggi, E. Novellino and P. Rovero, Bioorg. Med. Chem., 2002, 10, 3731–3739 CrossRef CAS PubMed.
- S. Foister, L. L. Taylor, J.-J. Feng, W.-L. Chen, A. Lin, F.-C. Cheng, A. B. Smith and R. Hirschmann, Org. Lett., 2006, 8, 1799–1802 CrossRef CAS PubMed.
- D. S. Pedersen and A. D. Abell, Huisgen cycloaddition in peptidomimetic chemistry, in Amino Acids, Peptides and Proteins in Organic Chemistry, 2011, 4, 101–127 Search PubMed.
- H. Li, R. Aneja and I. Chaiken, Molecules, 2013, 18, 9797–9817 CrossRef CAS PubMed.
- G. M. Williams, K. Lee, X. Li, G. J. S. Cooper and M. A. Brimble, Org. Biomol. Chem., 2015, 13, 4059–4063 CAS.
- M. Empting, O. Avrutina, R. Meusinger, S. Fabritz, M. Reinwarth, M. Biesalski, S. Voigt, G. Buntkowsky and H. Kolmar, Angew. Chem., Int. Ed., 2011, 50, 5207–5211 CrossRef CAS PubMed.
- V. Castro, H. Rodríguez and F. Albericio, ACS Comb. Sci., 2016, 18, 1–14 CrossRef CAS PubMed.
- W. Song, J. McDonald, V. Camarda, G. Calo, R. Guerrini, E. Marzola, J. P. Thompson, D. J. Rowbotham and D. G. Lambert, Naunyn Schmiedebergs Arch. Pharmacol., 2006, 373, 148–157 CrossRef CAS PubMed.
- A. Patel, R. A. Hirst, C. Harrison, K. Hirota and D. G. Lambert, Methods Mol. Biol., 2013, 937, 37–47 CAS.
- G. Grynkiewicz, M. Poenie and R. Y. Tsien, J. Biol. Chem., 1985, 260, 3440–3450 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Information on computational modelling and calculations, RP-HPLC chromatograms and MS spectra for each peptide. See DOI: 10.1039/c7ob00959c |
| This journal is © The Royal Society of Chemistry 2017 |