
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Construction of a polyproline structure with hydrophobic exterior using octahydroindole-2-carboxylic acid†
Vladimir
Kubyshkin
 * and
Nediljko
Budisa
*
* and
Nediljko
Budisa
*
Biocatalysis group, Institute of Chemistry, Technical University of Berlin, Müller-Breslau-Str., 10, 10623, Berlin, Germany. E-mail: kubyshkin@win.tu-berlin.de; nediljko.budisa@tu-berlin.de
First published on 8th December 2016
Abstract
The proline analogue (2S,3aS,7aS)-octahydroindole-2-carboxylic acid (Oic) has been previously applied as a proline substitute in pharmocologically active peptides and as a structural component of the antihypertensive drug Perindopril. Herein, we describe the formation of an oligoproline structure by an Oic oligomer. A series of Oic oligomers were investigated to show the structural and energetic contribution of appended residues to the structure. NMR investigation of these oligomers revealed an all-trans amide bond structure, and we provide evidence that a cascade-like mechanism is responsible for the all-trans folding cooperativity. X-ray crystallography of the Oic-hexapeptide clearly demonstrates that the all-trans structure of the Oic oligomer is a polyproline II helix. Thus, as a hydrophobic proline analog with a highly stable trans-amide bond, Oic represents an ideal building block for hydrophobic sites of polyproline II structures in biologically relevant contexts.
Introduction
Polyproline-II (PII) is a ubiquitous secondary structural class in peptides and proteins that occupies a unique region on the Ramachandran plot.1 The name PII was originally coined to describe the structure that emerged upon mutarotation of polyproline dissolved in water.2,3 Later discoveries showed the abundance of PII structures in a diversity of sequence folds. It was further shown that PII helices can consist of various poly-amino acid sequences and do not necessarily contain proline.4 As an extended structure, PII is common in unfolded proteins,5 and even single N-acylated amino acids exhibit spectroscopic signatures of this conformation.6Since PII structures do not necessarily contain proline in the linear sequences, some have proposed that PII be renamed ‘polypeptide-II’.7 However, as the most conformationally restricted among the canonical amino acids, proline (Fig. 1; 1) is uniquely effective at sustaining the PII structure backbone.8,9 Accordingly, proline-rich sequences can serve as scaffolds for functionalized PII motifs.10 For example, proline-rich antimicrobial11 and cell-penetrating peptides12 are known to adopt the PII fold with the help of amphipathic interactions in a membrane environment. Exploring this concept, Filon et al. constructed an amphipathic, cell-penetrating PII peptide by functionalizing the hydroxyl groups on an oligo-hydroxyproline scaffold with polar and hydrophobic side-chains at defined helical sites.13 In another example, Pujals et al. increased cellular uptake of the cell-penetrating peptide SAP by replacing proline with silaproline at the hydrophobic PII site.14,15 Notably, a simple PII structure (3.0 residues per turn) can serve as an effective molecular ruler in spectroscopic studies (e.g., FRET).16
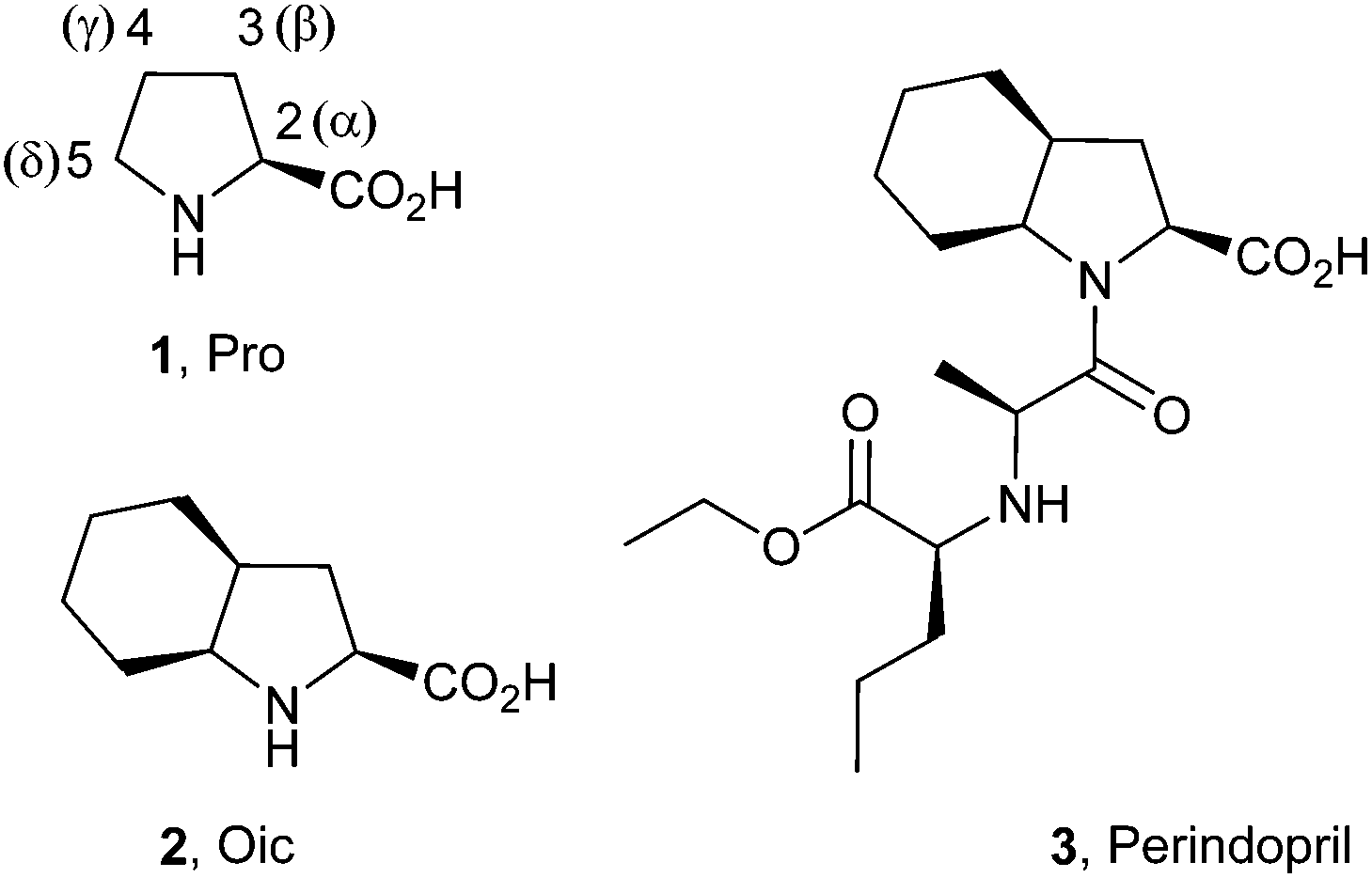 | ||
| Fig. 1 Structures of proline (1, Pro), (2S,3aS,7aS)-octahydroindole-2-carboxylic acid (2, Oic) and Perindopril (3). | ||
The kinetic and thermodynamic stability of the peptidyl-prolyl trans-peptide bond has a direct impact on the stability of proline-rich PII structures.17 Experiments with proline analogues showed that anchoring the proline ring in the exo-pucker conformation increases the preference for the trans-amide bond, which has enabled the construction of more stable PII structures.18,19 This effect has been attributed to the n → π* donative interaction between the carbonyl groups in the sequence (Fig. 2).20,21 Recently, we suggested that the donative effect may be enhanced through a cascade effect in the chain of the peptide bonds, leading to cooperative formation of the all-trans structural fold.22
 | ||
| Fig. 2 trans–cis amide equilibrium of proline residue (above), exo-pucker of the proline side chain (below). | ||
(2S,3aS,7aS)-Octahydroindole-2-carboxylic acid (2, commonly referred as Oic) is a proline analogue that has been used as a building block for the construction of pharmacologically relevant sequences, particularly bradykinin antagonists.23,24 Solution NMR structures25 have demonstrated that the Oic cyclohexane ring adopts a chair conformation in these peptides, which may anchor the proline in the exo-pucker conformation. A recent theoretical study of the methylamide of N-acetyl Oic suggested the ε backbone conformation for this residue is predominant in solution.26
In addition, Oic is a structural constituent of the antihypertension drug Perindopril (3). Early NMR studies have demonstrated a remarkably high trans-amide ratio (>90%) for the peptidyl-Oic constituent of Perindopril at high and low pH values.27 These results suggest Oic may be a good candidate for construction of a PII structure. Here, we demonstrate that Oic is capable of forming a stable and regular polyproline structure in solution, supported by the presence of a PII conformation in crystals of an Oic hexamer.
Results and discussion
Physicochemical comparison of Oic with (4S)- and (5S)-methylproline
We first aimed to physicochemically characterize Oic (2) in common model frames. Proline (1), (4S)-methylproline (4), and (5S)-methylproline (5) were used for comparison (Fig. 3). Reports have suggested that 4 exhibits a larger trans-content than proline due to the exo-pucker anchoring.18,28 In contrast, 5 is expected to sustain a trans-amide content similar to that of proline, despite the lower barrier of rotation for the amide due to the δ(cis)-substitution.29–31 | ||
| Fig. 3 Structures of the amino acids taken for comparison of their physicochemical properties: 1, 2, 4, 5. | ||
Our results indicate that Oic combines these structural impacts of γ(cis)- and δ(cis)-substitutions. The trans-amide content in Ac-Oic-OMe was among the highest reported to date (Table 1) for a proline analogue (e.g., higher than that in analogous (4R)-fluoro- and hydroxyproline models).21
| AA | pKa | ΔpKa |
|
K trans/cis | ||||
|---|---|---|---|---|---|---|---|---|
| AA (ammonium) | Ac-AA-OH (carboxyl) | Ac-AA-X | ||||||
| s-trans | s-cis | X = O− | X = OH | X = OMe | ||||
| 1 (Pro) | 10.68 | 3.55 | 2.85 | 0.70 | 0.67 | 0.81 ± 0.02 | 3.77 ± 0.05 | 4.95 ± 0.05 |
| 4 | 10.73 | 3.54 | 2.79 | 0.75 | 0.78 | 1.03 ± 0.01 | 6.24 ± 0.11 | 8.05 ± 0.13 |
| 5 | 10.54 | 3.83 | 3.03 | 0.80 | 0.76 | 0.65 ± 0.02 | 3.73 ± 0.06 | 4.64 ± 0.02 |
| 2 (Oic) | 10.57 | 3.84 | 2.98 | 0.86 | 0.83 | 1.09 ± 0.01 | 7.30 ± 0.16 | 9.38 ± 0.40 |

The high trans-amide content may be a result of a stabilized exo-pucker due to the restricted chair conformation of the cyclohexane ring and a stronger n → π* donation imposed by this conformational arrangement. Previously, we proposed ΔpKa and 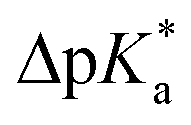 (eqn (1) and (2) for N-acetyl amino acids) as the descriptive parameters for this interaction.22 Here, we found that the ΔpKa and
(eqn (1) and (2) for N-acetyl amino acids) as the descriptive parameters for this interaction.22 Here, we found that the ΔpKa and  values are indeed larger for 4 and 5 than for proline, whereas Oic shows the largest values, indicating a strong n → π* alignment.
values are indeed larger for 4 and 5 than for proline, whereas Oic shows the largest values, indicating a strong n → π* alignment.
| ΔpKa = pKa(trans) − pKa(cis) | (1) |
 | (2) |
The crystal structure of Ac-Oic-OMe (Fig. 4A) illustrates the exo-pucker anchoring by the chair cyclohexane conformation.32 Careful inspection of the structure reveals that the methyl-group of the acetyl constituent restricts the possible flipping of the cyclohexane chair conformation, thus locking the pyrrolidine ring in the exo-pucker conformation. Furthermore, the close proximity of the acetyl oxygen to the carboxyl carbon stabilizes the n → π* contact.
 | ||
| Fig. 4 A. X-ray crystal structure of Ac-Oic-OMe (grey – carbon, blue – nitrogen, red – oxygen). B. Kinetics of the cis–trans isomerization of Ac-Oic-OMe in aqueous solution. | ||
The cis-to-trans rotational barriers in the Ac-AA-OMe models (Table 2) indicate that 5 and Oic have reduced rotational barriers due to the repulsion between the acetyl group and the 5-substituent of the proline ring in the ground state.33 However, this effect is more moderate for Oic, due to the compact size of the CH2-unit relative to the CH3-group. Regarding the trans-to-cis amide rotation, the reduction of the rotational barrier is countered by the stabilizing n → π* interaction, which contributes to the stability of the trans-ground state in Ac-Oic-OMe. Further analysis of the rotational velocities in Ac-Oic-OMe indicate an enthalpy driven transition (Fig. 4B), as expected.34 In the salt form, the rotational barriers are heightened due to the increased amide resonance, which is strengthened by the electrostatic interactions of the charges (CO2− ↔ +N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–O− attraction).
C–O− attraction).
| AA | In Ac-AA-OMe | ||||
|---|---|---|---|---|---|
| K trans/cis (at 298 K) | k 310 K, s−1 | E a, kJ mol−1 | |||
| c–t | t–c | c–t | t–c | ||
| 1 (Pro) | 4.95 ± 0.05 | 0.033 ± 0.002 | 0.0070 ± 0.0005 | 84.8 ± 0.2 | 88.8 ± 0.2 |
| 4 | 8.05 ± 0.13 | 0.041 ± 0.011 | 0.004 ± 0.001 | 84.3 ± 0.7 | 90.3 ± 0.7 |
| 5 | 4.64 ± 0.02 | 0.238 ± 0.011 | 0.055 ± 0.002 | 79.7 ± 0.1 | 83.5 ± 0.1 |
| 2 (Oic) | 9.38 ± 0.40 | 0.074 ± 0.013 | 0.008 ± 0.001 | 82.7 ± 0.5 | 88.5 ± 0.3 |
| AA | In Ac-AA-O− | ||||
|---|---|---|---|---|---|
| K trans/cis (at 298 K) | k 340 K, s−1 | E a, kJ mol−1 | |||
| c–t | t–c | c–t | c–t | ||
| 1 (Pro) | 0.81 ± 0.02 | 0.075 ± 0.003 | 0.093 ± 0.003 | 91.0 ± 0.4 | 90.4 ± 0.3 |
| 4 | 1.03 ± 0.01 | 0.071 ± 0.009 | 0.066 ± 0.004 | 91.1 ± 0.7 | 91.3 ± 0.5 |
| 5 | 0.65 ± 0.01 | 1.083 ± 0.072 | 0.645 ± 0.028 | 83.4 ± 0.5 | 84.9 ± 0.4 |
| 2 (Oic) | 1.09 ± 0.01 | 0.163 ± 0.003 | 0.153 ± 0.013 | 88.8 ± 0.3 | 88.9 ± 0.6 |
Oic in a peptide context
Next, we characterized Oic in a simple peptide context. To achieve this, Ac-GlyGlyOicGlyGly-NH2 and Ac-GlyGlyProGlyGly-NH2 were prepared by conventional Fmoc-solid phase peptide synthesis starting with Rink Amide resin (see ESI† for details). These peptides were then evaluated for their amide rotation properties (Table 3).| GGProGG | GGOicGG | ||
|---|---|---|---|
| K trans/cis (at 298 K) | 6.70 ± 0.20 | 12.3 ± 1.0 | |
| ΔGtrans/cis, kJ mol−1 | −4.71 ± 0.07 | −6.22 ± 0.21 | |
| E ≠, kJ mol−1 | c–t | 81.4 ± 0.3 | 76.8 ± 0.1 |
| (At 310 K) | t–c | 85.8 ± 0.5 | 82.1 ± 0.1 |
Our results indicate that the Pro-to-Oic mutation increased the relative stability of the trans-amide by approximately −1.5 kJ mol−1, in a similar fashion to the Ac-AA-OMe model comparison. However, the amide rotational barrier reduction with the Pro-to-Oic mutation was much stronger than that observed in Ac-AA-OMe models. This difference may be a result of the greater steric interference presented by the peptide chain. In addition, for the trans-amide fraction, the 1H{15N} single bond correlation spectra (Fig. 5) illustrate the fundamental similarity of the peptide chain conformations in the Pro- and Oic-containing peptides.
 | ||
| Fig. 5 1H{15N} sofast-HMQC spectra35 of AcGG-AA-GGNH2 peptides: blue – AA = Pro, red – AA = Oic. Recorded in aqueous medium at 277 K. | ||
Crystal structure of an Oic-hexapeptide
Recently, Wilhelm et al. reported a crystal structure of a proline hexapeptide p-BrBz-(Pro)6-OH in a PII conformation (p-BrBz = para-bromobenzoyl).36 We prepared an analogous hexapeptide composed of Oic residues with C-terminal amidation: p-BrBz-(Oic)6-NH2. The crystal structure of this peptide clearly demonstrates an all-trans PII fold with all pyrrolidine rings in the exo-pucker conformations and perfect 3 residues/1 turn periodicity.32 In contrast to the hexaproline structure (which shows 9.0 Å per turn (ref. 36 and 37)), the hexa-Oic structure was more compact, measuring approximately 8.6–8.7 Å per one helical turn (Fig. 6A). | ||
| Fig. 6 A. X-ray crystal structure of p-BrBz-(Oic)6-NH2 oligopeptide in PII conformation. B. α- and δ-CH fragment of the 1H cross-relaxation spectra of this peptide in deuteromethanol at 298 K and 700 MHz frequency. Only the exchange due to N-terminal rotation of para-bromobenzoyl fragment is seen in NOESY. In ROESY positive cross-peaks between α-CH and δ-CH spectral regions indicate the all-trans conformation. | ||
Regarding the N-terminal region, the para-bromobenzoyl moiety appeared tilted, with a N–C(O)–C–CH (ortho) torsion angle of approximately 68° (versus the 51° angle in the oligoproline structure). This tilting results from the strong spatial clash between the aromatic ring and the δ-CH pyrrolidine portion.38 In solution, this spatial clash substantially reduces the rotational barrier for this amide fragment. Thus, 1H NMR spectra of the peptide in methanol (at 298 K) demonstrate the presence of two rotameric forms (Ktrans/cis = 2.5) with relatively high transition velocities: 1.96 ± 0.20 and 0.84 ± 0.05 s−1 for cis-to-trans and trans-to-cis rotations, respectively. For other amides, the 1H ROESY spectra indicate an all-trans structure, consistent with the crystal observations (Fig. 6B). Our molecular modelling suggests that the cis-p-BrBz fragment folds into the first turn of the PII helix, while the trans-p-BrBz fragment is exposed in solution. The high content of the N-terminal cis-form thus results from the stabilizing hydrophobic contacts of p-BrBz with the first three residues Oic1–Oic3. The rotameric state of this fold also strongly influences the 1H chemical shifts of the first helical turn residues in the two rotameric species.
Oic oligopeptides
We next wished to study the events accompanying the growth of the polypeptide chain and the possibility of specific aggregation events. We prepared a series of oligopeptides with the structure Ac-(Oic)N-OH, where N was varied from 1 to 6. The peptides were synthesised on an Fmoc-Oic-pre-loaded 2-chlorotrityl resin, and the full-length peptides were cleaved mildly with 25 vol% hexafluoroisopropanol in dichloromethane, as described.39The peptides were well soluble in methanol (as well as other polar organic solvents). Peptides with N = 2–6 were very poorly soluble in water. Conversion to salts (with phosphate buffer, pH 7) enhanced their aqueous solubility. The NMR studies with the salt Ac-(Oic)N-O− species were then conducted in deuterium oxide. The concentration was 20 g l−1 for the Ac-(Oic)N-O− with N = 1–5, while the sample concentration of Ac-(Oic)N-O− with N = 6 was approximately 10 g l−1 due to its poor solubility. The NMR studies were also performed in deuteromethanol for the acidic Ac-(Oic)N-OH species at 20 g l−1 for all studied oligomers.
In deuteromethanol the 1H NOESY spectra showed a trans-amide bond with characteristic α-CH ↔ δ-CH nuclear Overhauser effect (NOE) for the internal positions and a CH3 ↔ δ-CH NOE for the N-terminal positions. The trans-amide constituted 80% of the population for N = 1 and ≥90% for N = 2, whereas in oligomers with N ≥ 3 only the all-trans conformation was observed. In this conformation, all the α-CH resonances appear as doublets of doublets with one J between 10.0 and 10.5 Hz and another between 7.1 and 8.2 Hz. Thus, the peptide backbone conformations all exhibit exceptional regularity, with the exo-pucker conformation predominant in the side-chain.40
The 1H NMR spectra for samples in deuterium oxide also indicated the dominance of the all-trans fold; however, minor populations of other folds were also observed (vide infra).
1H diffusion ordered spectra (DOSY) of the oligomeric peptides demonstrated a systematically decreasing diffusion, which correlated well with the increments in N. The diffusion coefficients obtained in DOSY were converted to molecular volumes using the Stokes–Einstein relationship according to eqn (3).41 These volumes were compared with theoretical COSMO volumes obtained in semi-empirical modelling (see eqn (4)). The resulting prediction correlated fairly well with the experimental values for both the methanol and deuterium oxide solutions (Fig. 7A).
 | (3) |
| V [Å3] = 10.9 + 1.223·MW [Da] | (4) |
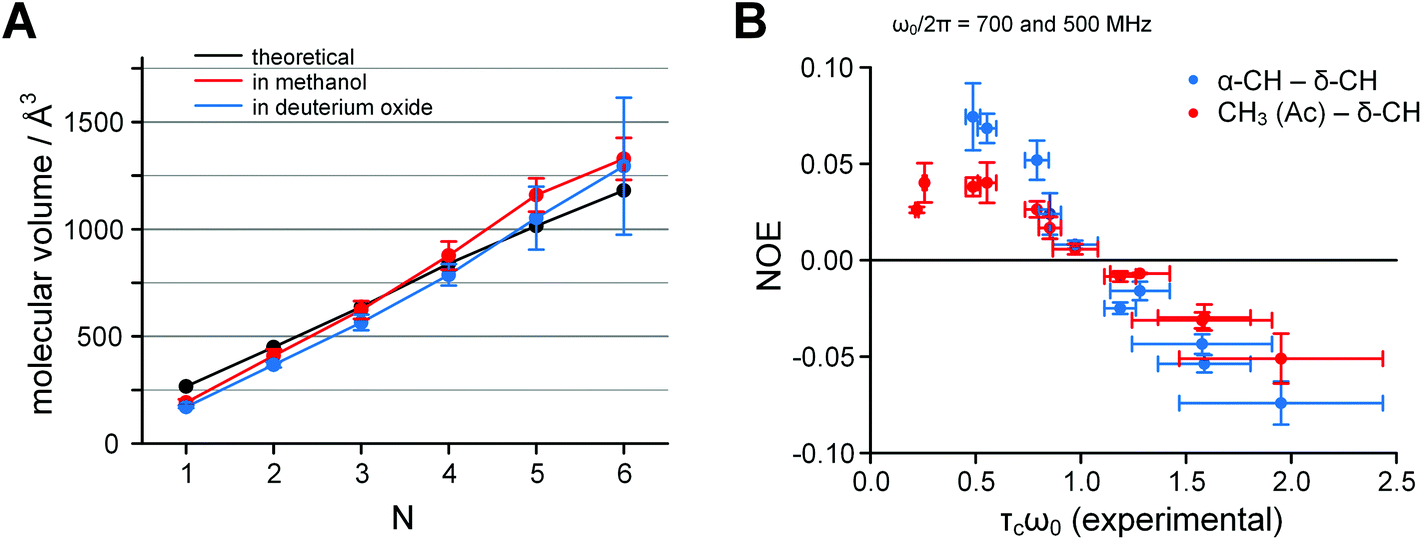 | ||
Fig. 7 A. Dependence of the molecular volume of Ac-(Oic)N-OH on the number of residues N: black – semi-empirical calculations, red – experimental in deuteromethanol (acidic form), blue – experimental in deuterium oxide (salt form); measured by 1H DOSY at 700 MHz frequency and 298 K. B. The correlation time problem in Ac-(Oic)N-O−. Inversion of the NOE sign in deuterium oxide samples occurs between lower and higher oligomers; the theoretical zero-cross point is at τcω0 = 1.12. The correlation time values were obtained from experimental log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) D values measured in 1H DOSY according to eqn (5). The data were collected at 700 and 500 MHz 1H frequency at 298 K. D values measured in 1H DOSY according to eqn (5). The data were collected at 700 and 500 MHz 1H frequency at 298 K. | ||
In the 1H NOESY spectra, the NOEs for samples in deuterium oxide appeared positive for the shorter oligomers and negative for the longer oligomers. The sign inversion occurred between N = 3 and 4 at 700 MHz and between N = 4 and 5 at 500 MHz, when measured at 298 K.
Next, we converted the logD values obtained in DOSY into correlation times τc according to eqn (5) (all SI units)41 and plotted NOE versus τcω0, where ω0 is the angular Larmour frequency. According to the theory, the sign inversion should occur at τcω0 = 1.12,42 which was indeed the experimental result (Fig. 7B). Thus, the molecular sizes derived by DOSY (spatial diffusion) were consistent with those derived by NOESY (rotational diffusion).
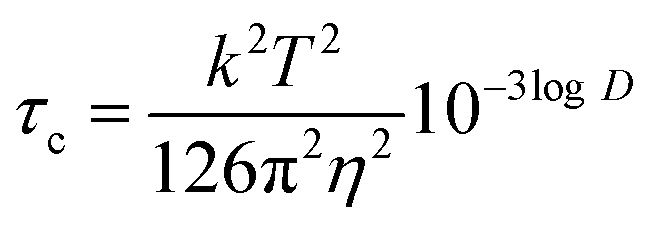 | (5) |
Overall the diffusion data indicated the absence of specific aggregation, despite the low aqueous solubility (≤10 g l−1) of the peptide with N = 6. However, the diffusion coefficient error is higher for the soluble fraction in aqueous conditions at N = 5 and 6. In addition, the applicability of the Stokes–Einstein equation, which assumes rigid spheres, clearly indicates sufficient molecular rigidity of the oligopeptide chains.43 We also found for the NOE (y-axis) that the errors in Fig. 7B are similar for both N-terminal (CH3 ↔ δ-CH1) and internal (α-CH1−(N−1) ↔ δ-CH2−N) positions, only for higher oligomers. This might indicate an increase in structural rigidity for longer oligomers.
Cooperativity of oligopeptide folding
Perhaps the most interesting question addressed by the oligopeptide studies is the degree of acidity of the C-terminal group. We previously observed that the acidity of the Ac-(Pro)2-OH peptide with the trans-Pro–Pro amide bond depends on the rotameric state of the upstream acetyl-fragment.22 Namely, the trans–trans peptide (pKa 3.59) exhibited a drop in acidity of approximately 0.1 pKa unit relative to the cis–trans isoform. This result suggests cooperativity of the all-trans fold, which we assign to a cascade of donative interactions established between the carbonyl groups in the peptide chain.We also measured the C-terminal acidity of the all-trans peptides in the Ac-(Oic)N-OH oligomers by analysing 1H NMR chemical shifts obtained at different pH values. In this case, the acidity of Ac-(Oic)N-OH (N = 1) with the cis-amide (pKa 2.97) can serve as a reference. The pKa increased by 0.8–0.9 units (see Table 1) in the trans-isoform of this compound, as the n → π* donation was stabilized by the geometry of the Oic residue. For higher oligomers with N ≥ 2, the pKa is further elevated by approximately 0.2–0.3 units (i.e., ∼1.1–1.7 kJ mol−1) due to the putative cascade effect (Fig. 8 and 9). The cascade effect should strengthen the alignment of the carbonyl groups throughout the peptide chain. The available data clearly indicate that the cascade effect is transmitted sufficiently over one residue and decays very quickly through the all-trans peptide chain. The cascade phenomenon might therefore represent a driving force for PII nucleation in peptide segments.
 | ||
| Fig. 8 Dependence of the C-terminal pKa on the oligomeric number N in all-trans Ac-(Oic)N-OH. Measured in buffered aqueous solutions at 298 K by 1H NMR. | ||
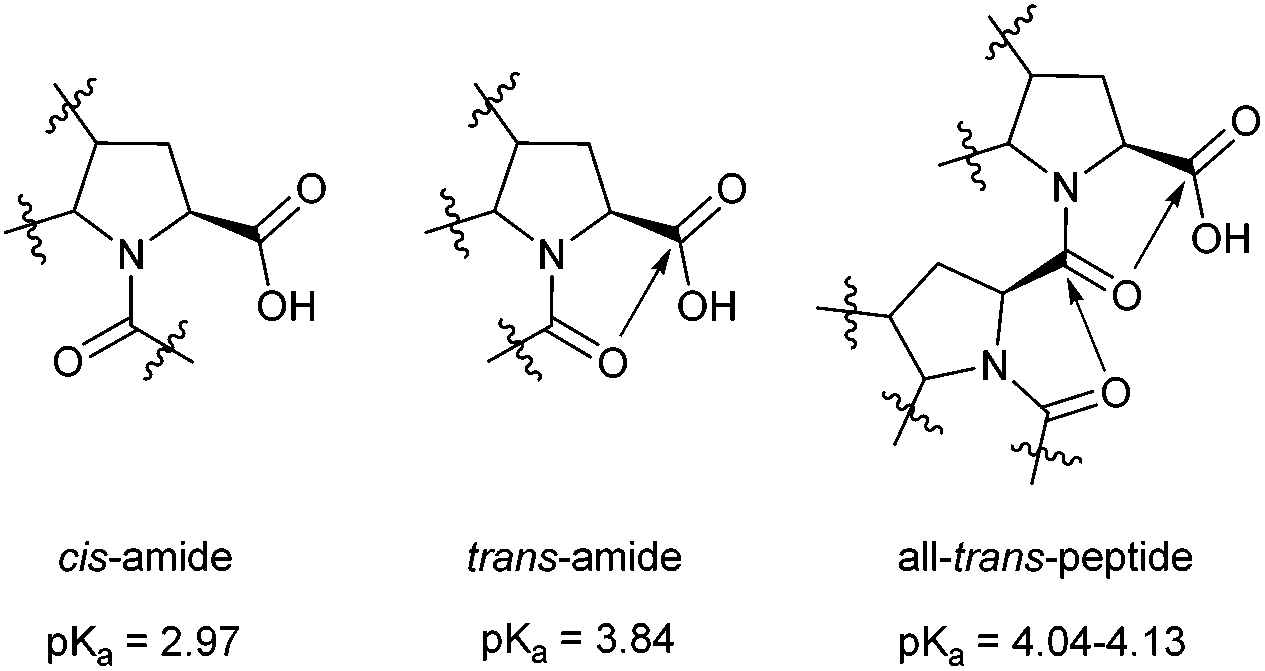 | ||
| Fig. 9 Manifestation of the cascade-effect in an all-trans polyproline peptide chain. | ||
Minor conformational forms in the oligopeptides
As noted previously, in methanol samples, only the all-trans conformation was observed in the 1H NMR spectra of Ac-(Oic)N-OH with N ≥ 3. A more complex situation occurred for Ac-(Oic)N-O− peptides in phosphate buffer, since the C-terminal group is a carboxylate, in which the carbonyl exhibits minimal electrophilicity. Whereas at N = 1 the cis-amide content was 48%, at N = 2 the distribution of trans–trans, cis–trans, cis–cis and trans–cis rotamers was 79:11:7:2, indicating a complex interdependence of the rotameric populations of two amides. For higher oligomers with N = 3–6, the all-trans conformation is dominant. Nevertheless, some minor fractions were persistently observed. We next aimed to identify the source of the minor conformational forms and their populations, which should provide a proper characterization of the relative stability of the peptide conformations.
In the aqueous samples of Ac-(Oic)N-O−, the most prominent minor resonance was δ-CH at 3.98 ppm (see Fig. 10). In order to assign this form, we evaluated 1H EXSY spectra at 330 K. In the EXSY spectra, this minor δ-CH signal exhibited an exchange with the N-terminal δ-CH but not the internal δ-CH resonances. In addition, one α-CH(major) ↔ α-CH(minor) and one CH3(major) ↔ CH3(minor) exchange together with an α-CH(minor) ↔ CH3(minor) NOE indicated that the minor form was a result of the N-terminal rotation. However, the intensity of this δ-CH (∼17–18% for N = 3–6) minor signal was higher than for the other minor resonances (α-CH and CH3, ∼6–10%), and it was remarkably higher than that of the Ac-Oic-OMe model (10%).
 | ||
| Fig. 10 α-CH, δ-CH region of the 1H NMR spectra of Ac-(Oic)6-O− (left) and Ac-(Oic)6-OMe (right) in deuteromethanol (bottom) and deuterium oxide (top). Measurements were performed at 298 K in stimulated echo pulse sequence used for solvent suppression. Residual OH resonance of deuteromethanol is marked with an asterisk. The resonances exchanged in 1H EXSY experiments (at 330 K) are marked with double-arrowed lines below the spectra. | ||
Furthermore, we compared the 1H NMR spectra of the methyl ester (Ac-(Oic)6-OMe) and salt (Ac-(Oic)6-O−) forms in methanol. The intensity of the (cis-Ac) δ-CH resonance increased when changing from the salt to methyl ester. This change was accompanied by the appearance of one additional CH3-resonance in 1H and 1H{13C} HSQC spectra. The dependence of the N-terminal rotation state on the C-terminal charge can be explained by a cooperative formation of the all-cis (polyproline I) structure, which is favoured in the case of charged peptide termini.44 The estimation of the s-cis-acetyl fragment originating from (a) the N-terminal rotation in the all-trans structure and (b) the formation of an all-cis conformation was performed by analysing three resolved CH3-resonances in 1H{13C} HSQC. These were: for Ac-(Oic)6-OMe 5 (a) and 1 (b) %, and for Ac-(Oic)6-O− 5 (a) and 3 (b) %, respectively (in the methanol solutions).
In deuterium oxide, the presumed all-cis fraction constituted about 2–3% for Ac-(Oic)6-O− and was not detected for Ac-(Oic)6-OMe. In these aqueous conditions, the cis-Ac content due to the N-terminal rotation in the all-trans form was 10–12%, and the trans-to-cis rotation rate was about 0.02 s−1 (for Ac-(Oic)N-O− with N = 3–6; at 330 K).
Therefore, similar to the p-BrBz-(Oic)6-NH2 peptide, the N-terminal rotation occurs in Ac-(Oic)N-OH/O− peptides. In addition, the all-cis fraction might be formed at a very low, yet detectable, level. The low percentage of the minor conformation explains why this fraction is obscured by the main resonances and is only visible when combined with other signals. Overall, the low percentage found for the minor conformational states indicates the high relative stability of the major cooperative all-trans fold for the oligo-Oic chain.
Conclusions
In summary, we have demonstrated that peptidyl-Oic is capable of creating a strong trans-amide carbonyl alignment in peptide structures. This rotameric state is stabilized by the anchoring of the pyrrolidine pucker in the exo-conformation by the cyclohexane ring. The all-trans backbone state in oligomeric structures is stabilized by the high trans-amide propensity of a single Oic-residue as well as the cascade effect established in the peptide chain. Furthermore, the residual rotational freedom of the N-terminal fragment (acetyl or para-bromobenzoyl) is the major source of alternative conformational states. The cascade effect is the enhancement of the original carbonyl alignment energy by a preceding residue. For Oic, the alignment energy is enhanced by approximately 1/3–1/4 (compared to an enhancement by 1/6 for a proline oligopeptide, as determined previously22). As a PII stabilizing residue, Oic represents an ideal candidate for construction of hydrophobic sites in a plethora of biologically relevant PII structures. We also envision that all-Oic structures might serve as a starting point for construction of a transmembrane PII helix, a motif non-existent in nature.Experimental section
(2S,3aS,7aS)-Octahydroindole-2-carboxylic acid (Oic) was obtained from commercial sources, (4S)-methylproline was synthesized as previously described,45 and (5S)-methylproline was synthesized according to Mohite et al.461-Acetyl-(2S,3aS,7aS)-octahydroindole-2-carboxylic acid
Oic (156 mg; 0.92 mmol; 1 equiv.) was mixed with acetic anhydride (200 μl; 2.1 mmol; 2.3 equiv.) in dichloromethane (2 ml). After 30 min, the solvent was removed under reduced pressure. The residue was taken up in water and freeze dried to give 192 mg Ac-Oic as a white powder (98% yield). 1H NMR (700 MHz, D2O, phosphate buffer), δ: 4.23 (dd, J = 9.5 and 7.9 Hz, s-cis) and 4.12 (dd, J = 10.1 and 8.7 Hz, s-trans) (1H, α-CH), 3.97 (m, s-cis) and 3.82 (m, s-trans) (1H, δ-CH), 2.28 (m, s-trans) and 2.19 (m, s-cis) (1H, γ-CH), 2.22 and 2.00 (two m, s-cis) and 2.11 and 1.89 (two m, s-trans) (2H, β-CH2), 2.03 (s, s-trans) and 1.83 (s, s-cis) (3H, CH3), 1.85 and 1.40 (two m, s-trans) and 1.82 and 1.26 (two m, s-cis) (2H, CH2), 1.66 and 1.57 (two m, CH2), 1.60 and 1.09 (two m, CH2), 1.39 and 1.23 (two m, CH2). 13C{1H} (126 MHz, D2O, phosphate buffer), δ: 179.9 (CO2−), 172.7 (s-cis, NCO) and 171.7 (s-trans, NCO), 63.3 (s-cis, α-CH) and 61.5 (s-trans, α-CH), 59.9 (s-trans, δ-CH) and 58.2 (s-cis, δ-CH), 36.6 (s-trans, γ-CH) and 35.5 (s-cis, γ-CH), 32.7 (s-cis, β-CH2) and 31.1 (s-trans, β-CH2), 27.0 (s-trans, CH2) and 26.0 (s-cis, CH2), 25.1 and 24.9 (CH2), 23.34 and 23.32 (CH2), 20.81 (s-trans, CH3) and 20.77 (s-cis, CH3), 19.8 and 19.6 (CH2). Mass-spectrum (ESI), Th: 212.13 [M + H]+, 234.11 [M + Na]+. [α]D = −60 (c = 1.0, methanol, 298 K).Methyl 1-acetyl-(2S,3aS,7aS)-octahydroindole-2-carboxylate
Ac-Oic (74 mg; 0.35 mmol) was dissolved in methanol (2 ml) and trimethylsilane (0.075 ml) was subsequently added. The mixture was stirred overnight, methanol was removed under reduced pressure and the residue was purified by silica gel column using a methanol–ethyl acetate mixture (1:19) as an eluent (Rf = 0.5). 79 mg of Ac-Oic-OMe was obtained as a colourless oil, which later crystallized (quantitative yield). 1H NMR (700 MHz, D2O), δ, only s-trans: 4.34 (dd, J = 10.0 and 8.5 Hz, 1H, α-CH), 3.89 (m, 1H, δ-CH), 3.69 (s, 3H, CH3O), 2.38 (m, 1H, γ-CH), 2.15 and 2.01 (two m, 2H, β-CH2), 2.06 (s, 3H, CH3CO), 1.91 and 1.37 (two m, 2H, CH2), 1.68 and 1.61 (two m, 2H, CH2), 1.63 and 1.14 (two m, 2H, CH2), 1.41 and 1.23 (two m, 2H, CH2). 13C{1H} (126 MHz, D2O), δ, only s-trans: 175.1 (CO2Me), 172.3 (NCO), 59.5 (δ-CH), 59.0 (α-CH), 36.9 (γ-CH), 30.3 (β-CH2), 27.0 (CH2), 24.9 (CH2), 23.2 (CH2), 20.5 (CH3), 19.5 (CH2). Mass-spectrum (ESI), Th: 226.14 [M + H]+. [α]D = −72 (c = 1.0, methanol, 298 K).
N-Acetyl derivatives and their methyl esters for (4S)- and (5S)-methylprolines were prepared in analogous procedures.
1-Fluorenylmethoxycarbonyl-(2S,3aS,7aS)-octahydroindole-2-carboxylic acid
Oic (0.75 g; 4.43 mmol; 1 equiv.) was dissolved in a 10% sodium carbonate solution (5 ml). Some water and acetone was added, and the solution was cooled down by an ice bath. A solution of Fmoc chloride (1.26 g; 4.87 mmol; 1.1 equiv.) in acetone (5 ml) was added dropwise upon stirring over 40 min. The mixture was stirred overnight at ambient temperature. The mixture was then diluted by water to reach a volume of 250 ml. It was then washed by diethyl ether (2 × 75 ml), acidified by addition of solid potassium hydrogen sulphate (to pH ≤ 2) and extracted by ethyl acetate (4 × 50 ml). Ethyl acetate fractions were dried over sodium sulphate and filtered, and the solvent was removed under reduced pressure. The residue was mixed with an acetonitrile–water mixture and freeze-dried. 1.72 g of Fmoc-Oic was obtained as a white powder (99% yield). 1H NMR (700 MHz, CD3OD), δ: 7.81 (m, 2H), 7.67–7.61 (m, 2H), 7.40 (m, 2H), 7.33 (m, 2H), 4.52 and 4.48 (two dd, J = 10.7, 5.7 Hz) and 4.34 and 4.26 (two m) (2H, CH2–O), 4.34 and 4.16 (two m, 1H, α-CH), 4.25 and 4.16 (two m, 1H, CH), 3.89 and 3.42 (two m, 1H, δ-CH), 2.36 and 2.20 (two m, 1H, γ-CH), 2.28 and 2.09, 2.24 and 1.94 (four m, 2H, β-CH2), 2.07, 180–1.68, 1.64–1.19 and 1.01 (series of multiplets, 6H, 3 × CH2). 13C{1H} NMR (126 MHz, CD3OD), δ: 175.0 and 174.6 (CO2H), 155.0 and 154.8 (NCO), 144.12, 144.08, 144.0, 143.7, 141.5, 141.4, 141.2 and 141.1 (all C), 127.43, 127.37, 127.33, 126.81, 126.75, 124.92, 124.85, 124.5, 119.54 and 119.49 (all CH), 67.6 and 66.8 (CH2–O), 58.9 and 58.8 (α-CH), 58.1 and 57.7 (CH), 47.2 and 47.1 (δ-CH), 36.7 and 36.3 (γ-CH), 32.4 and 31.3 (β-CH2), 30.4, 27.3, 27.0, 25.3, 23.4, 23.3, 20.1 and 20.1 (all CH2). Mass-spectrum (ESI), Th: 392.18 [M + H]+. [α]D = −28 (c = 1.0, methanol, 298 K).Acknowledgements
The authors would like to thank Paula Nixdorf and Dr Elisabeth Irran (TU Berlin) for performing crystallographic studies. DFG research group FOR 1805 is acknowledged for a postdoctoral fellowship (VK). Dr Marcie Jaffee (Atlanta, USA) is gratefully acknowledged for a revision of the manuscript.References
- S. A. Hollingsworth and P. A. Karplus, Biomol. Concepts, 2010, 1, 271–283 CrossRef CAS PubMed.
- J. Kurtz, A. Berger and E. Katchalski, Nature, 1956, 178, 1066–1067 CrossRef CAS.
- F. Conti, M. Piatelli and P. Viglino, Biopolymers, 1969, 7, 411–415 CrossRef CAS.
- A. A. Adzhubei, M. J. E. Sternberg and A. A. Makarov, J. Mol. Biol., 2013, 425, 2100–2132 CrossRef CAS PubMed.
- J. Makowska, S. Rodziewicz-Motowidło, K. Bagińska, J. A. Vila, A. Liwo, L. Chmurzyński and H. A. Sheraga, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 1744–1749 CrossRef CAS PubMed.
- I. Gokce, R. W. Woody, G. Anderluh and J. H. Lakey, J. Am. Chem. Soc., 2005, 127, 9700–9701 CrossRef CAS PubMed.
- S. A. Hollingsworth, D. S. Berkholz and P. A. Karplus, Protein Sci., 2009, 18, 1321–1325 CrossRef CAS PubMed.
- Z. Shi, K. Chen, Z. Liu, A. Ng, W. C. Bracken and N. R. Kallenbach, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 17964–17968 CrossRef CAS PubMed.
- A. M. Brown and N. J. Zondlo, Biochemistry, 2012, 51, 5041–5051 CrossRef CAS PubMed.
- M. P. Williamson, Biochem. J., 1994, 297, 249–260 CrossRef CAS PubMed.
- M. Scocchi, A. Tossi and R. Gennaro, Cell. Mol. Life Sci., 2011, 68, 2317–2330 CrossRef CAS PubMed.
- S. Pujals and E. Giralt, Adv. Drug Delivery Rev., 2008, 60, 473–484 CrossRef CAS PubMed.
- Y. A. Filon, J. P. Anderson and J. Chmielewski, J. Am. Chem. Soc., 2005, 127, 11798–11803 CrossRef PubMed.
- S. Pujals, J. Fernández-Carneado, M. J. Kogan, J. Martinez, F. Cavelier and E. Giralt, J. Am. Chem. Soc., 2006, 128, 8479–8483 CrossRef CAS PubMed.
- It was later demonstrated that oligo-silaproline is also able to form an all-trans structure, see: C. Martin, B. Legrand, A. Lebrun, D. Berthomieu, J. Martinez and F. Cavelier, Chem. – Eur. J., 2014, 20, 14240–14244 CrossRef CAS PubMed.
- (a) B. Schuler, E. A. Lipman, P. J. Steinbach, M. Kumke and W. A. Eaton, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 2754–2759 CrossRef CAS PubMed; (b) H. Sahoo, D. Roccatano, A. Hennig and W. M. Nau, J. Am. Chem. Soc., 2007, 129, 9762–9772 CrossRef CAS PubMed; (c) E. Dolghih, W. Ortiz, S. Kim, B. P. Krueger, J. L. Krause and A. E. Roitberg, J. Phys. Chem. A, 2009, 113, 4639–4646 CrossRef CAS PubMed.
- W. J. Wedemeyer, E. Welker and H. A. Scheraga, Biochemistry, 2002, 41, 14637–14644 CrossRef CAS PubMed.
- M. D. Shoulders, J. A. Hodges and R. T. Raines, J. Am. Chem. Soc., 2006, 128, 8112–8113 CrossRef CAS PubMed.
- M. Kümin, L.-S. Sonntag and H. Wennemers, J. Am. Chem. Soc., 2007, 129, 466–467 CrossRef PubMed.
- M. P. Hinderaker and R. T. Raines, Protein Sci., 2003, 12, 1188–1194 CrossRef CAS PubMed.
- M. D. Shoulders and R. T. Raines, Annu. Rev. Biochem., 2009, 78, 929–958 CrossRef CAS PubMed.
- V. Kubyshkin, P. Durkin and N. Budisa, New J. Chem., 2016, 40, 5209–5220 RSC.
- J. M. Stewart, Peptides, 2004, 25, 527–532 CrossRef CAS PubMed.
- S. Deekonda, D. Rankin, P. Davis, J. Lai, F. Porreca and V. J. Hruby, Bioorg. Med. Chem. Lett., 2015, 25, 4148–4152 CrossRef CAS PubMed.
- (a) D. J. Kyle, P. R. Blake, D. Smithwick, L. M. Green, J. A. Martin, J. A. Sinsko and M. F. Summers, J. Med. Chem., 1993, 36, 1450–1460 CrossRef CAS PubMed; (b) W. Guba, R. Haessner, G. Breipohl, S. Henke, J. Knolle, V. Santagada and H. Kessler, J. Am. Chem. Soc., 1994, 116, 7532–7540 CrossRef CAS; (c) J. Sejbal, J. R. Cann, J. M. Stewart, L. Gera and G. Kotovych, J. Med. Chem., 1996, 39, 1281–1292 CrossRef CAS PubMed; (d) J. Sejbal, Y. Wang, J. R. Cann, J. M. Stewart, L. Gera and G. Kotovych, Biopolymers, 1997, 42, 521–535 CrossRef CAS PubMed.
- J. Torras, J. G. Warren, G. Revilla-López, A. I. Jiménez, C. Cativiela and C. Alemán, Biopolymers, 2014, 102, 176–190 CrossRef CAS PubMed.
- J. P. Bouchet, J. P. Volland, M. Laubie, M. Vincent, B. Marchand and N. Platzer, Magn. Reson. Chem., 1992, 30, 1186–1195 CrossRef CAS.
- C. Siebler, R. S. Erdmann and H. Wennemers, Angew. Chem., Int. Ed., 2014, 53, 10340–10344 CrossRef CAS PubMed.
- N. G. Delaney and V. Madison, Int. J. Pept. Protein Res., 1982, 19, 543–548 CrossRef CAS PubMed.
- Y. K. Kang, J. Mol. Struct. (THEOCHEM), 2002, 585, 209–221 CrossRef CAS.
- K. Zhang, R. B. Teklerbhan, G. Schreckenbach, S. Wetmore and F. Schweizer, J. Org. Chem., 2009, 74, 3735–3743 CrossRef CAS PubMed.
- The crystal structures are deposited in Cambridge Crystallographic Data Centre under deposition numbers: Ac-Oic-OMe CCDC 1510246, p-BrBz-(Oic)6-NH2 CCDC 1510247.
- E. Beausoleil, R. Sharma, S. W. Michnick and W. D. Lubell, J. Org. Chem., 1998, 63, 6572–6578 CrossRef CAS.
- C. Renner, S. Alefelder, J. H. Bae, N. Budisa, R. Huber and L. Moroder, Angew. Chem., Int. Ed., 2001, 40, 923–925 CrossRef CAS PubMed.
- P. Schanda, Ē. Kupče and B. Brutscher, J. Biomol. NMR, 2005, 33, 199–211 CrossRef CAS PubMed.
- P. Wilhelm, B. Lewandowski, N. Trapp and H. Wennemers, J. Am. Chem. Soc., 2014, 136, 15829–15832 CrossRef CAS PubMed.
- Similarly, a 9.1 Å periodicity was recently described for the crystal structure of cis-4,5-methanoproline tetramer, see: G. Berger, M. Vilchis-Reyes and S. Hanessian, Angew. Chem., Int. Ed., 2015, 54, 13268–13272 CrossRef CAS PubMed.
- V. Kubyshkin, Y. Kheylik and P. K. Mykhailiuk, J. Fluorine. Chem., 2015, 175, 73–83 CrossRef CAS.
- R. Bollhagen, M. Schmiedberger, K. Barlos and E. Grell, J. Chem. Soc., Chem. Commun., 1994, 2559–2560 RSC.
- M. Cai, Y. Huang, J. Liu and R. Krishnamoorthi, J. Biomol. NMR, 1995, 6, 123–128 CrossRef CAS PubMed.
- Legend for eqn (3)–(5): D – diffusion coefficient in m2 s−1, T – absolute temperature in K, η – dynamic viscosity in Pa s−1, V – molecular volume in Å3, MW – molecular weight in Da, k – Boltzmann constant in m2 kg s−2 K−1, τc – correlation time in s rad−1, ω0 – angular frequency in rad s−1.
- B. Luy, A. Frank and H. Kessler, Conformational Analysis of Drugs by Nuclear Magnetic Resonance Spectroscopy, in Molecular Drug Properties - Measurement and Prediction, ed. R. Mannhold, Wiley, 2008, pp. 207–254 Search PubMed.
- R. Evans, Z. Deng, A. K. Rogerson, A. S. McLachlan, J. J. Richards, M. Nilsson and G. A. Morris, Angew. Chem., Int. Ed., 2013, 52, 3199–3202 CrossRef CAS PubMed.
- M. Kuemin, S. Schweizer, C. Ochsenfeld and H. Wennemers, J. Am. Chem. Soc., 2009, 131, 15474–15482 CrossRef CAS PubMed.
- D. Dietz, V. Kubyshkin and N. Budisa, ChemBioChem, 2015, 16, 403–406 CrossRef CAS PubMed.
- A. R. Mohite and R. G. Bhat, J. Org. Chem., 2012, 77, 5423–5428 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Extended experimental details and analytical data; copies of the NMR spectra. CCDC 1510246 and 1510247. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6ob02306a |
| This journal is © The Royal Society of Chemistry 2017 |