
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Structural insights into the ene-reductase synthesis of profens†
J.
Waller‡
a,
H. S.
Toogood‡
a,
V.
Karuppiah
a,
N. J. W.
Rattray
a,
D. J.
Mansell
a,
D.
Leys
a,
J. M.
Gardiner
 a,
A.
Fryszkowska§
b,
S. T.
Ahmed
a,
R.
Bandichhor
c,
G. P.
Reddy
c and
N. S.
Scrutton
*a
a,
A.
Fryszkowska§
b,
S. T.
Ahmed
a,
R.
Bandichhor
c,
G. P.
Reddy
c and
N. S.
Scrutton
*a
aManchester Institute of Biotechnology, University of Manchester, 131 Princess Street, Manchester M1 7DN, UK. E-mail: nigel.scrutton@manchester.ac.uk
bDr Reddy's Laboratories, Chirotech Technology Centre, 410 Cambridge Science Park Milton Rd, Cambridge CB4 0PE, UK
cInnovation Plaza, IPDO Bachupally, Dr Reddy's Laboratories, Hyderabad-500090, India
First published on 28th April 2017
Abstract
Reduction of double bonds of α,β-unsaturated carboxylic acids and esters by ene-reductases remains challenging and it typically requires activation by a second electron-withdrawing moiety, such as a halide or second carboxylate group. We showed that profen precursors, 2-arylpropenoic acids and their esters, were efficiently reduced by Old Yellow Enzymes (OYEs). The XenA and GYE enzymes showed activity towards acids, while a wider range of enzymes were active towards the equivalent methyl esters. Comparative co-crystal structural analysis of profen-bound OYEs highlighted key interactions important in determining substrate binding in a catalytically active conformation. The general utility of ene reductases for the synthesis of (R)-profens was established and this work will now drive future mutagenesis studies to screen for the production of pharmaceutically-active (S)-profens.
Introduction
Biocatalysis is an important tool in sustainable chemicals production by enabling mild reaction conditions and often high stereo-, regio- and enantio-selectivity.1 The ene reductases (ERs) are identified as important biocatalysts for asymmetric reduction of activated C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds.2 The flavin-containing NAD(P)H-dependent Old Yellow Enzyme (OYE) family of enzymes have been studied extensively for their biocatalytic potential due to their ability to catalyse the asymmetric reduction of activated CC bonds to generate up to two stereogenic centres. e.g.ref. 2a and 3. Typical activating groups include keto, aldehyde and nitro-moieties,2a in contrast to α,β-unsaturated mono-carboxylic acids and monoesters, which are typically poor substrates. In the latter case, reduction requires an additional electron-withdrawing group(s) conjugated to the double bond.3b,4
C bonds.2 The flavin-containing NAD(P)H-dependent Old Yellow Enzyme (OYE) family of enzymes have been studied extensively for their biocatalytic potential due to their ability to catalyse the asymmetric reduction of activated CC bonds to generate up to two stereogenic centres. e.g.ref. 2a and 3. Typical activating groups include keto, aldehyde and nitro-moieties,2a in contrast to α,β-unsaturated mono-carboxylic acids and monoesters, which are typically poor substrates. In the latter case, reduction requires an additional electron-withdrawing group(s) conjugated to the double bond.3b,4
Profens (2-arylpropanoic acids), such as naproxen ((S)-1), are important class of non-steroidal anti-inflammatory drugs (NSAIDS), widely used to treat pain and inflammatory diseases such as osteo- and rheumatoid arthritis.5 There has been interest in developing new routes to the enantiomerically-pure 2-arylpropanoic acids. e.g.ref. 6. Recently, the use of ERs in the chemoenzymatic synthesis of (R)-flurbiprofen (2) has been demonstrated,7 where the asymmetric CC reduction of the precursor was achieved by the OYE YqjM from Bacillus subtilis. Several alternative biocatalytic strategies were proposed such as (i) self-sufficient H-borrowing cascades in which OYEs coupled to aldehyde dehydrogenases to convert α,β-unsaturated aldehydes to a diverse range of (chiral/achiral) α-substituted carboxylic acids8 or (ii) use of FMN and ferredoxin [4Fe–4S]-dependent clostridial enoate reductases, which can reduce weakly activated enoates.9
Here, we explored the reaction scope of ene reductases from OYEs and other enzyme classes2a in profen synthesis from profen precusors (Scheme 1b and c). The experimental data was underpinned by the co-crystal structures of two oxidised OYEs XenA (from Pseudomonas putida)10 and NerA (from Agrobacterium radiobacter)11 with 2-phenylacrylic acid 5b. This revealed the active site structural features crucial for substrate binding that will drive future structure-driven evolution of the enzymes in the synthesis of non-steroidal anti-inflammatory profens and related compounds.
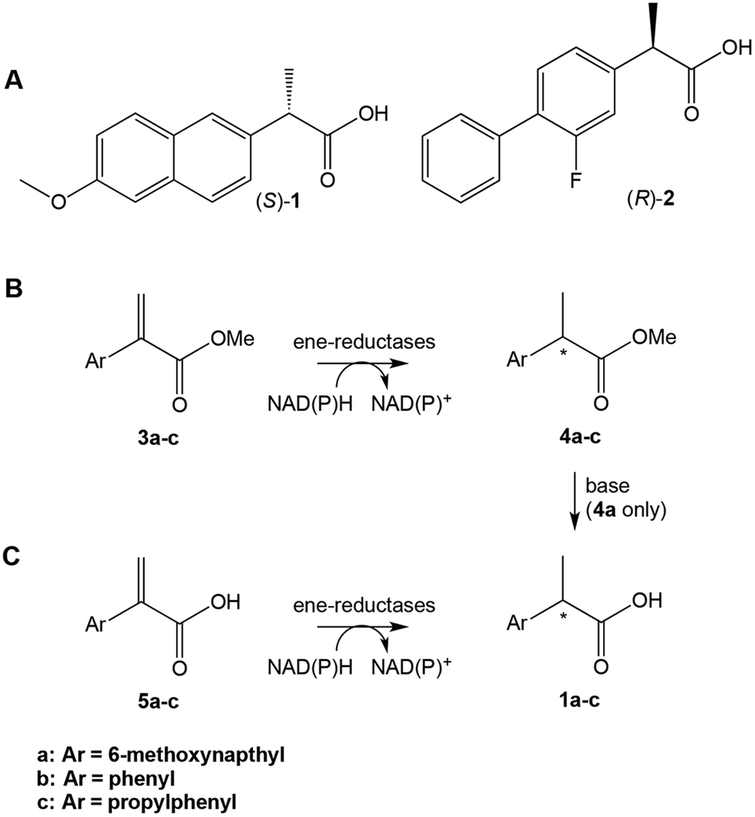 | ||
| Scheme 1 (A) Profens: (S)-naproxen 1a, (R)-flurbiprofen 2. (B) and (C) Proposed ene-reductase-catalysed steps in the chemoenzymatic synthesis of profens. | ||
Results and discussion
General
We studied the reduction of α,β-unsaturated carboxylic acids 5a–c by eight OYE family members12 and a flavin-independent, medium chain reductase, NtDBR, from Nicotiana tabacum.13 A prior study showed only a few ene-reductases were capable of reducing 5b.14 We ran 6 OYE classical subclass members: PETNR,15 OYE2 and OYE3,16 GYE,17 LeOPR1,18 NerA19 as well as 2 thermophilic-like members XenA,20 and TOYE21 (ESI Table S1† and Table 1 legends). These two subclasses differ in sequence length, key residue substitutions, oligomeric state and display distinct structural motifs.2 Thermophilic OYEs have tetrameric or higher oligomeric states, shorter sequence lengths due to the loss of some surface loops and contain a highly-conserved, arginine finger involved in substrate binding.12 Each enzyme contained either a N- or C-terminal His6-tag (C-His8 for PETNR) to enable rapid protein purification.| Substrate | Product | Enzyme | Conv.a (%) | eeb (%) |
|---|---|---|---|---|
| Reactions (1 mL) were performed in buffer (K2HPO4/KH2PO4 pH 7.5) containing the alkene (5 mM in 100% DMF), NADP+ (10 μM), D-glucose (15 mM), glucose dehydrogenase (GDH; 10 U) and enzyme (2 μM). Reactions were shaken at 30 °C for 24 h at 130 rpm.a Conv. = conversions. Quantitative analysis was performed on reactions extracted with ethyl acetate (0.9 mL) and derivatised with trimethylsilyl diazomethane to produce the respective methyl ester. Samples were analysed by GC using a ZB-semi volatiles column.b Enantiomeric excess was determined on reactions diluted 10-fold with acetonitrile, and analysed by HPLC using a Chiralpak AS-RH or Chiralpak AD-H column for 1a or 4a, respectively.c Reactions were performed in K2HPO4/KH2PO4 buffer pH 6.0.d Reactions were performed in 20 mM phosphate buffer containing 2-methyl-tetrahydrofuran. OYEs tested were PETNR = pentaerythritol tetranitrate reductase from Enterobacter cloacae PB2; LeOPR1 = 12-oxophytodienoate reductase 1 from Solanum lycopersicum; NerA = GTN reductase from Agrobacterium radiobacter; OYE2 from Saccharomyces cerevisiae; OYE3 from Saccharomyces cerevisiae; GYE from Gluconobacter oxydans; XenA = xenobiotic reductase from Pseudomonas putida; and TOYE = thermophilic Old Yellow Enzyme from Thermoanaerobacter pseudethanolicus E39. Due to quantity limitations of substrates 5a to 5c, they were only tested with OYEs active towards other profen precursors. | ||||
| 3a |
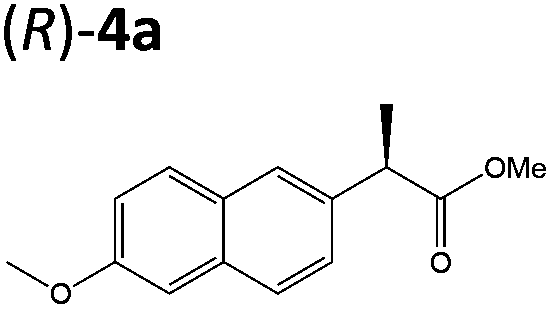
|
XenA | 57 | >99 (R) |
| GYE | 24 | >99 (R) | ||
| LeOPR1 | 21 | >99 (R) | ||
| TOYE | 17 | >99 (R) | ||
| PETNR | 9 | >99 (R) | ||
| NerA | 3 | >99 (R) | ||
| 5a |
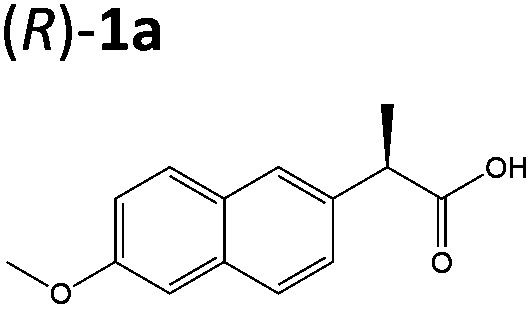
|
XenA | 94 | >99 (R) |
| GYE | 95 | >99 (R) | ||
| 5b |
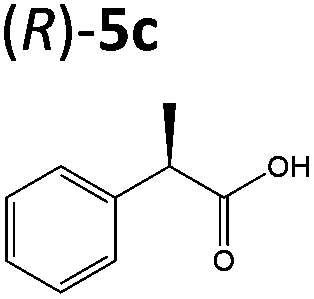
|
XenA | 97c | >99 (R) |
| GYE | 44b | >99 (R) | ||
| YqjMd | 95d (ref. 7) | >99 (R) | ||
| 5c |
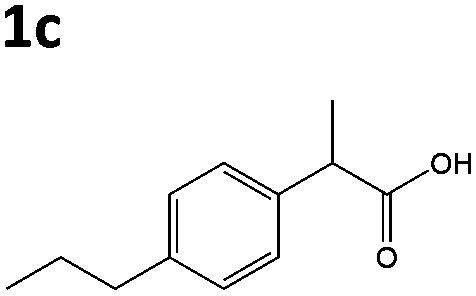
|
XenA | 7c,d | N/D |
| GYE | 27c,d | N/D | ||
Enzymatic activity
Steady-state turnover reactions were performed with each purified enzyme using the known substrates ketoisophorone, 2-cyclohexen-1-one and nitrocyclohexene to check for native activity (ESI Table S1†). The specific activities of PETNR and NtDBR matched literature values, while TOYE and XenA showed a 40% and 30% reduction in activity, respectively. Subsequently two naproxen (1) precursors 2-(6-methoxynaphthalen-2-yl)acrylic acid 5a and its methyl ester 3a were tested (Table 1). Carboxylic acid products were converted into the equivalent methyl esters using trimethylsilyl diazomethane prioir the GC analysis (ESI Fig. 1†). Out of 9 ene-reductases tested with substrate 5a only XenA and GYE were active, showing 94–95% conversion to 1a. Subsequently, we ran the reactions on 50 mg-scale with both XenA and GYE to isolate the product by preparative HPLC to confirm its structure by LCMS, 1H NMR and chiral HPLC (ESI Fig. 2 and 3†). Unfortunately both XenA and GYE generated (R)-1a with high enantiopurity (>99% ee; ESI Fig. 4 and 5†), rather than the pharmacologically active (S)-enantiomer.Six enzymes displayed activity towards methyl ester 3a to give (R)-4a (Table 1 and ESI Fig. 6†). The highest conversion (54%) was obtained with XenA, while GYE, LeOPR1 and TOYE were 2–3-fold less active. PETNR and NerA showed <10% conversion. No (S)-selective enzyme was identified in this study.
OYEs XenA and GYE were subsequently tested with 2-(4-propylphenyl)acrylic acid 5b and 2-phenylacrylic acid 5c. Low conversions were obtained with substrate 5b (Table 1), as determined previously with GYE in previous studies.14 No activity was detected with LeOPR1, however earlier work showed a slight (15% conversion) reaction with 5b.14 In contrast, XenA and YqjM quantitatively reduced 2-phenylpropenoic acid 5b to 5c, with moderate conversion (44%) obtained with GYE (Table 1; ESI Fig. 7†). Reactions with the best enzymes were performed on a 50 mg-scale and the products were isolated by preparative HPLC. Structural identity was confirmed by 1H NMR and UHPLC-MS (ESI Fig. 8–10†). In comparison, prior studies of the reduction of the methyl ester derivative of 3b by the OYE ClER from Clavispora lusitaniae showed no activity.22 We tested all the OYEs and NtDBR with (Z)-but-2-enoic acid and methacrylic acid, but no activity was detected.
Structures of XenA and NerA bound to 2-phenylacrylic acid 5b
We performed sequence (Fig. 1) and structural (Table 2 and Fig. 2) comparisons of XenA, NerA and other OYEs to explain the differences in their reactivity towards the substrates 3a and 5a–c. While XenA and YqjM belong to the thermophilic-like class, GYE is a classical OYE, indicating that the activity towards profens is not restricted to a single subclass of OYEs. We determined the co-crystal structures of XenA and NerA with 2-phenylacrylic acid 5b, the latter inactive against this substrate. In spite of extensive crystallisation trials, no co-crystal structure of XenA or NerA could be obtained with naproxen precursor 5a due to poor compound solubility in crystallisation solution. The data summary and refinement parameters for these structures are listed in Table 2. The structures enabled a comparison of substrate binding modes in both active and inactive conformations. | ||
| Fig. 1 Sequence alignment, using PROMALS3D,23 showing selected regions covering the active site residues of ‘classical’ and ‘thermophilic-like’ class of OYEs. Active site residues are highlighted in green and red for classical and thermophilic-like OYEs respectively. Residue numbers are displayed for NerA and XenA. | ||
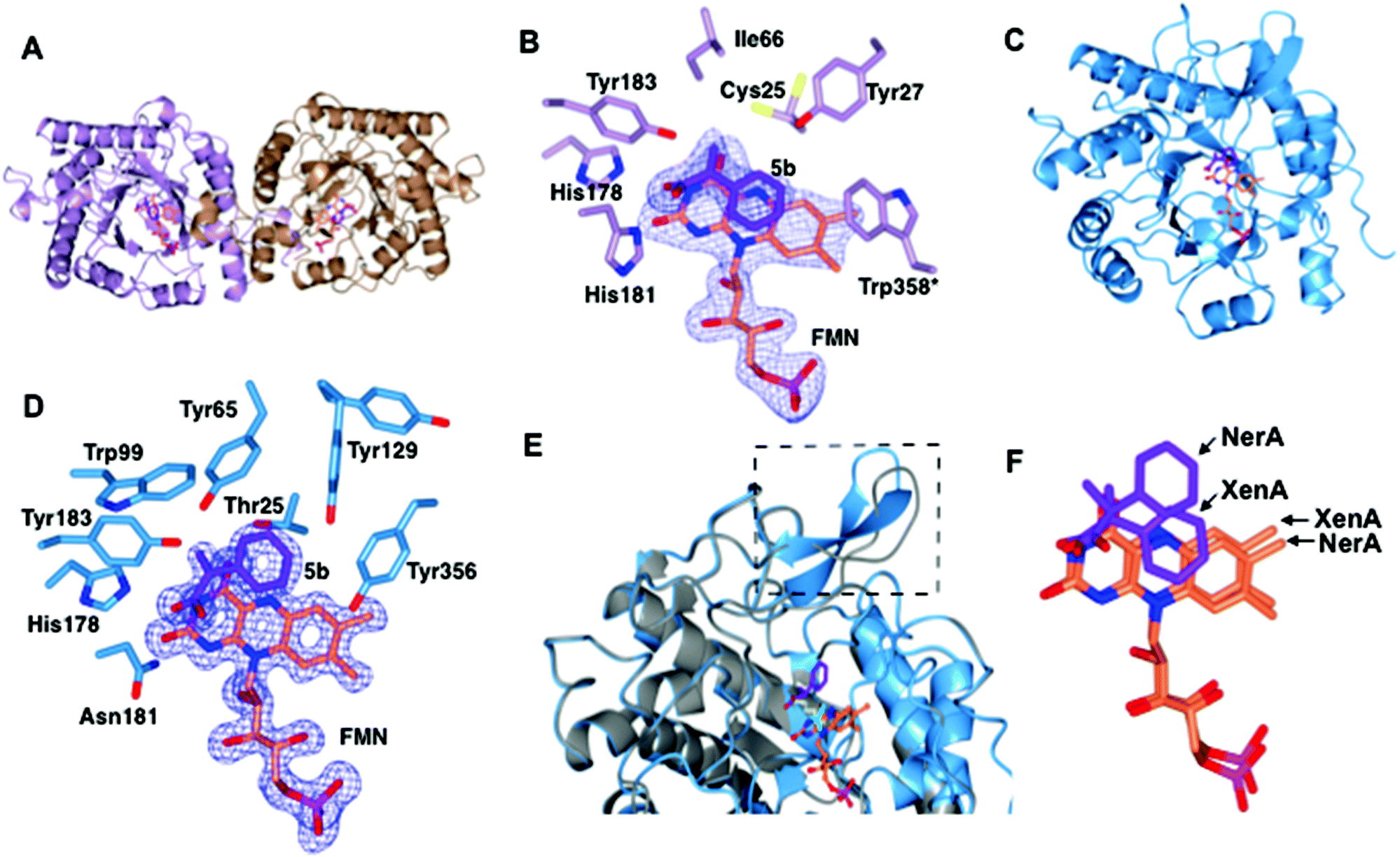 | ||
| Fig. 2 Structural comparisons of the co-crystal structures of XenA-5b and NerA-5b. (A) Overall structure of XenA, with the amino acids shown as ribbons with FMN (coral) and 5b (purple) as sticks. (B) Active site of XenA bound to ligand 5b. The side chains of residues that are up to 4 Å distance surrounding 5b are displayed. Trp358 in XenA, which is from the adjacent monomer, is labelled with an asterisk. The oxygen, nitrogen and phosphorous atoms are coloured red, blue and magenta respectively. The Fo–Fc omit maps (blue mesh) of FMN and 5b are contoured at 3σ. (C) Overall structure of NerA, with amino acids, FMN and 5b shown as in A. (D) Active site of NerA bound to ligand 5b with amino acids, FMN, 5b and omit maps as shown as in B. (E) Overlay of apo-NerA (4JIC, grey) and NerA-5b complex. The loop β3 that shifts and adopts an anti-parallel β-strand in the NerA-5b complex is indicated by dotted lines. The PEG molecule (yellow) in the active site of apo-NerA was replaced by 5b (purple) and acetate (cyan) in the NerA-5b complex. (F) Orientation of 5b and FMN when XenA and NerA co-crystal structures were superimposed. The figure was generated using ccp4 mg.31 | ||
| Parameters | XenA-5b (PDB 5N6Q) | NerA-5b (PDB 5N6G) |
|---|---|---|
| a Diamond light source. b Values in parentheses correspond to the higher resolution shell. | ||
| Data collection | ||
| Space group | P22121 | P212121 |
| Unit cell dimensions | a = 57.39 Å, b = 84.04, | a = 60.09 Å, b = 69.16, |
| c = 155.97 Å; α = β = γ = 90° | c = 91.89 Å; α = β = γ = 90° | |
| X-ray source | DLSa-I24 | DLSa-I03 |
| Wavelength (Å) | 0.96862 | 0.97625 |
| Resolution range (Å) | 77.99–2.20 (2.27–2.20)b | 91.89–1.58 (1.62–1.58)b |
| Multiplicity | 3.5 (3.5) | 6.3 (6.2) |
| I/σI | 5.6 (1.8) | 12.1 (1.5) |
| Completeness (%) | 98.4 (98.6) | 99 (98.1) |
| Rmerge | 0.195 (0.982) | 0.088(1.14) |
| Rmeas | 0.230 (1.153) | 0.106 (1.357) |
| Rpim | 0.118 (0.592) | 0.042 (0.535) |
| CC1/2 | 0.982 (0.514) | 0.998 (0.543) |
| Total observations | 133![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 897 (11604) 897 (11604) |
332942 (23776) |
| Total unique observations | 38419 (3290) |
52569 (3809) |
| Wilson B factor | 18.269 | 14.638 |
| Refinement | ||
| R-work | 0.1658 | 0.1404 |
| R-free | 0.2254 | 0.1641 |
| RMS (bonds) | 0.009 | 0.008 |
| RMS (angles) | 0.94 | 0.97 |
| Average B-factor | 25.6 | 23.9 |
| Ramachandran plot statistics (%) | ||
| Favored | 95.46 | 97.82 |
| Allowed | 4.26 | 1.91 |
| Outliers | 0.28 | 0.27 |
The overall crystal structure of XenA (Fig. 2A) is similar to the ‘thermophilic-like’ OYEs namely GkOYE from Geobacillus kaustophilus24 (rmsd of 1.1 Å over 333 residues), YqjM12 (rmsd of 1.1 Å over 332 residues), TsOYE from Thermus scotoductus SA-0125 (rmsd of 1.2 Å over 348 residues), and TOYE from Thermoanaerobacter pseudethanolicus E3921 (rmsd of 1.2 Å over 331 residues). In this OYE subclass the functional unit is a homodimer, composed of two sets of monomeric active sites with the addition of a highly conserved ‘arginine finger’ residue (R333 in TOYE; Fig. 1) from an adjacent monomer.21 In the case of XenA, this conserved arginine is replaced by a tryptophan (W358; Fig. 2B). Comparisons with our previously determined XenA structures bound to nicotinamide biomimetics,3b showed that the majority of the active site residues were relatively unchanged in position. The exception was W358 that was oriented away from the FMN, likely due to the presence of the bulky ligand 5b (Fig. 2B). Interestingly, the position of ligand 5b in XenA is roughly equivalent to the location of NADPH mimics in other crystal structures.3b
The aromatic ring and unsaturated carbons of 5b are oriented parallel and facing the si-face of the non-covalently bound isoalloxazine ring of FMN (Fig. 2B). The carbonyl oxygen and hydroxyl group are positioned to enable hydrogen bonds with residue atoms H178 NE, H181 ND and Y183 OH (Fig. 2B). Additionally, the carbonyl oxygen of 5b is 3.1 and 3.2 Å away from N3 and O2 atoms of FMN, respectively. Finally, the aromatic ring of 5b is stabilised by hydrogen bonding with residue Y27. Given that the Cα and Cβ of the substrate is only 3.8 Å from the N5 atom of FMN, this suggests that the structure represents an active confirmation for CC reduction.
The overall co-crystal structure of NerA (Fig. 2C), bound to 5b, is similar to the ‘classical’ OYEs such as PETNR26 (rmsd of 1.3 Å over 354 residues), MR from Pseudomonas putida M1027 (rmsd of 1.3 Å over 354 residues), NCR from Zymomonas mobilis28 (rmsd of 1.3 Å over 350 residues), previous NerA structure29 and SYE1 from Shewanella oneidensis30 (rmsd of 1.6 Å over 355 residues). Similar to the XenA-5b structure, the monomeric NerA-5b complex looks similar to its respective apo structure.19 The residues lining the active site (Fig. 2D) are in a similar position as in the apo structure.19
As previously noted19 the long loop β3, which partially caps the active site, is highly flexible as measured by the higher B-factors and the observation of weaker electron density for this region. Compared to the apo-structure, the loop β3 in the phenylacrylic acid-bound structure has significantly moved away from the active site (Fig. 2E).
This rearrangement has caused the side chain of residue T129 to be shifted closer to the aromatic ring of phenylacrylic acid 5b (Fig. 2D), a position occupied by F139 in the apo-structure. However the density for the side chain of Y129 indicates it likely occupies at least two different conformations, the second position located further away from the active site.
Phenylacrylic acid 5b is bound to the active site of NerA with the carbonyl oxygen and hydroxyl group position similar to the XenA-5b complex. As expected, the carbonyl oxygen of 5b forms hydrogen bonds with catalytic residues H178, N181 and Y183. Also, the hydroxyl moiety forms a hydrogen bond with a nearby water molecule. More importantly, the orientation of unsaturated carbons and the aromatic ring of 5b differ significantly from XenA-5b complex (Fig. 2F). This part of the molecule has flipped upwards and twisted away from the N5 atom of FMN, causing the aromatic ring of 5b to no longer be parallel to the isoalloxazine ring of FMN. This alternate orientation results in the formation of hydrogen bonds between the methylene group of 5b and OG atom of T125, and one of the aromatic carbon atoms of phenylacrylic acid 5b with the hydroxyl group of Y356. Crucially, this conformation in NerA has increased the distance between Cβ of 5b and N5 of FMN to 4.12 Å, and positions it at a non-optimal angle for hydride attack. Therefore, this structure suggests that the absence of activity with 5b could be due to ligand binding in an orientation not favoured for hydride transfer. The presence of the bulky side chain of Y27 in XenA would prevent substrate binding in a NerA-like conformation due to a clash with the aromatic ring of phenylacrylic acid 5b (N27 in NerA).
A sequence alignment of different OYEs shows that Tyr27 is conserved only among the ‘thermophilic-like’ subgroup (Fig. 1), in which XenA, TOYE and YqjM belong. As TOYE is inactive towards 5b, this suggests other residues and/or subtle active site features must play a role in determining if an OYE can bind 5b in an active conformation, particularly when noting that GYE (active with 5b) is a member of the classical subgroup of OYEs. Unfortunately, structural comparisons and/or docking models of GYE (pdb code: 3WJS) and other classical OYEs with 5b were not possible (results not shown) as the known low resolution X-ray crystal structure of GYE is lacking its FMN cofactor.
Conclusions
OYEs are traditionally known to be inactive towards α,β-unsaturated carboxylic acids or methyl esters unless there is a second activating group conjugated to the double bond, such as halides or a second acid/methyl ester functionality.2a We demonstrated that selected ene reductases from the OYE family are active towards α,β-unsaturated carboxylates 5a–c. Analysis of the crystals structures XenA (active) and NerA (inactive) with 5b highlighted subtle differences in active site and conformation, determining their ability to catalyse the double bond reduction.Recent reports have highlighted the potential use OYEs in the semi-synthetic biosynthesis of medicinally-important chirally active profens, namely (R)-2.5b,32 Given that the pharmacologically active form in most cases are (S)-profens, knowledge of the substrate binding mode of precursor substrates could lead to rationally-guided OYE active site mutagenesis studies to generate (S)-selective profen products.
Experimental
General reagents and equipment
All solvents used were Fisher Optima LCMS grade, and formic acid was Aristar grade from VWR. Compounds naproxen 1a, profen precursors methyl 2-(6-methoxynaphthalen-2-yl)propanoate 4a, 2-phenylacrylic acid 5b and 2-phenylpropionic acid 1b, 2-(4-propylphenyl)acrylic acid 5c, 2-(4-propylphenyl)propanoic acid 1c, and the Codexis glucose dehydrogenase enzyme were kindly supplied by Dr Reddy's Laboratories EU via commercial suppliers. The concentration of nicotinamide coenzymes (Melford) was determined by the extinction coefficient method (ε340 = 6220 M cm−1). Steady-state kinetic analyses were performed on a Cary UV-50 Bio UV/Vis scanning spectrophotometer using a quartz cuvette (1 mL; Hellma) with a 1 cm path length. Anaerobic kinetics and biotransformation reactions were set up and/or monitored within an anaerobic glove box (Belle Technology Ltd) under a nitrogen atmosphere (<5 ppm oxygen). Prior to anaerobic reactions, enzymes were deoxygenated by passage through a BioRad 10DG column equilibrated in anaerobic reaction buffer. Samples for single molecule 1H NMR analysis were dissolved in CDCl3 and analysed on a Bruker 400 Hz NMR spectrophotometer. All spectra were compared to authentic standards.Compound synthesis
:EtOAc 4:1, Rf = 0.45) to afford the title compound as white crystals (0.162 g, 30%). 1H NMR (400 MHz, CDCl3) δ ppm: 7.88 (s, 1H, H-1), 7.44–7.79 (m, 2H), 7.52–7.55 (dd, 1H, J = 8, 4 Hz, H-5), 7.16–7.21 (m, 2H), 6.44 (d, 1H, J = 2 Hz, CCH2), 6.02 (d, 1H, J = 2 Hz, CCH2), 3.95 (s, 3H), 3.89 (s, 3H). 13C NMR (100 MHz, CDCl3) δ ppm: 167.5 (CO), 158.1 (C-6), 141.2, 134.3, 131.9, 129.8, 128.5, 127.3, 126.6, 119.1, 105.6, 55.3 (OCH3), 52.2 (COOCH3). NMR traces are located in the ESI (Fig. S15†).
CH2), 6.06 (d, 1H, 2J = 1 Hz, CCH2), 3.86 (s, 3H, OCH3). 13C NMR (100 MHz, CDCl3) ∂ ppm: 168.0 (CO), 157.6 (C-6), 141.4, 133.8, 131.7, 129.7, 127.9, 126.8, 126.5, 126.3, 125.5, 118.8, 105.6, 55.2 (OCH3). NMR traces are located in the ESI (Fig. S15†).
Enzyme production and purification
The OYEs investigated were the following: (i) PETNR from Enterobacter cloacae PB2, (ii) TOYE from Thermoanaerobacter pseudethanolicus E39, (iii–iv) OYE2 and OYE3 from Saccharomyces cerevisiae, (v) NerA from Agrobacterium radiobacter, (vi) XenA from Pseudomonas putida, (vii) GYE from Gluconobacter oxydans and (viii) LeOPR1 from Solanum lycopersicum.33 The flavin-independent double bond reductase NtDBR from Nicotiana tabacum was also used in this study.13 The enzymes XenA, NerA and LeOPR1 were cloned into plasmid pET21a, while OYE2, OYE3, TOYE and NtDBR were expressed in pET21b. GYE was supplied in a pET28b plasmid, while the C-terminally His8-tagged PETNR gene34 was cloned into pBluescript SK+ (Stratagene) under the control of a native lac promoter.35 All constructs contained a C-terminal His6/8-tag, and were expressed in the Escherichia coli strain BL21(DE3) except for PETNR and TOYE which were expressed in JM109 and Arctic Express strains, respectively.All ene-reductases were produced and purified using the same general protocol. Starter cultures (5 mL and 20 mL) were produced overnight in lysogeny broth (LB) containing glucose (0.2%) and ampicillin (100 μg mL−1; 15 μg mL−1 kanamycin for GYE). The starter cultures were used to inoculate terrific broth (TB; 12 × 1 L per enzyme) containing glucose (2%), and incubated at 37 °C and 190 rpm until mid log phase (OD 600 nm ∼ 0.5). Recombinant protein expression was induced by the addition of IPTG (10 μM), followed by incubation overnight at 25 °C (18 °C for TOYE), at 190 rpm. Cells were harvested by centrifugation for 10 min at 5000g at 4 °C, and the supernatant discarded. The cell pellets were frozen in liquid nitrogen and stored at −80 °C.
The cells were resuspended in lysis buffer (50 mM KH2PO4/K2HPO4 pH 8.0) containing 1× protease inhibitor cocktail (Roche) and DNase (10 μg mL−1) and lysozyme (10 μg mL−1). Excess free FMN was added to the OYE cell slurries to increase the degree of flavination of the enzymes. The cells were lysed using a sonicator (Bandelin) with a probe set at 40% amplitude with cycles of 10 s ON and 10 s OFF for 12 min. The lysed cells were centrifuged at 18000g for 1 h and the supernatant was passed through a 0.2 micron filter. NaCl (300 mM) and imidazole (10 mM) were added, and the extracts were passaged through Nickel Sepharose affinity column (20 mL), pre-equilibrated in equilibration buffer (50 mM KH2PO4/K2HPO4 pH 8.0, 300 mM NaCl, 10 mM imidazole). The column was washed with wash buffer (50 mM KH2PO4/K2HPO4 pH 8.0, 300 mM NaCl, 20 mM imidazole; 200 mL) followed by elution buffer (50 mM KH2PO4/K2HPO4 pH 8.0, 300 mM NaCl, 300 mM imidazole, 150 mL). In some cases an additional nickel Sepharose purification step was performed, as detailed above, to increase the protein purity. Protein purity was assessed by SDS-PAGE, using 10–12% Mini-PROTEAN® TGX Stain-Free™ gels and Precision Plus protein unstained markers (BioRad) according to the manufacturer's instructions. Purified protein was desalted and concentrated using Vivaspin tubes (10000 MW cut-off). Protein concentrations of each OYE was determined using the extinction coefficient method with the following values (M cm−1): PETNR ε464 = 11300; TOYE ε456 = 11300; OYE2 ε462 = 10600; OYE3 ε464 = 10600; GYE ε450 = 11300; XenA ε450 = 11300; NerA ε450 = 11300 and LeOPR1 ε450 = 11300. Enzyme NtDBR protein concentration was determined by the Bradford method.36 Aliquots of enzyme were flash frozen in liquid nitrogen and stored in fractions at −80 °C.
Steady state kinetics
Standard ene-reduction reactions (1 mL) were performed anaerobically in buffer (50 mM K2HPO4/KH2PO4) containing alkene (1 mM) and NADPH (100 μM) at 25 °C at the reported pH optimum for each enzyme. The reaction was initiated by addition of the oxidative substrate and the loss of NADPH was monitored continuously at OD 340 nm. The substrates tested were ketoisophorone, 2-cyclohexen-1-one and nitrocyclohexene, and the results were compared to literature values where available. Initial rates were determined using Cary WinUV software and expressed as specific activity (μmol min−1 mg−1).Biotransformation reactions
Biotransformation reactions (1.0 mL) were performed anaerobically in buffer (K2HPO4/KH2PO4 pH 7.5) containing the alkene (5 mM in 100% DMF), NADP+ (10 μM), D-glucose (15 mM), glucose dehydrogenase (GDH; 10 U) and enzyme (2 μM). Reactions were shaken at 30 °C for 24 h at 130 rpm and terminated by extraction with ethyl acetate (0.9 mL) for achiral GC analysis, or diluted 10-fold with acetonitrile for chiral HPLC analysis. Scaled up reactions (50 mg) were performed using the same molarities as standard reactions and were incubated for 48 h. All biotransformation reactions were performed in at least duplicates, and the results are averages of the data.Analysis of biotransformations
Carboxylic acids 1a–b and 5a–b were converted to methyl esters using trimethylsilyl diazomethane prior to analysis. The biotransformation mixture (500 μL) was acidified to pH 5 using 1 M HCl and extracted into ethyl acetate (500 μL). Methanol (50 μL) was then added followed by trimethylsilyl diazomethane (10 μL). The mixtures were incubated for 20 min at 25 °C then quenched by the addition of glacial acetic acid (5 μL).The conversions and yields were determined by GC (Agilent Technologies 7890A system with FID detector) using limonene as an internal standard (0.5%): ZB-semi volatiles column (30 m; 0.25 mm; 0.25 μm film thickness; Phenomenex); injector 220 °C, split ratio of 20:1; 1 μL injection, 5 psi, flow 1 mL min−1 (helium); oven: 80 °C (2 min), 30 °C min−1 to 300 °C, hold (3 min). Quantitative analysis was carried using calibration curves.
Chiral analysis of the products was performed by HPLC (Agilent 1100 m with diode array detection, λ 220 nm) and compared to authentic standards of enantiomers. Compounds 1a and 5a: Chiralpak AS-RH (150 × 4.6 mm ID), 20 °C, MeCN:H2O (40:60), 0.8 mL min−1, Rt: (S)-1a 4.1 min; (R)-1a 4.6 min. Compound 5b: Chiralpak AD-H (150 × 4.6 mm ID), 20 °C, hexane:isopropanol (95:5), 1 mL min−1, Rt: (R)-1b 8.1 min; (S)-1b 9.2 min. Preparative HPLC purification of 1a was performed using Phenomenex Luna C18 column (5 μ × 21.2 × 250 mm) at 15 mL min−1 using a gradient of MeCN:H2O 10:90 to 50:50 over 15 min. 1H NMR analysis is presented in ESI Fig. S10–S13.†
UHPLC-MS analysis was performed using a Dionex U3000 RSLC system equipped with a Thermo-Fisher Q-Exactive Plus detector, 5 μL injection. All analysis was carried out in positive ionisation mode using Thermo Fisher Accucore C18 column (2.6 μm × 2.1 mm × 100 mm); average peak 6 s; 0.5 mL min−1; mobile phase: A (95% H2O 5% MeOH 0.1% formic acid) and B (95% MeOH 5% H2O 0.1% formic acid); gradient A:B: 95:5 (hold 2 min) to 5:95 over 3 min (hold 2 min), to 95:5 over 1 min (hold 2 min).
Protein crystallography
XenA and NerA were concentrated to 15 mg mL−1 and 23 mg mL−1 respectively. Crystallisation trials were set up using a Mosquito robot (TTP Labtech) by dispensing 200 nL of both protein and crystallisation solution. XenA and NerA crystals were obtained in crystallisation solution A (0.2 M sodium iodide, 0.1 M Bis–Tris propane pH 7.5 and 20% (w/v) PEG 3350) and B (0.1 M sodium acetate, 1 M lithium chloride, 30% (w/v) PEG 6000), respectively. Crystals were incubated with 25 mM 5b dissolved in their respective mother liquors and supplemented with 20% glycerol for 10 min. Crystals were cryo-cooled in liquid nitrogen prior to data collection.Structure solution for XenA and NerA
X-ray diffraction data were collected at Diamond Light Source beamlines I03 and I24. The NerA dataset was processed using the automated data reduction pipeline in xia237 using XDS41 to 1.58 Å. The XenA dataset was manually processed using MOSFLM38 and Aimless,39 as implemented in ccp4,40 to 2.2 Å. The 5b-bound structures of XenA and NerA were solved by molecular replacement using XenA (modified 5bPM) and NerA (modified 4JIC) coordinates, respectively, as search models in Phaser.41 The models were built and refined using Phenix.42 The structures were completed by iterative cycles of manual model building in Coot43 and refinement using phenix.refine.44 The structures were validated using PDB_REDO45 and Molprobity.46 The data summary and refinement parameters are listed in Table 2. The atomic coordinates and structure factors have been deposited in the Protein Data Bank (PDB codes 5N6Q (XenA) and 5N6G (NerA)).Acknowledgements
We thank Diamond Light Source for access to beamlines I03 and I24 (proposal number mx12788). We thank Dr Colin Levy, Manchester Protein Structure Facility (MPSF), for help with X-ray data collection. We thank Alexander Geddes for help with protein purification. This work was funded and supported by the UK Biotechnology and Biological Sciences Research Council (BB/I015779/1), Dr Reddy's Laboratories and Centre for Synthetic Biology of Fine and Speciality Chemicals (SynBioChem; BBSRC: BB/M017702/1). NSS was a Royal Society Wolfson Merit Award holder and is an Engineering and Physical Sciences Research Council (EPSRC; EP/J020192/1) Established Career Fellow.Notes and references
- P. J. Dunn, Chem. Soc. Rev., 2012, 41, 1452 RSC.
- (a) H. S. Toogood, J. M. Gardiner and N. S. Scrutton, ChemCatChem, 2010, 2, 892 CrossRef CAS; (b) H. S. Toogood, D. Mansell, J. M. Gardiner and N. S. Scrutton, in Comprehensive Chirality Vol. 7, Elsevier Science, Oxford, 1., 2011, p. 216 Search PubMed.
- (a) E. Brenna, G. Fronza, C. Fuganti, D. Monti and F. Parmeggiani, J. Mol. Catal. B: Enzym., 2011, 73, 17 CAS; (b) T. Knaus, C. E. Paul, C. W. Levy, S. de Vries, F. G. Mutti, F. Hollmann and N. S. Scrutton, J. Am. Chem. Soc., 2016, 138, 1033 CrossRef CAS PubMed; (c) H. S. Toogood and N. S. Scrutton, Curr. Opin. Chem. Biol., 2014, 19, 107 CrossRef CAS PubMed; (d) M. Hall and A. S. Bommarius, Chem. Rev., 2011, 111, 4088 CrossRef CAS PubMed.
- (a) S. Koul, D. H. G. Crout, W. Errington and J. Tax, J. Chem. Soc., Perkin Trans. 1, 1995, 2969 RSC; (b) H. S. Toogood, A. Fryszkowska, V. Hare, K. Fisher, A. Roujeinikova, D. Leys, J. M. Gardiner, G. M. Stephens and N. S. Scrutton, Adv. Synth. Catal., 2008, 350, 2789 CrossRef CAS PubMed; (c) C. K. Winkler, G. Tasnadi, D. Clay, M. Hall and F. Faber, J. Biotechnol., 2012, 162, 381 CrossRef CAS PubMed.
- (a) A. M. Evans, J. Clin. Pharmacol., 1996, 36, 7S CAS; (b) K. C. Duggan, D. J. Hermanson, J. Musee, J. J. Prusakiewicz, J. L. Scheib, B. D. Carter, S. Banerjee, J. A. Oates and L. J. Marnett, Nat. Chem. Biol., 2011, 7, 803 CrossRef PubMed.
- (a) H. Alper, A. Eisenstat and N. Satyanarayana, J. Am. Chem. Soc., 1990, 112, 7060 CrossRef CAS; (b) C. Giordano, S. Castaldi, S. Cavicchioli and M. Villa, Tetrahedron, 1989, 45, 4243 CrossRef CAS; (c) P. J. Harrington and E. Lodewijk, Org. Process Res. Dev., 1997, 1, 72 CrossRef CAS; (d) I. T. Harrison, B. Lewis, P. Nelson, W. Rooks, A. Roszkowski, A. Tomolonis and J. H. Fried, J. Med. Chem., 1970, 13, 203 CrossRef CAS PubMed; (e) T. Hiyama, M. Wakasa and T. Kutumoso, Synlett, 1991, 569 CrossRef CAS; (f) T. Ohta, H. Takaya, M. Kitamura, K. Nagai and R. Noyori, J. Org. Chem., 1987, 52, 3174 CrossRef CAS; (g) T. V. RajanBabu and A. L. Casalnuovo, J. Am. Chem. Soc., 1992, 114, 6265 CrossRef CAS; (h) H. R. Sonawane, N. S. Bellur, J. R. Ahuja and D. G. Kulkami, Tetrahedron: Asymmetry, 1992, 3, 163 CrossRef CAS.
- J. Pietruszka and M. Schölzel, Adv. Synth. Catal., 2012, 354, 751 CrossRef CAS.
- T. Knaus, F. G. Mutti, L. D. Humphreys, N. J. Turner and N. S. Scrutton, Org. Biomol. Chem., 2014, 13, 223 Search PubMed.
- (a) S. Nizam, R. K. Gazara, S. Verma, K. Singh and P. K. Verma, PLoS One, 2014, 9, e95989 Search PubMed; (b) F. Rohdich, A. Wiese, R. Feicht, H. Simon and A. Bacher, J. Biol. Chem., 2001, 276, 5779 CrossRef CAS PubMed; (c) A. Fryszkowska, K. Fisher, J. M. Gardiner and G. Stephens, Org. Biomol. Chem., 2010, 8, 533 RSC.
- D. S. Blehert, B. G. Fox and G. H. Chambliss, J. Bacteriol., 1999, 181, 6254 CAS.
- J. R. Snape, N. A. Walkley, A. P. Morby, S. Nicklin and G. F. White, J. Bacteriol., 1997, 179, 7796 CrossRef CAS PubMed.
- K. Kitzing, T. B. Fitzpatrick, C. Wilken, J. Sawa, G. P. Bourenkov, P. Macheroux and T. Clausen, J. Biol. Chem., 2005, 280, 27904 CrossRef CAS PubMed.
- D. J. Mansell, H. S. Toogood, J. Waller, J. M. X. Hughes, C. W. Levy, J. M. Gardiner and N. S. Scrutton, ACS Catal., 2013, 3, 370 CrossRef CAS PubMed.
- T. Reß, W. Hummel, S. P. Hanlon, H. Iding and H. Gröger, ChemCatChem, 2015, 7, 1302 CrossRef.
- A. Fryszkowska, H. Toogood, M. Sakuma, J. M. Gardiner, G. M. Stephens and N. S. Scrutton, Adv. Synth. Catal., 2009, 351, 2976 CrossRef CAS PubMed.
- M. Hall, C. Stueckler, B. Hauer, R. Stuermer, T. Friedrich, M. Breuer, W. Kroutil and K. Faber, Eur. J. Org. Chem., 2008, 1511 CrossRef CAS.
- N. Richter, H. Gröger and W. Hummel, Appl. Microbiol. Biotechnol., 2011, 89, 79 CrossRef CAS PubMed.
- C. Stueckler, M. Hall, H. Ehammer, E. Pointner, W. Kroutil, P. Macheroux and K. Faber, Org. Lett., 2007, 9, 5409 CrossRef CAS PubMed.
- G. Oberdorfer, A. Binter, S. Wallner, K. Durchschein, M. Hall, K. Faber, P. Macheroux and K. Gruber, ChemBioChem, 2013, 14, 836 CrossRef CAS PubMed.
- Y. Yanto, H. H. Yu, M. Hall and A. S. Bommarius, Chem. Commun., 2010, 46, 8809 RSC.
- B. V. Adalbjörnsson, H. S. Toogood, A. Fryszkowska, C. R. Pudney, T. A. Jowitt, D. Leys and N. S. Scrutton, ChemBioChem, 2010, 11, 197 CrossRef PubMed.
- Y. Ni, H.-L. Yu, G.-Q. Lin and J.-H. Xu, Enzyme Microb. Technol., 2014, 56, 40 CrossRef CAS PubMed.
- J. M. Pei and N. V. Grishin, Methods Mol. Biol., 2014, 1079, 263 Search PubMed.
- M. Schittmayer, A. Glieder, M. K. Uhl, A. Winkler, S. Zach, J. H. Schrittwieser, W. Kroutil, P. Macheroux, K. Gruber, S. Kambourakis, J. D. Rozzell and M. Winkler, Adv. Synth. Catal., 2011, 353, 268 CrossRef CAS.
- D. J. Opperman, B. T. Sewell, D. Litthauer, M. N. Isupov, J. A. Littlechild and E. van Heerden, Biochem. Biophys. Res. Commun., 2010, 393, 426 CrossRef CAS PubMed.
- H. S. Toogood, A. Fryszkowska, M. Hulley, M. Sakuma, D. Mansell, G. M. Stephens, J. M. Gardiner and N. S. Scrutton, ChemBioChem, 2011, 12, 738 CrossRef CAS PubMed.
- C. R. Pudney, S. Hay, J. Y. Pang, C. Costello, D. Leys, M. J. Sutcliffe and N. S. Scrutton, J. Am. Chem. Soc., 2007, 129, 13949 CrossRef CAS PubMed.
- S. Reich, H. W. Hoeffken, B. Rosche, B. M. Nestl and B. Hauer, ChemBioChem, 2012, 13, 2400 CrossRef CAS PubMed.
- G. Oberdorfer, A. Binter, S. Wallner, K. Durchschein, M. Hall, K. Faber, P. Macheroux and K. Gruber, ChemBioChem, 2013, 14, 836 CrossRef CAS PubMed.
- D. van den Hemel, A. Brige, S. N. Savvides and J. Van Beeumen, J. Biol. Chem., 2006, 281, 28152 CrossRef CAS PubMed.
- S. McNicholas, E. Potterton, K. S. Wilson and M. E. Noble, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2011, 67, 386 CrossRef CAS PubMed.
- J. L. Eriksen, S. A. Sagi, T. E. Smith, S. Weggen, P. Das, D. C. McLendon, V. V. Ozols, K. W. Jessing, K. H. Zavitz, E. H. Koo and T. E. Golde, J. Clin. Invest., 2003, 112, 440 CrossRef CAS PubMed.
- J. Straßner, A. Fürholz, P. Macheroux, N. Amrhein and A. Schaller, J. Biol. Chem., 1999, 274, 35067 CrossRef.
- M. E. Hulley, H. S. Toogood, A. Fryszkowska, D. J. Mansell, G. M. Stephens, J. M. Gardiner and N. S. Scrutton, ChemBioChem, 2010, 11, 2433 CrossRef CAS PubMed.
- R. E. Williams and N. C. Bruce, Microbiology, 2002, 148, 1607 CrossRef CAS PubMed.
- G. L. Peterson, Methods Enzymol., 1983, 91, 95 CAS.
- G. Winter, C. M. Lobley and S. M. Prince, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2013, 69, 1260 CAS.
- H. R. Powell, O. Johnson and A. G. W. Leslie, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2013, 69, 1195 CrossRef CAS PubMed.
- P. R. Evans and G. N. Murshudov, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2013, 69, 1204 CrossRef CAS PubMed.
- M. D. Winn, C. C. Ballard, K. D. Cowtan, E. J. Dodson, P. Emsley, P. R. Evans, R. M. Keegan, E. B. Krissinel, A. G. Leslie, A. McCoy, S. J. McNicholas, G. N. Murshudov, N. S. Pannu, E. A. Potterton, H. R. Powell, R. J. Read, A. Vagin and K. S. Wilson, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2011, 67, 235 CrossRef CAS PubMed.
- A. J. McCoy, R. W. Grosse-Kunstleve, P. D. Adams, M. D. Winn, L. C. Storoni and R. J. Read, J. Appl. Crystallogr., 2007, 40, 658 CrossRef CAS PubMed.
- P. D. Adams, P. V. Afonine, G. Bunkoczi, V. B. Chen, I. W. Davis, N. Echols, J. J. Headd, L. W. Hung, G. J. Kapral, R. W. Grosse-Kunstleve, A. J. McCoy, N. W. Moriarty, R. Oeffner, R. J. Read, D. C. Richardson, J. S. Richardson, T. C. Terwilliger and P. H. Zwart, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2010, 66, 213 CrossRef CAS PubMed.
- P. Emsley, B. Lohkamp, W. G. Scott and K. Cowtan, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2010, 66, 486 CrossRef CAS PubMed.
- P. V. Afonine, R. W. Grosse-Kunstleve, N. Echols, J. J. Headd, N. W. Moriarty, M. Mustyakimov, T. C. Terwilliger, A. Urzhumtsev, P. H. Zwart and P. D. Adams, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2012, 68, 352 CrossRef CAS PubMed.
- R. P. Joosten, F. Long, G. N. Murshudov and A. Perrakis, IUCrJ, 2014, 1, 213 CrossRef CAS PubMed.
- V. B. Chen, W. B. Arendall III, J. J. Headd, D. A. Keedy, R. M. Immormino, G. J. Kapral, L. W. Murray, J. S. Richardson and D. C. Richardson, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2010, 66, 12 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ob00163k |
| ‡ These authors contributed equally to the work. |
| § Current address: Merck Research Laboratories, Merck & Co., Inc., P.O. Box 2000, Rahway, New Jersey 07065, USA. |
| This journal is © The Royal Society of Chemistry 2017 |