DOI:
10.1039/C7NR06985E
(Paper)
Nanoscale, 2018,
10, 222-230
Size controllable DNA nanogels from the self-assembly of DNA nanostructures through multivalent host–guest interactions†
Received
19th September 2017
, Accepted 21st November 2017
First published on 23rd November 2017
Abstract
Nanogels made of biomolecules are one of the potential candidates as a nanocarrier for drug delivery applications. The unique structural characteristics and excellent biocompatibility of DNA suggest that DNA nanogels would be an ideal candidate. Herein, a general design strategy for the crafting of DNA nanogels with controllable size using the multivalent host–guest interaction between β-CD functionalized branched DNA nanostructures as the host and a star-shaped adamantyl-terminated 8-arm poly(ethylene glycol) polymer as the guest is reported. Our results reveal that multivalent host–guest interactions are necessary for the nanogel formation. Nanogels exhibit excellent biocompatibility, good cell permeability and high drug encapsulation ability, which are promising features for their application as a drug carrier. The encapsulation of doxorubicin, an anticancer drug, inside the hydrophobic network of the nanogel and its delivery into cancer cells are also reported. We hope that the general design strategy demonstrated for the creation of DNA nanogels may encourage other researchers to use this approach for the design of DNA nanogels of other DNA nanostructures, and explore the potential of DNA nanogels in drug delivery applications.
1. Introduction
Nanogels are physically or chemically crosslinked hydrogel nanoparticles.1 The characteristic structural features of nanogels such as nanosize, porosity, stimuli responsiveness and high guest encapsulation ability have greatly attracted researchers to use them as nanocarriers in several biomedical applications, particularly in drug delivery.2 Even more beneficial would be the design of nanogels derived from biomolecules such as DNA due to its excellent biocompatibility and unique structural features. DNA nanostructures offer the possibility of efficient encapsulation of the hydrophilic/hydrophobic payload and its stimuli-responsive release upon demand.3 Reversible integration of targeting ligands/aptamers onto the surface of DNA nanostructures is easily achievable through sequence specific DNA hybridization and thereby offers the opportunity for targeted drug delivery.4 Most importantly, DNA nanostructures exhibit excellent cell permeability and also show enhanced resistance towards enzymatic degradation.5 Despite these promising features of DNA nanostructures, less attention has been paid to the design of DNA nanogels and their applications in drug delivery.6
The synthesis of nanogels is routinely achieved through either chemical or physical crosslinking of the monomer chains. Though chemical crosslinking yields stable nanogels, it is often laborious and produces insoluble polymeric materials. On the other hand, physical crosslinking through noncovalent interactions such as hydrogen bonding, metal coordination, π-stacking or host–guest interactions is advantageous in many ways over chemical crosslinking. These include the ease of synthesis, reversibility and stimuli-responsive nature, which are extremely advantageous for drug delivery applications. Hence, physically crosslinked nanogels have received much attention over the covalently crosslinked nanogels in recent years.7 Among the various non-covalent interactions,8 the host–guest interaction between β-cyclodextrin (β-CD) and adamantane has been extensively explored in supramolecular chemistry for the design of various functional nanostructures owing to their efficient molecular recognition with a reasonable association constant (Ka = 104–105 M−1).9 We have recently explored the β-CD-adamantane host–guest interaction for the design of a series of supramolecular DNA amphiphiles10 and reported their amphiphilicity-driven self-assembly into nano-to-micro sized DNAsomes.11 Our findings revealed that molecular recognition between β-CD functionalized DNA (host) and a adamantane modified hydrophobic segment (guest) exhibits excellent selectivity and a high association constant. These results motivated us to explore the β-CD-adamantane noncovalent interaction for the design of DNA nanogels.
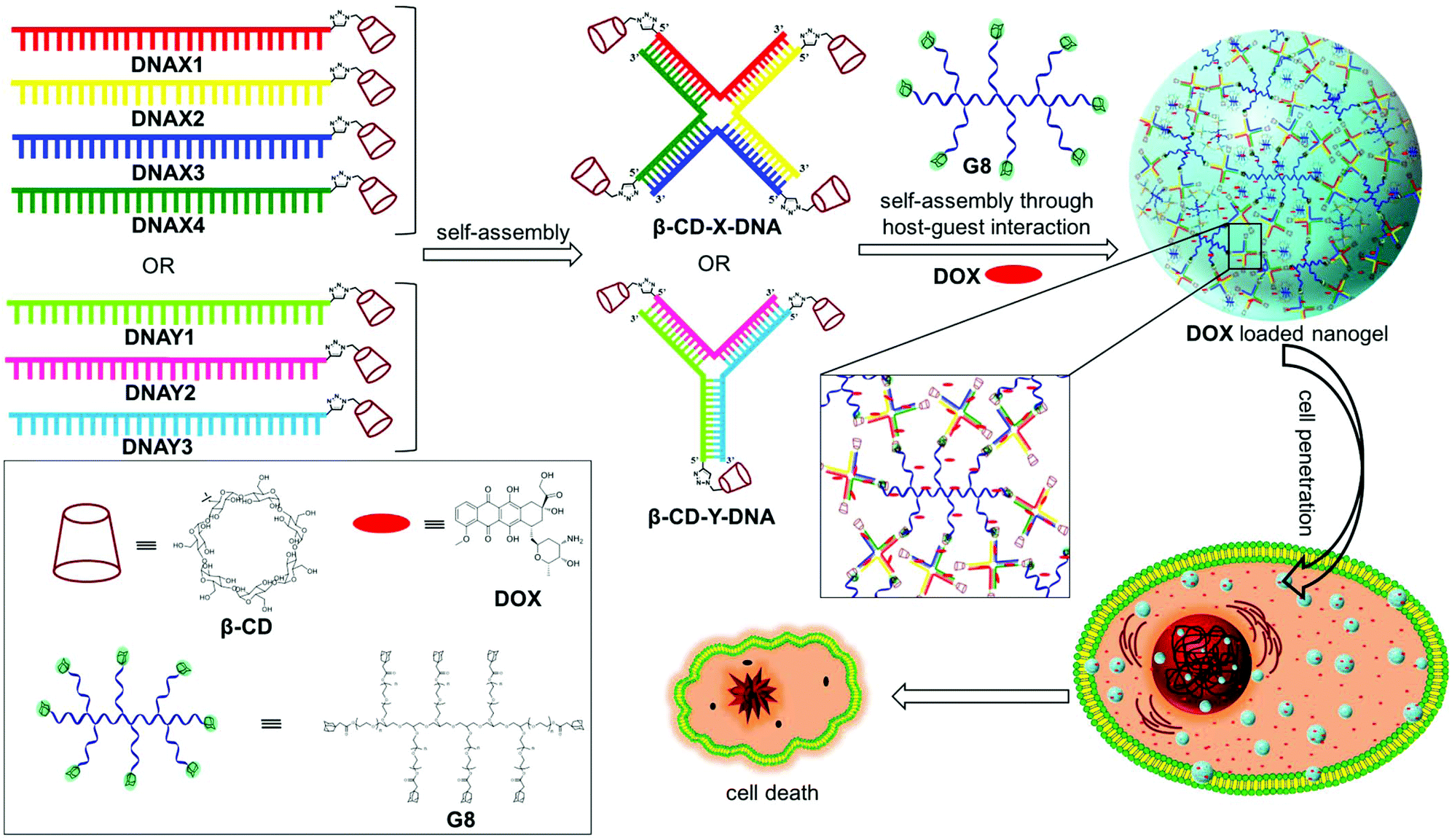
Herein, we report a general strategy for the design of DNA nanogels with a defined size using the multivalent host–guest interaction between β-CD functionalized branched DNA nanostructures as the host and a star-shaped adamantyl-terminated 8-arm poly(ethylene glycol) (PEG) polymer as the guest. It is to be noted that both the host and guest selected in our study exhibit excellent biocompatibility. As a proof-of-concept, we have chosen branched X-DNA and Y-DNA as representative examples in our study.12 Our approach involves the initial self-assembly of β-CD modified oligonucleotides to form branched X-DNA (β-CD-X-DNA) or Y-DNA (β-CD-Y-DNA) having β-CD tethered at the end of all oligonucleotides, which then serves as multivalent host DNA in the nanogel formation. The subsequent self-assembly of the host DNA nanostructure with an 8-arm star PEG polymer (G8) through the formation of a β-CD-adamantane inclusion complex leads to the formation of nanogels. One of the interesting features of the nanogel design is the concentration dependency in gelation. Nanogel formation is observed at very low concentrations of the host and guest, and with the increase in concentration, a hierarchical self-assembly of the nanogels is observed that results in the formation of a hydrogel. The importance of multivalent host–guest interactions in nanogel formation is also studied using guest molecules with two (G2), three (G3) and four (G4) adamantane modifications, which shows that multivalency is necessary for gelation. Very interestingly, nanogels exhibit excellent cell permeability due to their nanosize. The potential of the nanogels as a drug carrier is also demonstrated with the encapsulation of doxorubucin (DOX) in the nanogel network and its release inside cancerous cells, which leads to the killing of cancer cells (Scheme 1).
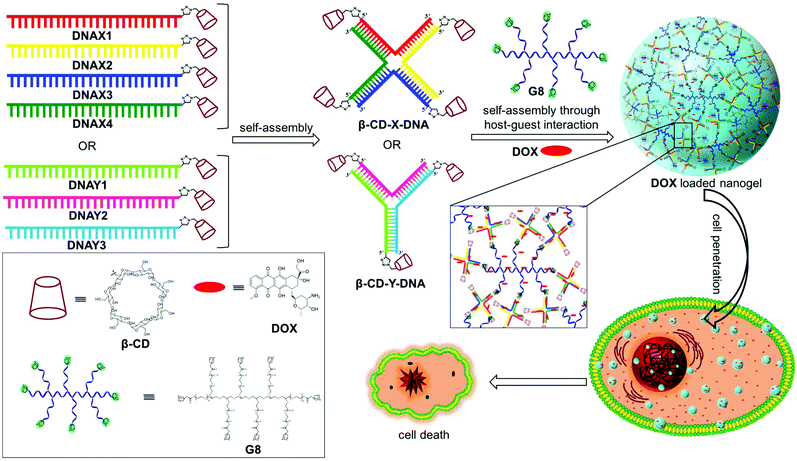 |
| | Scheme 1 Schematic representation showing the structures of DNAX1–4, DNAY1–3 and their self-assembly into β-CD tethered branched X- and Y-DNA. Self-assembly of β-CD-X-DNA or β-CD-Y-DNA with G8 in the presence of DOX leading to the formation of DOX encapsulated DNA nanogels through multivalent host–guest interactions. The cell penetration of DOX loaded DNA nanogels, drug release and cell death are also shown. Chemical structures of β-CD, DOX and G8 are shown in the box. | |
2. Experimental section
2.1 Materials and methods
All chemicals used for the organic syntheses were purchased from Sigma Aldrich and were used as received. Alkyne modified phosphoramidites for automated solid phase DNA synthesis were purchased from Glen Research. CY3 modified oligonucleotides (DNAX-CY3 and DNAY-CY3) were purchased from Integrated DNA Technologies (IDT). TLC analyses were performed on aluminium plates coated with silica gel 60 F254, and column chromatography was performed on 200–400 mesh silica gel. Melting points were measured on Stuart SMP30 melting point apparatus and are uncorrected. 1H and 13C NMR spectra were recorded on a Bruker Avance 500 MHz spectrometer using 1,1,1,1-tetramethylsilane (TMS) as an internal standard. The water used for all studies was Milli-Q deionised water (18.2 MΩ cm). Mass measurements were performed on a Shimadzu GC-MS, QP-2010. HRMS analyses were performed on a Waters Xevo G2 QTof LC/MS system. Measurements were done in negative ion mode. Oligonucleotides were synthesised on a H-8 K&A DNA synthesizer. The alkyne-modified oligonucleotide was purified on a reverse column (Phenomenex Luna C-18 column) on a Shimadzu HPLC system. Native PAGE analyses were performed on an SCIE–PLAS TV400YK vertical electrophoresis unit. Ethidium bromide stained gels were imaged using a Syngene Chemi XR5 gel documentation system. AFM analyses were carried out on a Multimode SPM (Veeco Nanoscope V). Samples were prepared by drop-casting 5 μL solution of the sample on a freshly cleaved mica surface and dried under air. The probe used for imaging was an antimony doped silicon cantilever with a resonant frequency of 300 kHz and a spring constant of 40 Nm−1. TEM analyses were carried out on an FEI Tecnai G2 F20 (200 kV) high-resolution TEM. Samples were prepared by depositing 2 μL of the sample on a 400 mesh carbon coated copper grid (Ted Pella, Inc.) which was negatively glow discharged (PELCO easiGlow, glow discharge cleaning system) for 1 min prior to use. The samples were allowed to adsorb on the grid for 2 min and then excess sample was wicked with a piece of filter paper and the sample was stained with 0.7% uranyl formate stain solution. FE-SEM was carried out on an FEI Nova NanoSEM 450 (FEG type). Samples were prepared for FE-SEM by drop-casting 5 μL of the sample on a freshly cleaved mica surface and allowed to air dry. CLSM was carried out on a Leica TCS SP5 X. The cells were plated on a 12 mm cover slip (Hi-media) at a seeding density of 0.05 × 106 and grown for 24 h. Subsequently, the cells were treated with 200 nM CY3 tagged DNA nanogels and incubated for 24 h. The cells were fixed using 4% paraformaldehyde solution for 10 minutes, followed by three times washing with 1 × PBS. The cells were further permeabilized using 0.01% Triton X for 10 minutes, stained with DAPI (10 μg mL−1) and then washed off. The cover slips were then mounted on a glass slide with Prolong Gold Anti-fade Reagent for confocal imaging. After air drying the samples, an aliquot of hardset mounting medium (Vectashield, with and without DAPI) was dropped over the sample, a cover slip was mounted on a sample without air bubbles and the samples were kept undisturbed for 30 minutes. Then slides were imaged in the presence of immersion oil. Absorption spectra were recorded using a quartz cuvette of 10 mm path length on a Shimadzu UV-3600 Vis-NIR Spectrophotometer having a Peltier controlled cell holder. Steady state fluorescence spectra were recorded on a Horiba Jobin Yvon Fluorimeter equipped with a thermostat Peltier cell holder, in a quartz cuvette of 10 mm path length. Dynamic Light Scattering (DLS) was performed on a Malvern Zetasizer Nano ZS equipped with a 655 nm laser. Experiments were performed at 25 °C at a back-scattering angle of 173°. DSC experiments were carried out on Q20 (TA Instruments) under a nitrogen atmosphere at the heating and cooling rates of 2 °C per minute. FACS analyses were carried out on a FACS LSR Fortessa flow cytometer (BD, USA). Cell lines were borrowed from NCCS, Pune (A549 and HeLa) and RGCB, Trivandrum (MCF-7), and were obtained from American Type Culture Collection (ATCC). DMEM, RPMI, antibiotic solution 100X liquid-1000U penicillin and 10 mg streptomycin per mL were obtained from Hi-media Lab Pvt Ltd, India, and 10% FBS was obtained from PAN-Biotech GmbH, Germany.
3. Results and discussion
3.1 Synthesis of nanogels
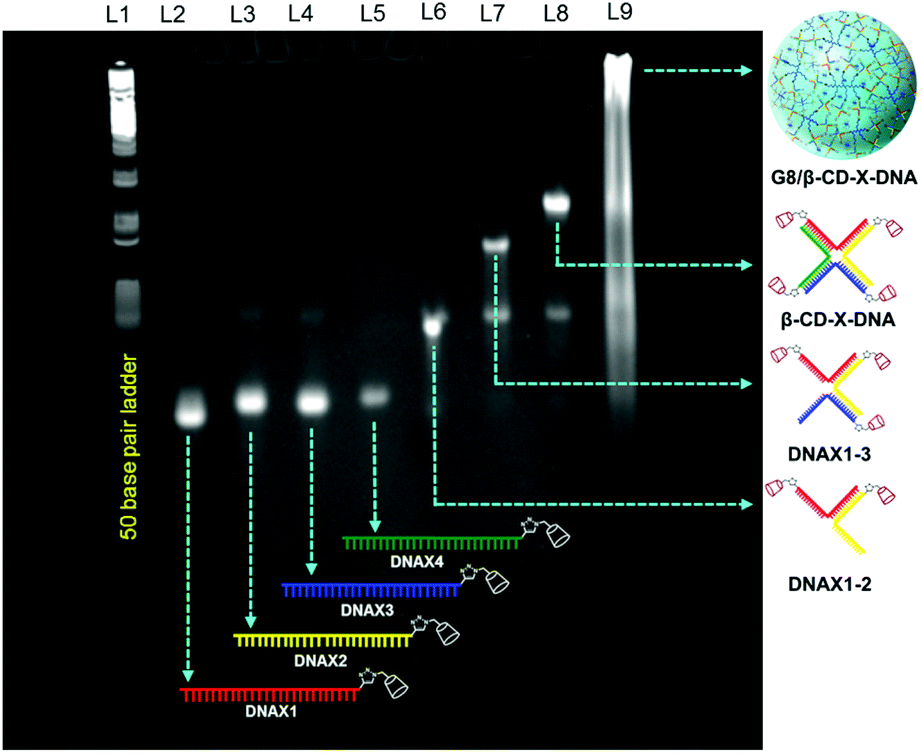
Adamantane modified guest molecules G2–G4 and G8 were synthesized through multistep organic reactions and the details are provided in the ESI.† The guest molecules G2–G4 and G8 differ primarily in the number of adamantane modifications, which is increased from two in G2 to three in G3 to four in G4 to eight in G8. These guest molecules were designed to study the importance of multivalency in nanogel formation. The synthesis of β-CD modified DNAs was achieved following our recently published procedure.11b Briefly, azide functionalized β-CD was synthesized following a reported procedure,13 which was then conjugated to 5′-alkyne modified oligonucleotides using the copper assisted alkyne–azide cycloaddition (CuAAC) reaction, yielding β-CD conjugated oligonucleotides (DNAX1–4 and DNAY1–3, Table 1). Details of the synthesis and characterization of β-CD functionalized oligonucleotides are provided in the ESI.† The sequences of oligonucleotides DNAX1–4 are complementary to form a branched X-DNA with β-CD protruding on each of the four arms (β-CD-X-DNA) as shown in Scheme 1. Similarly, oligonucleotides DNAY1–3 are complementary to form a branched Y-DNA with β-CD protruding on each of the three arms of Y-DNA (β-CD-Y-DNA). Cyanine dye (CY3) tagged oligonucleotides were also synthesized in order to characterize the nanoparticle formation and also to monitor the cell permeability of the nanogels using the fluorescence property of CY3. For this purpose, CY3 was incorporated instead of β-CD into one of the complementary DNA strands (DNAX4 for β-CD-X-DNA and DNAY3 for β-CD-Y-DNA) to give X-DNA-CY3 and Y-DNA-CY3, respectively. Initially, we studied the self-assembly of β-CD functionalized X-DNA and Y-DNA formation (host DNAs) through native polyacrylamide gel electrophoresis (PAGE) analyses (Fig. 1). For the synthesis of host DNAs, equimolar quantities of DNAX1–4 (for β-CD-X-DNA) or DNAY1–3 (for β-CD-Y-DNA) were mixed in 10 mM PBS buffer (pH 8.0) and annealed from 90 °C to room temperature following the reported annealing protocol.12 Native PAGE analyses clearly show that the sequential addition of complementary oligonucleotides for β-CD-X-DNA (Fig. 1) and β-CD-Y-DNA (Fig. S1†) leads to the formation of β-CD-X-DNA and β-CD-Y-DNA, respectively as the major products. This is evident from the reduced electrophoretic mobility of the resultant DNA nanostructure compared with the corresponding individual complementary DNA strands. This also demonstrates that the covalent incorporation of β-CD onto the oligonucleotides is not affecting the self-assembly property of DNAs to form the designed structures in high yields. After establishing the self-assembling condition for the branched host DNAs, we studied the host–guest interaction between the host DNAs and the adamantane modified guest molecules. Interestingly, the self-assembly of G8 and β-CD-X-DNA (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 molar ratio) through the multivalent host–guest interaction between the β-CD and adamantane leads to the formation of nanoassembly as is evident from the further reduction in the gel mobility of the G8/β-CD-X-DNA complex when compared with the gel mobility of β-CD-X-DNA alone. As expected, a band corresponding to the β-CD-X-DNA is also seen in lane L9. This can be attributed to the partial dissociation of the inclusion complex due to the reversible nature of the host–guest interactions. Similar self-assembly behaviours were observed in the case of the G8/β-CD-Y-DNA inclusion complex as well (Fig. S1†). Very importantly, no nanostructure formation is observed when the self-assembly of β-CD-X-DNA or β-CD-Y-DNA was performed with G2, G3 and G4 (Fig. S2†), which clearly demonstrates the importance of multivalent interactions in the nanogel formation.
:2 molar ratio) through the multivalent host–guest interaction between the β-CD and adamantane leads to the formation of nanoassembly as is evident from the further reduction in the gel mobility of the G8/β-CD-X-DNA complex when compared with the gel mobility of β-CD-X-DNA alone. As expected, a band corresponding to the β-CD-X-DNA is also seen in lane L9. This can be attributed to the partial dissociation of the inclusion complex due to the reversible nature of the host–guest interactions. Similar self-assembly behaviours were observed in the case of the G8/β-CD-Y-DNA inclusion complex as well (Fig. S1†). Very importantly, no nanostructure formation is observed when the self-assembly of β-CD-X-DNA or β-CD-Y-DNA was performed with G2, G3 and G4 (Fig. S2†), which clearly demonstrates the importance of multivalent interactions in the nanogel formation.
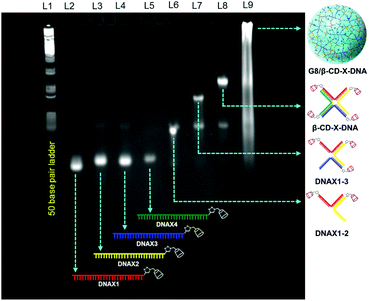 |
| | Fig. 1 Native PAGE (14%) analysis of the self-assembly of G8/β-CD-X-DNA. L1 for 50 base pair ladder, L2–L5 for DNAX1–4, respectively, L6 for the assembly of DNAX1 and DNAX2, L7 for the assembly of DNAX1, DNAX2 and DNAX3, L8 for the assembly of DNAX1, DNAX2, DNAX3 and DNAX4, L9 for the assembly of G8 and β-CD-X-DNA. | |
Table 1 Sequence of DNAs
| DNA |
Sequence (5′ → 3′) |
|
DNAX1
|
βCD-TCGACCGATGAATAGCGGTCAGATCCGT ACCTACTCGA |
|
DNAX2
|
βCD-TCGAGTAGGTACGGATCTGCGTATTGCGAACGACTCGA |
|
DNAX3
|
βCD-TCGAGTCGTTCGCAATACGGCTGTACGTATGGTCTCGA |
|
DNAX4
|
βCD-TCGAGACCATACGTACAGCACCGCTATTCATCGGTCGA |
|
DNAX-CY3
|
Cy3-TCGAGACCATACGTACAGCACCGCTATTCATCGGTCGA |
|
DNAY1
|
βCD-TTGGATCCGCATGACATTCGCCGTAAGA |
|
DNAY2
|
βCD-TCTTACGGCGAATGACCGAATCAGCCTA |
|
DNAY3
|
βCD-TAGGCTGATTCGGTTCATGCGGATCCAA |
|
DNAY-CY3
|
Cy3-TAGGCTGATTCGGTTCATGCGGATCCAA |
3.2 Morphological analyses
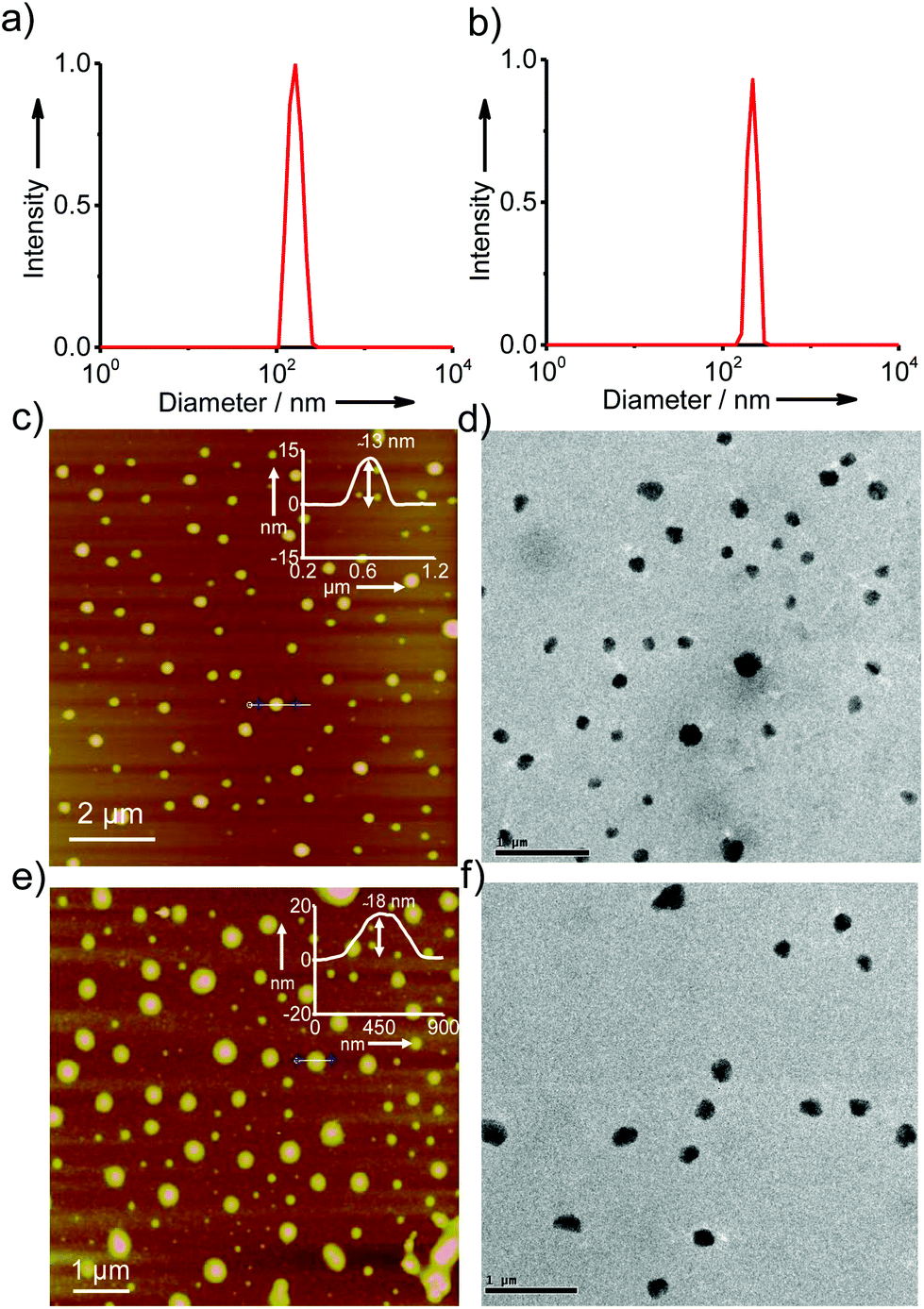
In order to gain better insight into the morphology of the inclusion complexes of G8/β-CD-X-DNA and G8/β-CD-Y-DNA, detailed dynamic light scattering (DLS), atomic force microscopy (AFM), scanning electron microscopy (SEM), transmission electron microscopy (TEM) and confocal microscopy analyses were carried out. DLS analyses of G8/β-CD-X-DNA at a stoichiometry of 1:2 μM (G8:β-CD-X-DNA) show the unimodal distribution of spherical particles with sizes ranging from 105 nm to 250 nm (Fig. 2a). At the same concentration, G8/β-CD-Y-DNA also reveals the formation of spherical particles with a size distribution of 140 nm to 290 nm (Fig. 2b). These particle size distributions imply that the hierarchical self-assembly of G8 with β-CD-X-DNA and β-CD-Y-DNA through the multiple host–guest interactions leads to the formation of spherical nanoparticles (nanogels) of controlled size in aqueous medium. Furthermore, zeta potential (ζ) measurements show values of −16.1 mV and −16.6 mV for G8/β-CD-X-DNA and G8/β-CD-Y-DNA nanoparticles, respectively (Fig. S3†). This indicates that the particle surface is decorated with negatively charged DNA in their respective nanogels. Interestingly, particle formation is observed when the self-assembly is carried out with β-CD-X-DNA and G4 as is evident from the DLS analyses (Fig. S4†). However, no particle formation is observed for the self-assembly of β-CD-X-DNA with G2 or G3 (Fig. S4†). Different microscopy analyses were further performed to confirm the nanogel formation in different concentration ranges. Initially, the microscopy analyses were carried out at a concentration of 1:2 molar ratio for G8:β-CD-X-DNA and G8:β-CD-Y-DNA. The height images of the self-assemblies of G8/β-CD-X-DNA obtained from tapping mode-AFM show the formation of nanoparticles on the mica surface with size distribution in the range of 75–300 nm (Fig. 2c). The cross-sectional analysis of a representative nanoparticle reveals that the height of the particle is 13 nm (Fig. 2c inset). Similarly, nanoparticles with diameters in the range of 150–300 nm are observed for G8/β-CD-Y-DNA (Fig. 2e). The height of a representative particle is 18 nm (Fig. 2e inset). The spherical morphology of the G8/β-CD-X-DNA and G8/β-CD-Y-DNA complexes were further confirmed by TEM analyses, which show the formation of spherical nanoparticles for the G8/β-CD-X-DNA (Fig. 2d) and G8/β-CD-Y-DNA complexes (Fig. 2f). The average diameters of the particles calculated from TEM are ∼200 nm and ∼250 nm for G8/β-CD-X-DNA and G8/β-CD-Y-DNA, respectively. Nanoparticle formation for G8/β-CD-X-DNA and G8/β-CD-Y-DNA is also confirmed by the SEM analyses (Fig. S9 and S10†). Self-assembly with DNAX-CY3 and DNAY-CY3 (CY3 modified oligonucleotides) instead of DNAX4 and DNAY3, respectively, yielded the corresponding CY3 tagged β-CD-X-DNA (X-DNA-CY3) and β-CD-Y-DNA (Y-DNA-CY3) as the hosts. Confocal microscopy analyses (λex = 514 nm) of CY3 tagged X-DNA and Y-DNA with G8 reveal the formation of fluorescent nanoparticles for the inclusion complexes (Fig. S11†). Though DLS analyses revealed oligomerization for the G4:β-CD-X-DNA inclusion complex, no particle formation is observed in the microscopic analyses, suggesting the weak association of G4 and β-CD-X-DNA. Subsequently, we have investigated the self-assembly behaviours of the inclusion complexes at higher concentrations. As expected, hierarchical aggregation of nanoparticles is observed with the increase in the concentration of the host and guest. At a molar ratio of 5:10 μM for G8:β-CD-X-DNA and G8:β-CD-Y-DNA, fusion of the nanoparticles is seen as is evident from AFM (Fig. 3a) and SEM (Fig. 3b) analyses. This leads to the formation of bigger nanoparticles due to the hierarchical self-assembly of the nanoparticles with the increase in the concentration of the host and guest molecules. Very interestingly, a hydrogel is formed at a concentration of 1:2 mM of G8:β-CD-X-DNA (Fig. 3c) and G8:β-CD-Y-DNA. Fig. 3d shows the SEM image of the G8:β-CD-X-DNA hydrogel, which clearly shows the fused nanoparticles in the gel matrix. Furthermore, the hydrogelation of G8:β-CD-X-DNA was studied using differential scanning calorimetry (DSC) analysis, which shows an endothermic peak at 41.7 °C in the heating curve. This corresponds to the melting of the hydrogel (Tgel) to the sol (Fig. S12†). These results collectively suggest that the self-assembly of β-CD-X-DNA or β-CD-Y-DNA and the guest with two (G2) and three (G3) adamantanes is unable to form particles in solution, whereas the guest with four adamantanes (G4) undergoes oligomerization and forms weakly associated particles in solution. Self-assembly of β-CD-X-DNA or β-CD-Y-DNA with the guest having eight adamantanes (G8) leads to the formation of nanoparticles of size in the range of 100–300 nm at a molar ratio of 1:2 μM for guest:host and with the increase in concentration, the hierarchical assembly of the particles leads to the fusion of nanoparticles, which eventually forms hydrogel at a concentration of 1:2 mM of guest:host. These results confirm our hypothesis on the importance of multivalency in the self-assembly and nanogel formation.
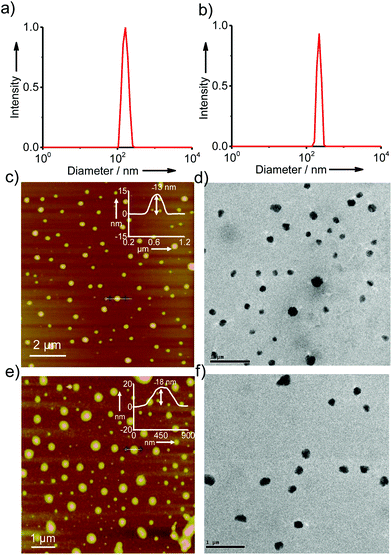 |
| | Fig. 2 Size distribution graphs from DLS analysis of (a) G8/β-CD-X-DNA and (b) G8/β-CD-Y-DNA. Tapping-mode AFM height images of (c) G8/β-CD-X-DNA (z-scale = 22 nm) and (e) G8/β-CD-Y-DNA (z-scale = 33 nm). The corresponding insets of AFM show section analysis of a representative nanoparticle. TEM images of (d) G8/β-CD-X-DNA and (f) G8/β-CD-Y-DNA. | |
 |
| | Fig. 3 (a) Tapping-mode AFM height image of G8/β-CD-X-DNA at [G8:β-CD-X-DNA] = 5:10 μM (z-scale = 56 nm) and (b) the corresponding SEM image. (c) Photograph of hydrogel of G8/β-CD-X-DNA at [G8:β-CD-X-DNA] = 1:2 mM and (d) SEM image of hydrogel of G8/β-CD-X-DNA. | |
3.3 Drug encapsulation studies with doxorubicin
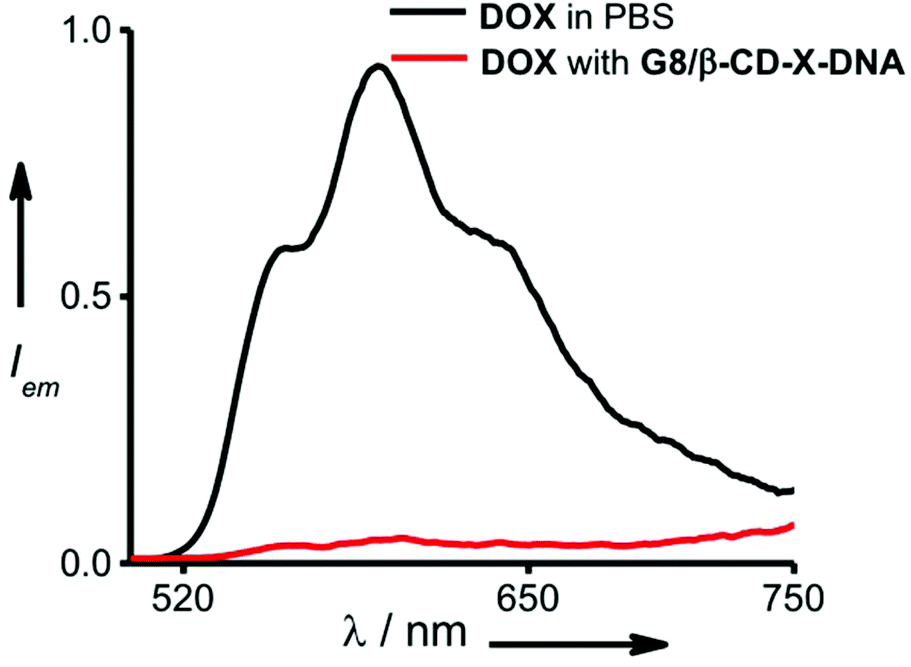
One of the potential applications of nanogels is to act as a nanocarrier in drug delivery applications. Having established the self-assembly conditions for the formation of nanoparticles of controlled size, we have investigated the potential of the nanogel as a nanocarrier for drug delivery. The nanogel network consists of hydrophobic domains of ethylene glycol and hydrophilic domains of DNA. The drug encapsulation capability of the nanogels of G8/β-CD-X-DNA and G8/β-CD-Y-DNA was investigated using DOX, a hydrophobic chemotherapeutic drug, as a representative example. The fluorescence changes of DOX were monitored in order to study the loading of the drug into the nanogel network. The emission spectrum of DOX shows a maximum centred at 596 nm with shoulder bands at 558 nm and 642 nm (λex = 470 nm). Loading of the drug was achieved by incubating DOX during the self-assembly of the nanogels. The emission spectrum of DOX after loading into the G8/β-CD-X-DNA (Fig. 4) and G8/β-CD-Y-DNA (Fig. S13†) nanogels show 95% and 96% quenching of fluorescence, respectively when compared to the emission of the absorption matched solution of free DOX. This significant quenching of the fluorescence of DOX after its loading into the nanogel network can be attributed to the self-quenching of fluorescence due to the efficient loading of DOX into the nanogel network, most likely inside the hydrophobic domain (ethylene glycol region) of the nanogels. However, the intercalation of DOX into ds-DNA cannot be completely ruled out as observed in similar systems.14 The loading efficiency of DOX into the G8/β-CD-X-DNA and G8/β-CD-Y-DNA nanogels is calculated to be 95%. Our results indicate that the DOX loading efficiency of the present nanogel formulation is comparable to many of the DNA based drug delivery systems.15 This high efficiency of DOX loading can be attributed to the availability of dual binding regions (hydrophobic domains of ethylene glycol and intercalation sites of ds-DNA) for DOX.
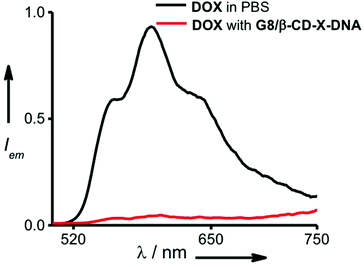 |
| | Fig. 4 Comparison of fluorescence spectra of DOX encapsulated in the gel network of G8/β-CD-X-DNA and free DOX (λex = 470 nm). | |
3.4 Cell permeability studies
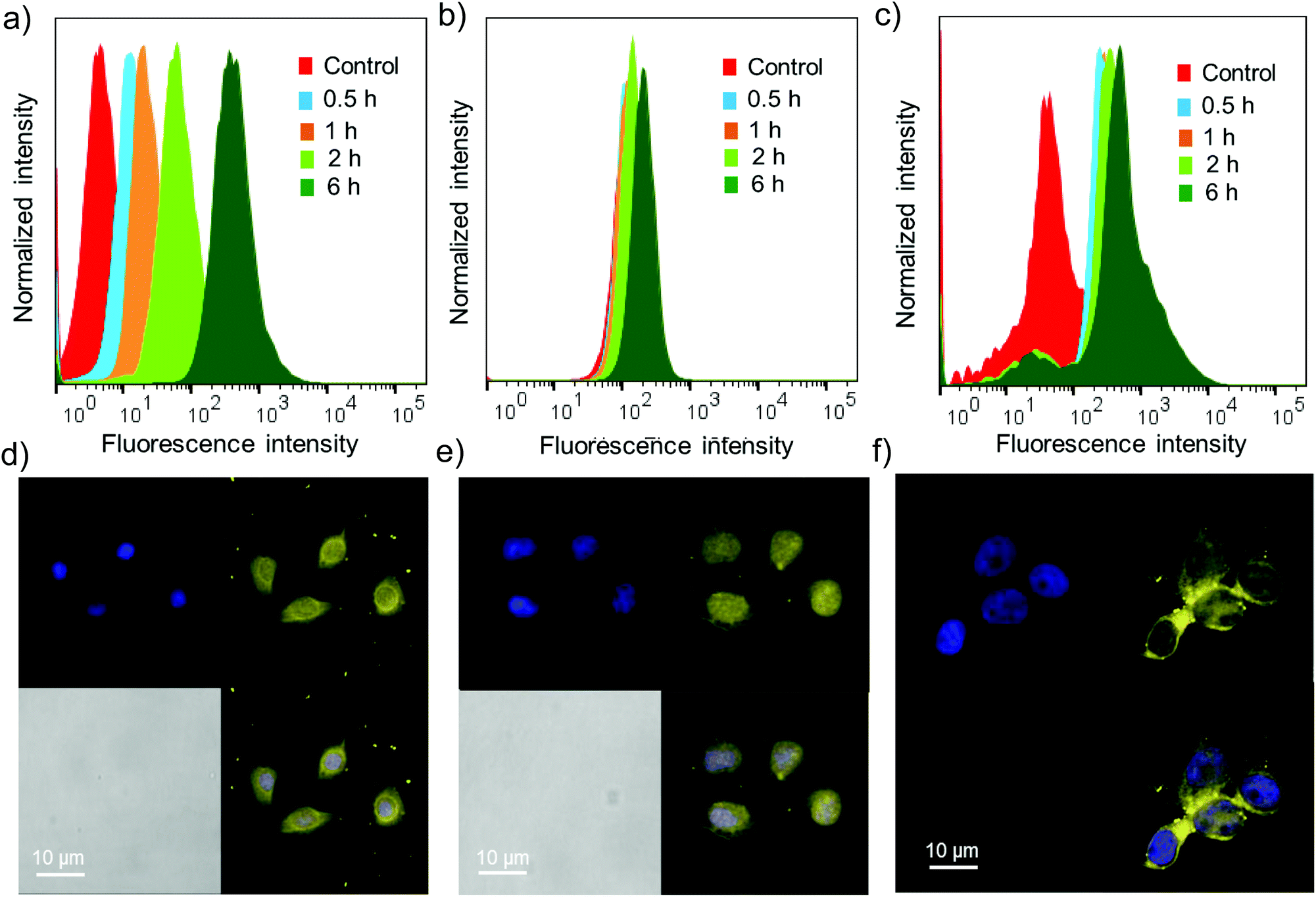
Cell permeability studies were carried out on different cancerous cell lines including HeLa, A549 and MCF-7. To investigate the cell permeability, mammalian cells were treated with G8/X-DNA-CY3 nanogels and their presence in the cells was analysed using laser scanning confocal microscopy (LSCM) and fluorescence activated cell sorting (FACS) techniques. The fluorescence of CY3 was used to monitor the presence of nanogels inside the cells. The cells were treated with nanogels of G8/X-DNA-CY3 for different time intervals (0.5 h, 1 h, 2 h and 6 h). FACS analyses clearly show that fluorescence is associated with A549, HeLa and MCF-7 cells and reveal a mean fluorescence intensity (MFI) of 512, 293, 890 for the 6 h treated samples when compared with the untreated samples (control) that have MFIs of 8, 128, 78, respectively (Fig. 5a–c). Treatment for intermediate time scales exhibit the MFI accordingly. Subsequently, we have carried out experiments in the cells treated with 4′,6-diamidino-2′-phenylindole (DAPI) that selectively stains the nucleus of the cells to assess the internal localization of the nanoparticles. Confocal microscopic images show that internalization of nanogels occurs both in the nucleus and cytoplasm of A549 (Fig. 5d), HeLa (Fig. 5e) and MCF-7 (Fig. 5f) cells. Similar results were observed with G8/Y-DNA-CY3 nanogels as well (Fig. S14†). Microscopic and FACS analyses collectively demonstrate the excellent cell penetration of nanogels, most likely through the endocytosis mechanism as observed with similar systems.16
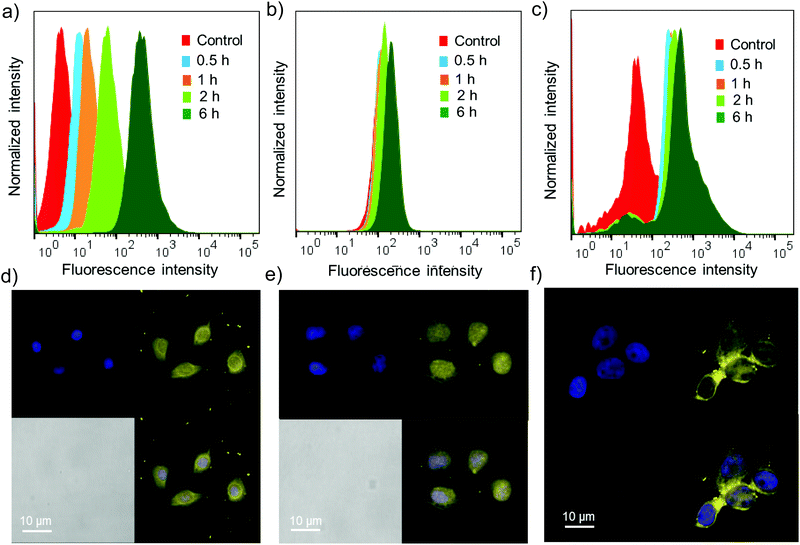 |
| | Fig. 5 Flow cytometry analyses to monitor the binding of G8/X-DNA-CY3 nanogel with (a) A549, (b) HeLa and (c) MCF-7 cell lines at different time intervals. CLSM analyses of (d) A549 cells, (e) HeLa cells and (f) MCF7 cells after their incubation with G8/X-DNA-CY3 nanogels for 24 h. | |
3.5 Drug delivery studies
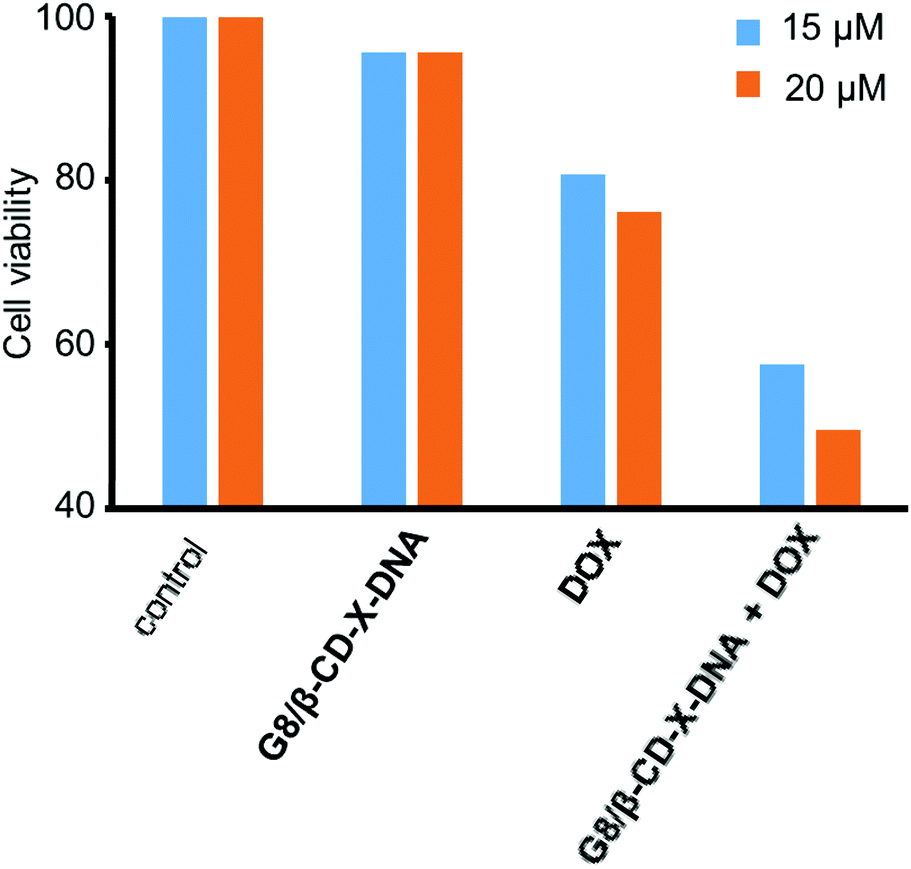
Having demonstrated the drug encapsulation and cell penetration of the nanogels, we have investigated the drug delivery potential of the nanogels using G8/β-CD-X-DNA as the representative example. The in vitro cytotoxicity analysis of the DOX-loaded G8/β-CD-X-DNA nanogel was carried out on a DOX-resistant MCF-7 cell line. For this purpose, the DOX-loaded G8/β-CD-X-DNA nanogel and DOX-free G8/β-CD-X-DNA nanogel were incubated with the MCF-7 cell line for 48 h. Cell viability of the drug-loaded nanogel was studied by means of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. The viability of the untreated MCF-7 cells was taken as 100%. In order to assess the biocompatibility of the nanogel as a drug carrier, the MCF-7 cells were incubated with DOX-free G8/β-CD-X-DNA nanoparticles. As expected, the MTT assay reveals cell viability above 95% demonstrating the excellent biocompatibility of the nanogels (Fig. 6). Subsequently, we have investigated the in vitro cytotoxicities of DOX-loaded G8/β-CD-X-DNA nanogels and DOX alone towards the MCF-7 cell line. The MTT assay reveals that the DOX-loaded G8/β-CD-X-DNA nanogel shows higher cytotoxicity towards the DOX-resistant MCF-7 cells than DOX alone. The half-maximal inhibitory concentration (IC50) of DOX/G8/β-CD-X-DNA is 18.3 μM, which is significantly lower when compared with DOX alone (48.6 μM) under the same experimental conditions. These results suggest that the DOX loaded nanogel efficiently penetrates into the cell, distributes in the cytoplasm as well as the nucleus without much selectivity, and releases the encapsulated drug inside the cell. Though the exact mechanism of drug release from the nanogel is not clear at this stage, the cellular environments facilitate the slow release of the encapsulated drug and its distribution inside the cell, which leads to cell death as observed in similar systems.6b,14 However, the selectivity of the nanogel towards the cancer cells and the efficient release of the encapsulated drugs in the cancerous environment need to be improved for the effective application of the nanogel as a drug carrier, which is the current focus of our laboratory.
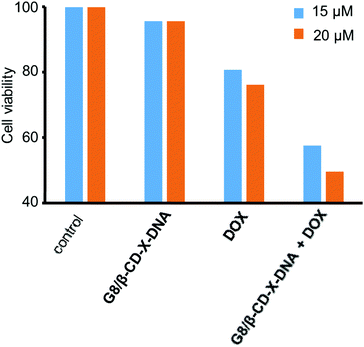 |
| | Fig. 6 Comparison of the viability of MCF-7 cells treated with DOX/G8/β-CD-X-DNA and free DOX. [DOX/G8/β-CD-X-DNA] = 1 μM and [DOX] = 15 μM and 20 μM. | |
4. Conclusions
In summary, we have reported a supramolecular approach based on the host–guest interaction between β-CD and adamantane (physical crosslinking) for the design of a new class of DNA nanogels with controllable size. Our approach is universal and modular in nature, and in principle can be applied as a general strategy for the design of nanogels of any 2D and 3D DNA nanostructures by appropriate functionalization. Nanogel formation was observed only with branched DNA nanostructures and a star-shaped adamantyl-terminated 8-arm PEG polymer with eight adamantane modifications as the guest, which demonstrated the importance of multivalent interactions in nanogel formation and hydrogelation. Microscopy analyses have shown that the nanoparticle formation (100–300 nm) was observed at low concentrations of the host and guest, and the hierarchical fusion of nanoparticles was observed with the increase in the concentration of the components that finally led to hydrogelation at very high concentrations of the host and guest components. One of the most attractive features of DNA nanogels is that they act as a nanocarrier for cargo delivery applications due to their remarkable structural features including excellent biocompatibility, efficient drug encapsulation ability, stimuli-responsive nature and DNA-based surface addressability. We have demonstrated the excellent biocompatibility and efficient drug (DOX) encapsulation ability of the DNA nanoparticles. Confocal microscopy imaging and MTT assay analyses have shown the excellent cell permeability of the DOX loaded nanogel, and the delivery of the loaded drug inside the cell. These results clearly suggest that DNA nanogel of this kind is a promising candidate for the drug delivery applications. However, cell targeting and efficient release of the encapsulated drug need to be improved for their effective use as a drug carrier for cancer therapy. These features can easily be incorporated onto DNA nanoparticles as demonstrated in similar DNA based systems.17 For instance, cell targeting ligands/aptamers could be incorporated onto the nanoparticle surface by taking advantage of the sequence specific hybridization of DNA. Similarly, the stimuli responsive nature for the nanoparticle can be achieved by the incorporation of an acid cleavable linker that could be efficiently cleaved in the acidic cellular environment of cancerous cells.6b We hope the novel design strategy reported here might encourage other researchers to design new DNA nanogels for addressing the challenges in nanomedicine.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
Financial support from DBT (BT/PR7030/NNT/28/63/2012) is gratefully acknowledged. H. V. P. T. and N. K. thank UGC and S. K. A. thanks CSIR for research fellowships. We thank the University of Madras for TEM, and MGU and CSIR-NIIST for HR-MS analyses. The help of Sarika Mohan S. is acknowledged for the FACS analyses.
References
- A. V. Kabanov and S. V. Vinogradov, Angew. Chem., Int. Ed., 2009, 48, 5418–5429 CrossRef CAS PubMed.
- T. Nochi, Y. Yuki, H. Takahashi, S.-i. Sawada, M. Mejima, T. Kohda, N. Harada, G. Kong, A. Sato, N. Kataoka, D. Tokuhara, S. Kurokawa, Y. Takahashi, H. Tsukada, S. Kozaki, K. Akiyoshi and H. Kiyono, Nat. Mater., 2010, 9, 572–578 CrossRef CAS PubMed.
-
(a) D. Bhatia, S. Surana, S. Chakraborty, S. P. Koushika and Y. Krishnan, Nat. Commun., 2011, 2, 339 CrossRef PubMed;
(b) Q. Jiang, C. Song, J. Nangreave, X. Liu, L. Lin, D. Qiu, Z.-G. Wang, G. Zou, X. Liang, H. Yan and B. Ding, J. Am. Chem. Soc., 2012, 134, 13396–13403 CrossRef CAS PubMed;
(c) A. Banerjee, D. Bhatia, A. Saminathan, S. Chakraborty, S. Kar and Y. Krishnan, Angew. Chem., Int. Ed., 2013, 52, 6854–6857 CrossRef CAS PubMed;
(d) S. M. Douglas, I. Bachelet and G. M. Church, Science, 2012, 335, 831–834 CrossRef CAS PubMed;
(e) F. Huang, W.-C. Liao, Y. S. Sohn, R. Nechushtai, C.-H. Lu and I. Willner, J. Am. Chem. Soc., 2016, 138, 8936–8945 CrossRef CAS PubMed.
-
(a) F. A. Aldaye, A. L. Palmer and H. F. Sleiman, Science, 2008, 321, 1795–1799 CrossRef CAS PubMed;
(b) B. Saccà and C. M. Niemeyer, Chem. Soc. Rev., 2011, 40, 5910–5921 RSC;
(c) J. Fu, Y. R. Yang, A. Johnson-Buck, M. Liu, Y. Liu, N. G. Walter, N. W. Woodbury and H. Yan, Nat. Nanotechnol., 2014, 9, 531–536 CrossRef CAS PubMed; M. R. Jones, N. C. Seeman and C. A. Mirkin, Science, 2015, 347, 1260901–1260911 Search PubMed;
(d) C. Zhou, X. Duan and N. Liu, Nat. Commun., 2015, 6, 8102 CrossRef CAS PubMed;
(e) W. Meng, R. A. Muscat, M. L. McKee, P. J. Milnes, A. H. El-Sagheer, J. Bath, B. G. Davis, T. Brown, R. K. O'Reilly and A. J. Turberfield, Nat. Chem., 2016, 8, 542–548 CrossRef CAS PubMed;
(f) F. N. Gür, F. W. Schwarz, J. Ye, S. Diez and T. L. Schmidt, ACS Nano, 2016, 10, 5374–5382 CrossRef PubMed;
(g) M. J. Urban, P. K. Dutta, P. Wang, X. Duan, X. Shen, B. Ding, Y. Ke and N. Liu, J. Am. Chem. Soc., 2016, 138, 5495–5498 CrossRef CAS PubMed.
- A. M. Rush, M. P. Thompson, E. T. Tatro and N. C. Gianneschi, ACS Nano, 2013, 7, 1379–1387 CrossRef CAS PubMed.
-
(a) H. Kang, A. C. Trondoli, G. Zhu, Y. Chen, Y.-J. Chang, H. Liu, Y.-F. Huang, X. Zhang and W. Tan, ACS Nano, 2011, 5, 5094–5099 CrossRef CAS PubMed;
(b) W. Sun, T. Jiang, Y. Lu, M. Reiff, R. Mo and Z. Gu, J. Am. Chem. Soc., 2014, 136, 14722–14725 CrossRef CAS PubMed;
(c) J. Li, C. Zheng, S. Cansiz, C. Wu, J. Xu, C. Cui, Y. Liu, W. Hou, Y. Wang, L. Zhang, I.-t. Teng, H.-H. Yang and W. Tan, J. Am. Chem. Soc., 2015, 137, 1412–1415 CrossRef CAS PubMed;
(d) K. Matsuura, T. Yamashita, Y. Igami and N. Kimizuka, Chem. Commun., 2003, 376–377 RSC.
- S. Yu, J. Hu, X. Pan, P. Yao and M. Jiang, Langmuir, 2006, 22, 2754–2759 CrossRef CAS PubMed.
-
(a) K. Jalani, M. Kumar and S. J. George, Chem. Commun., 2013, 49, 5174–5176 RSC;
(b) C. Rest, M. J. Mayoral, K. Fucke, J. Schellheimer, V. Stepanenko and G. Fernández, Angew. Chem., Int. Ed., 2014, 53, 700–705 CrossRef CAS PubMed;
(c) X. J. Loh, J. del Barrio, T.-C. Lee and O. A. Scherman, Chem. Commun., 2014, 50, 3033–3303 RSC;
(d) Y. Taniguchi, T. Takishita, T. Kawai and T. Nakashima, Angew. Chem., Int. Ed., 2016, 55, 2083–2086 CrossRef CAS PubMed.
-
(a) A. Harada, A. Hashidzume, H. Yamaguchi and Y. Takashima, Chem. Rev., 2009, 109, 5974–6023 CrossRef CAS PubMed;
(b) X. Zhang and C. Wang, Chem. Soc. Rev., 2011, 40, 94–101 RSC.
-
(a) Z. Li, Y. Zhang, P. Fullhart and C. A. Mirkin, Nano Lett., 2004, 4, 1055–1058 CrossRef CAS;
(b) F. E. Alemdaroglu, K. Ding, R. Berger and A. Herrmann, Angew. Chem., Int. Ed., 2006, 45, 4206–4210 CrossRef CAS PubMed;
(c) X.-J. Chen, B. L. Sanchez-Gaytan, S. E. N. Hayik, M. Fryd, B. B. Wayland and S.-J. Park, Small, 2010, 6, 2256–2260 CrossRef CAS PubMed;
(d) M. P. Thompson, M.-P. Chien, T.-H. Ku, A. M. Rush and N. C. Gianneschi, Nano Lett., 2010, 10, 2690–2694 CrossRef CAS PubMed;
(e) T. G. W. Edwardson, K. M. M. Carneiro, C. K. McLaughlin, C. J. Serpell and H. F. Sleiman, Nat. Chem., 2013, 5, 868–875 CrossRef CAS PubMed;
(f) Z. Zhao, C. Chen, Y. Dong, Z. Yang, Q.-H. Fan and D. Liu, Angew. Chem., Int. Ed., 2014, 53, 13468–13470 CrossRef CAS PubMed;
(g) S. E. Averick, S. K. Dey, D. Grahacharya, K. Matyjaszewski and S. R. Das, Angew. Chem., Int. Ed., 2014, 53, 2739–2744 CrossRef CAS PubMed;
(h) Y. Vyborna, M. Vybornyi and R. Häner, J. Am. Chem. Soc., 2015, 137, 14051–14054 CrossRef CAS PubMed;
(i) R. J. Banga, B. Meckes, S. P. Narayan, A. J. Sprangers, S. T. Nguyen and C. A. Mirkin, J. Am. Chem. Soc., 2017, 139, 4278–4281 CrossRef CAS PubMed.
-
(a) S. K. Albert, H. V. P. Thelu, M. Golla, N. Krishnan, S. Chaudhary and R. Varghese, Angew. Chem., Int. Ed., 2014, 53, 8352–8357 CrossRef CAS PubMed;
(b) S. K. Albert, H. V. P. Thelu, M. Golla, N. Krishnan and R. Varghese, Nanoscale, 2017, 9, 5425–5432 RSC;
(c) S. K. Albert, M. Golla, H. V. P. Thelu, N. Krishnan and R. Varghese, Chem. – Eur. J., 2017, 23, 8348–8352 CrossRef CAS PubMed.
- S. H. Um, J. B. Lee, N. Park, S. Y. Kwon, C. C. Umbach and D. Luo, Nat. Mater., 2006, 5, 797–801 CrossRef CAS PubMed.
- W. Tang and S.-C. Ng, Nat. Protoc., 2008, 3, 691–697 CrossRef CAS PubMed.
- M. Chang, C.-S. Yang and D.-M. Huang, ACS Nano, 2011, 5, 6156–6163 CrossRef CAS PubMed.
- M. I. Setyawati, R. V. Kutty and D. T. Leong, Small, 2016, 12, 5601–5611 CrossRef CAS PubMed.
- L. L. Lock, C. D. Reyes, P. Zhang and H. Cui, J. Am. Chem. Soc., 2016, 138, 3533–3540 CrossRef CAS PubMed.
- F. E. Alemdaroglu, N. C. Alemdaroglu, P. Langguth and A. Herrmann, Adv. Mater., 2008, 20, 899–902 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details and figures referenced throughout the text. See DOI: 10.1039/c7nr06985e |
|
| This journal is © The Royal Society of Chemistry 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *a
*a