
A protected excitation-energy reservoir for efficient upconversion luminescence†
Kai
Huang‡
 a,
Haichun
Liu‡
b,
Marco
Kraft
c,
Swati
Shikha
a,
Xiang
Zheng
d,
Hans
Ågren
b,
Christian
Würth
c,
Ute
Resch-Genger
c and
Yong
Zhang
*ad
a,
Haichun
Liu‡
b,
Marco
Kraft
c,
Swati
Shikha
a,
Xiang
Zheng
d,
Hans
Ågren
b,
Christian
Würth
c,
Ute
Resch-Genger
c and
Yong
Zhang
*ad
aDepartment of Biomedical Engineering, Faculty of Engineering, National University of Singapore, 4 Engineering Drive 3, 117583 Singapore. E-mail: biezy@nus.edu.sg
bDivision of Theoretical Chemistry and Biology, Royal Institute of Technology, S-10691 Stockholm, Sweden
cFederal Institute of Materials Research and Testing (BAM), Division of Biophotonics, Richard-Willstätter-Str. 11, 12489 Berlin, Germany
dNUS Graduate School for Integrative Sciences & Engineering, National University of Singapore, , 117456 Singapore
First published on 10th November 2017
Abstract
Lanthanide-doped upconversion nanoparticles (UCNPs) are of great interest for biomedical applications. Currently, the applicability of UCNP bionanotechnology is hampered by the generally low luminescence intensity of UCNPs and inefficient energy transfer from UCNPs to surface-bound chromophores used e.g. for photodynamic therapy or analyte sensing. In this work, we address the low-efficiency issue by developing versatile core–shell nanostructures, where high-concentration sensitizers and activators are confined in the core and shell region of representative hexagonal NaYF4:Yb,Er UCNPs. After doping concentration optimization, the sensitizer-rich core is able to harvest/accumulate more excitation energy and generate almost one order of magnitude higher luminescence intensity than conventional homogeneously doped nanostructures. At the same time, the activator ions located in the shell enable a ∼6 times more efficient resonant energy transfer from UCNPs to surface-bound acceptor dye molecules due to the short distance between donor–acceptor pairs. Our work provides new insights into the rational design of UCNPs and will greatly increase the general applicability of upconversion nanotechnologies.
Introduction
Lanthanide ion-doped upconversion nanoparticles (UCNPs) are able to convert low power continuous-wave near-infrared (NIR) light into higher-energy and multicolor UV/visible light by energy transfer between two types of doping ions, termed sensitizers and activators, respectively.1 In recent years, UCNPs have been exploited intensively to achieve various NIR-triggered diagnostic and therapeutic implementations, including bioimaging,2–4 bioassays,5,6 photodynamic therapy (PDT),7 and controlled drug release.8–10 In these applications, UCNPs are mainly employed as energy relay nanosystems that convert and transfer excitation light energy and activate other chromophores, except in a very few examples where UCNPs were utilized as direct imaging tools and therapeutic agents (e.g., in bioimaging).11–13 In spite of numerous proof-of-concept studies, the general applicability of UC nanobiotechnology remains low because of efficiency problems. In order to enhance the overall efficacy of this technique, two key tasks must be performed, i.e., increasing the upconversion luminescence (UCL) intensity and promoting the efficiency of subsequent energy transfer processes from UCNPs to surface-bound substances like stimuli-responsive dyes.14,15UCNPs are generally comprised of inorganic hosts, doped with rare earth (RE) ions. Spatial arrangement of the dopant ions, including dopant concentrations and their spatial distribution, plays a fundamental role in regulating their optical properties.16–21 In addition, the nanoparticle surface is a critical issue that needs to be seriously considered when constructing UCNPs. On one hand, UCL is very sensitive to the immediate environment of the emissive RE ions, because of the high probability of energy dissipation from UCNPs to high energy vibrational modes of surface ligands and solvent molecules.22 Thus, to maximize UCL, UCNPs are preferred to be rationally constructed in a way that optically active ions (especially sensitizers) are protected from the quenching environment.23,24 On the other hand, in all applications where UCNPs act as indirect energy-relay materials, the nanoparticle surface directly interacts with surface-bound chromophores and activates them, e.g., by nonradiative resonant energy transfer. To make this distance-dependent process efficient, a close proximity between the donor (i.e., activator ions) and acceptor (i.e., organic dye molecules) is mandatory.14,15 Currently, there lacks a general approach to rationalize UCNP architectures that meet both requirements, although many studies have been dedicated to resolving these challenges, particularly to enhance the UCL intensity.25–30
In this work, we report a versatile core–shell UCNP architecture that addresses both challenges and will facilitate UCNP-mediated applications. In our design, high concentrations of sensitizer and activator ions, here Yb3+ and Er3+, are spatially separated in the core and shell region of a β-NaYF4 host material, respectively, enabling a previously unattainable breakthrough in the concentration quenching threshold of UCL for both sensitizer and activator ions.31,32 The sensitizer-rich core is able to harvest more excitation energy from the light source, and more importantly, it is able to accumulate the energy since it is protected from sensitizer de-excitation channels caused by surface effects like lattice defects as well as surface ligands and solvent molecules containing chemical groups with high energy vibrations.24,33 Such a well-protected excitation-energy reservoir proves to be able to generate significantly enhanced UCL. Additionally, the emissive lanthanide ions located in the shell region enable a high energy transfer efficiency to surface-bound organic acceptors required e.g., for UCNP-mediated biomedical as well as sensing applications due to the close proximity between the activator ions and the dye molecules. Our work provides a paradigm design concept for UCNPs to promote the success of UC nanobiotechnology.
Experimental
Synthesis of UCNPs
Core and core–shell nanoparticles were synthesized following a previously reported protocol with modifications.34 Typically, 1 mmol lanthanide chlorides were mixed with 6 mL of oleic acid and 15 mL of 1-octadecene in a 100 mL flask. The solution was heated to 150 °C to form a homogenous solution and then cooled down to room temperature. A solution of 4 mmol NH4F and 2.5 mmol NaOH in 10 mL of methanol was added to the flask and stirred for 30 min. Subsequently, the solution was heated to 100 °C to remove the methanol. After methanol was evaporated, the solution was heated to 300 °C and incubated at that temperature for 1 hour under an argon atmosphere and then cooled to room temperature. The UCNPs were precipitated with 10 mL of acetone, collected after centrifugation, then washed thrice with ethanol/water (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v) and finally dispersed in cyclohexane as the core nanoparticles for subsequent use. Core–shell UCNPs were synthesized subsequently on the basis of core UCNPs by growing a shell layer on it. Detailed preparation of core–shell UCNPs with different compositions is stated in the ESI.†
:1 v/v) and finally dispersed in cyclohexane as the core nanoparticles for subsequent use. Core–shell UCNPs were synthesized subsequently on the basis of core UCNPs by growing a shell layer on it. Detailed preparation of core–shell UCNPs with different compositions is stated in the ESI.†
Removal of capping ligands from the surfaces of UCNPs
Ligand-free colloidally stable water-dispersible UCNPs were prepared using the HCl treatment method reported previously.35 In a typical process, 1 mL (∼5 mg mL−1) UCNP cyclohexane suspension was mixed with 1 mL 0.1 M HCl. The mixture was vortexed and then sonicated for 60 min, and most nanoparticles were transferred to the bottom aqueous layer. The top layer was discarded. The nanoparticles in the bottom layer were purified with water, ethanol, and acetone, followed by centrifugation (12000 rpm) for 60 min. The supernatant was discarded and the resulting pellets were dried before they were redispersed in D2O by sonication.
Synthesis of Rose Bengal-doped UCNP@SiO2 nanospheres
Rose Bengal (RB)-doped UCNP@SiO2 nanospheres were prepared following a previously reported protocol with modifications.36 CO-520 (0.75 ml), 11 mL of cyclohexane, and 2 mL of 0.05 M (concentration of total rare earth ions) UCNP cyclohexane solution were mixed and sonicated for 1 min. The mixture was stirred for 10 min before 0.08 mL ammonia (30 wt%) was added. After 30 min, 0.02 mL TEOS was added to the mixture and stirred for 1 day at a speed of 700 rpm. UCNP@SiO2 nanospheres were precipitated by adding acetone, and the nanoparticles were washed with ethanol/water (1:1 v/v) twice and then dispersed in 20 mL ethanol. 0.2 mL ammonia (30 wt%) was added to the solution and stirred for 30 min before Rose Bengal solution was added for loading.
The Rose Bengal dye molecules were first grafted onto APTES to improve their stability, and then the mixture was co-hydrolyzed with TEOS to form silica coatings on the upconversion nanospheres using the reverse microemulsion method. In a typical process, 2 mg Rose Bengal was mixed with 1 mg EDC and 2 mg NHS in 1 mL ethanol. The mixture solution was vortexed for 30 min before adding 0.02 mL APTES. The mixture was vortexed for another 1 hour before adding into the UCNP/ammonia ethanol solution. 0.02 mL TEOS was added into the mixture solution after 4 hours and stirred for 1 day at a speed of 700 rpm. The nanoparticles were washed with ethanol/water (1:1 v/v) twice and then dispersed in 20 mL ethanol for subsequent use.
Rose Bengal photobleaching by UV irradiation
RB-doped UCNP@SiO2 nanospheres dispersed in ethanol were irradiated with a UV lamp (LED UV system Uvata Precision Optoelectronics Co. Ltd, Shanghai, China) with an intensity of 0.8 W cm−2 for 2 hours until the Rose Bengal was photobleached (no emission under 532 nm excitation).Characterization
Transmission electron microscopy (TEM) images were recorded on a JEOL 2010F transmission electron microscope (Jeol Ltd, Tokyo, Japan) operating at an acceleration voltage of 200 kV. X-ray powder diffraction (XRD) measurements were performed on a Siemens D5005 X-ray powder diffractometer with Co Kα radiation (λ = 1.78897 Å). Fluorescence spectra were recorded on a Hitachi F-500 fluorescence spectrophotometer (Hitachi High-Technologies Corporation, Tokyo, Japan) under excitation of a 980 nm continuous-wave laser (Photonitech (Asia) Pte. Ltd, Singapore) or a 532 nm solid state laser (Photonik Singapore Pte. Ltd, Singapore). The UV-visible absorption measurement of a suspension of RB-doped UCNP@SiO2 nanospheres was performed using a UV-visible spectrophotometer (Shimadzu spectrophotometer UV 2401). In the time-dependent emission profile measurements, pulsed laser light output was achieved by modulating the laser driving current using a SFG-2120 synthesized function generator (GW INSTEK). The decay profiles of upconversion emission were recorded by using a digital storage oscilloscope (Tektronix TDS2024C) coupled to the output of a photomultiplier tube (Hamamatsu R928).Results and discussion
Recent studies revealed that excited Yb3+ sensitizer ions can strongly interact with molecules with high energy vibrational modes in the immediate neighborhood of the UCNP surface and dissipate excitation energy.24,37 Due to the nonlinear nature of UC and the energy transfer-mediated interaction between the excited Yb3+ ions, quenching of Yb3+ ions has a much stronger effect on the resulting UCL intensity than other non-radiative deactivation pathways. Thus, the suppression of sensitizer de-excitation is of vital importance for efficient UC.24,37 In this regard, our new core–shell UCNP architecture, in which Yb3+ sensitizers are confined in the core region and spatially separated from the surrounding environment by a shell doped with activator ions (Fig. 1a), has significant potential to elegantly inhibit Yb3+ de-activation. In order to verify this, UCNPs of this new core–shell design (NaYF4:20% Yb@NaYF4:2% Er, denoted as Yb@Er UCNPs) were synthesized using a previously reported protocol,38 together with their counterparts, including conventional homogeneously doped UCNPs (NaYF4:20% Yb,2% Er, referred to as YbEr UCNPs) and another core–shell architecture where sensitizers and activators were spatially inversely distributed (NaYF4:2% Er@NaYF4:20% Yb, denoted as Er@Yb UCNPs), and their UC properties were studied. These three types of differently structured UCNPs, containing the same amounts of Yb3+ and Er3+ ions, display similar physical dimensions with particle diameters of ∼23 nm of the core and (24 × 33) nm of the core–shell UCNPs (Fig. S1a–c†) and lattice structures (Fig. S1d†).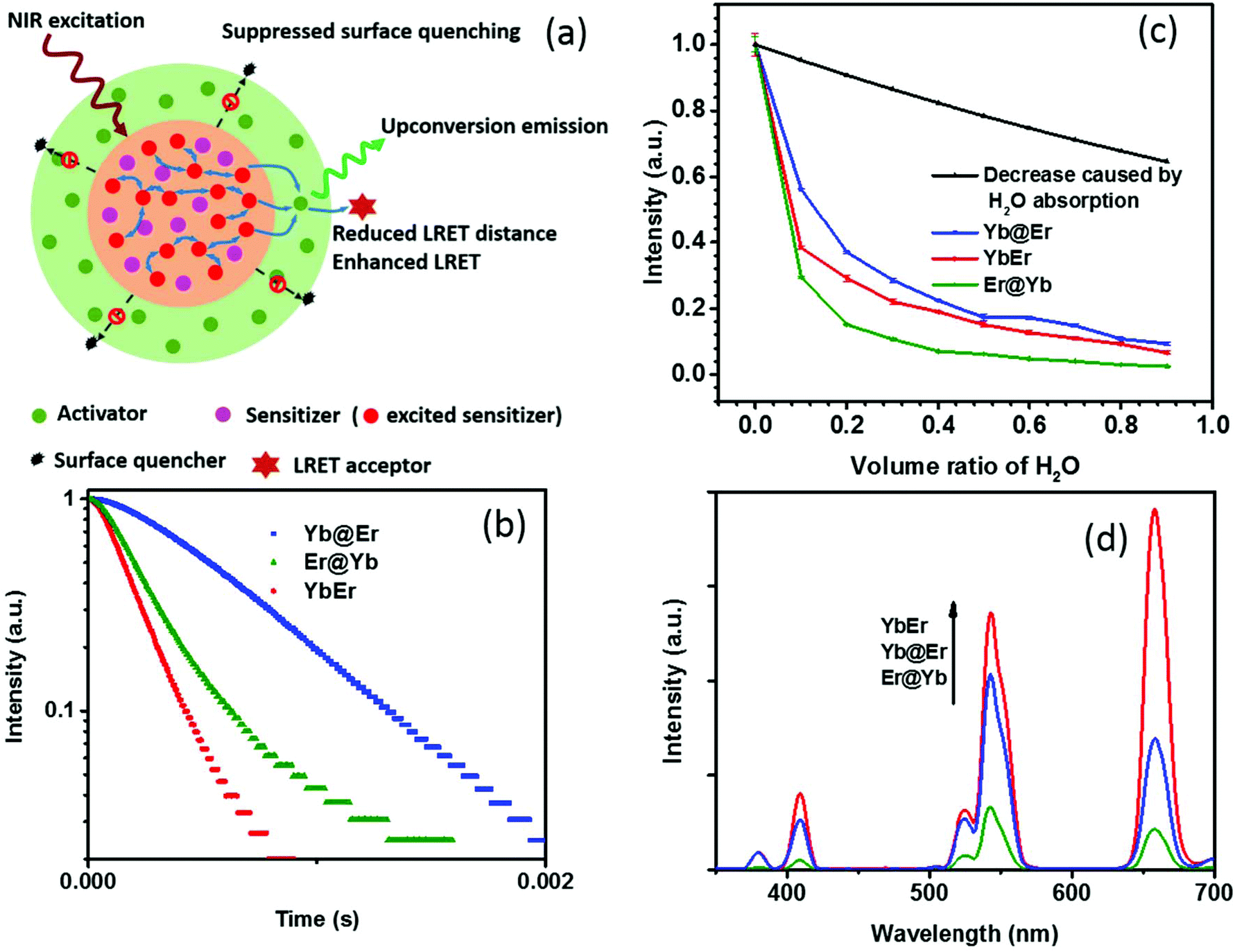 | ||
| Fig. 1 (a) Schematic illustration of the rationale of the proposed Yb@Er UCNP architecture. (b) Temporal behavior of the 543 nm UC of Yb@Er, YbEr, and Er@Yb UCNPs (λexc = 980 nm) in cyclohexane. (c) Quenching effect of the increasing content of H2O on the green Er3+ emission at 543 nm upon 980 nm excitation. The violet line represents the predicted decrease in UC intensity originating from the excitation intensity attenuation due to H2O absorption at 980 nm. (d) Comparison of the UC spectra of Yb@Er, YbEr, and Er@Yb UCNPs with similar physical dimensions (see the ESI†) and lattice structures which contain all the same amounts of Yb3+ and Er3+ ions; the spectra were acquired for nanoparticles dispersed in cyclohexane. | ||
Emission decay profiles at 543 nm of these samples were measured under excitation at 980 nm, as shown in Fig. 1b. Since decay profiles measured under upconversion excitation conditions often involve contribution from the excited state lifetime of directly excited Yb3+ ions to a different extent depending on the measurement conditions,4,39 two implications can be obtained from Fig. 1b. First, the comparison of the decay profiles of the Yb@Er and Er@Yb samples indicates that the Yb3+ ions in the Yb@Er structure have longer lifetimes than the Yb3+ ions in the Er@Yb structure; otherwise, the Yb@Er sample would exhibit a shorter decay time than the Er@Yb sample, given that Er3+ ions in the former structure should have shorter intrinsic lifetimes due to direct exposure to the environment.24 Second, the apparently longer decay time of the Yb@Er sample, exceeding that of the YbEr sample under 980 nm excitation indicates that the Yb3+ ions in the Yb@Er structure also have longer lifetimes than in the YbEr structure, respectively, since the Er3+ ions in these two structures should have similar intrinsic lifetimes due to their similar exposure to the environment. These results suggest that our Yb@Er structure has considerable potential to inhibit undesired Yb3+ de-activation.
In order to investigate the interactions of these UCNPs with different quenching environments, we recorded their UC spectra in D2O/H2O solutions, utilizing that high energy O–H vibrations can strongly interact with excited Yb3+ ions and quench the UC by efficiently coupling to the 2F5/2 energy level of Yb3+ and the 4S3/2/2H11/2 and 4I11/2 energy levels of Er3+, respectively, thereby increasing the nonradiative decay rates of these levels.24,32,37 The oleic acid (OA) capping of UCNPs was initially removed using an acid-wash method from a previously reported protocol with some modifications,40 after which the UCNPs were redispersed in D2O/H2O solutions with varying content of H2O. As shown in Fig. 1c, with increasing H2O amount, a significant decrease of the UCL intensity of the green Er3+ band was observed for YbEr and Er@Yb UCNPs, especially for Er@Yb UCNPs, disclosing that this sensitizer-rich-shell structure (Er@Yb)41 is prone to quenching by H2O molecules and hence not favorable for applications in the aqueous environment. The UCL of Yb@Er UCNPs, however, was much less affected (Fig. 1c). For instance, the retained emitting ability of Yb@Er nanoparticles in the H2O–D2O mixture (relative to their original emission in D2O) with a H2O volume ratio of 30% and 70% was 29.6% and 35.0%, respectively, stronger than that of YbEr nanoparticles under the same conditions. This result is in accordance with the previous studies that the H2O-induced quenching was mainly attributed to the interaction of the O–H vibrations with excited Yb3+ ions.24,37 It should be noted here that water absorbs 980 nm light and thus attenuates the excitation light intensity reaching the UCNPs resulting in a decrease in UCL.42 Therefore, the predicted UCL intensity evolution solely due to attenuation of the excitation intensity by absorption of the increasing water content is also given in Fig. 1c. Here, the absorption effect of H2O molecules is evaluated by the Beer–Lambert law,43
| Iex = I0e−γαL, | (1) |
| If ∝ Iex2 = I02e−2γαL. | (2) |
Thus, the attenuation factor e−2γαL is plotted in Fig. 1c to estimate the absorption effect of H2O. The absorption of D2O at 980 nm was neglected in this calculation. This reveals that the Yb@Er particle architecture makes UCNPs significantly less susceptible to environmental quenching effects than conventional homogeneously doped (YbEr) and sensitizer-rich-shell (Er@Yb) systems due to better protection of excited Yb3+ ions.
One drawback of the proposed structure compared to the homogeneously doped Yb3+–Er3+ UCNP structure may be the increased Yb3+⋯Er3+ interionic distance, which could lead to less efficient Yb3+ → Er3+ interionic energy transfer and thus weaker UCL. Surprisingly, the Yb@Er nanoparticles have a well-matched UC performance, much better than UCNPs with a sensitizer-rich shell (Fig. 1d), indicating the pivotal role of the suppression of the de-excitation of Yb3+ ions. Although the pre-optimized Yb@Er nanoparticles showed a weaker UCL intensity compared to the homogeneously doped YbEr nanoparticles due to the increased average Yb3+⋯Er3+ distance, there is much room to maximize their upconversion output by optimizing the doping concentrations of the sensitizer and activator ions and incorporating bridging ions in the shell, which will be presented in the following.
The effect of confining Yb3+ ions in the core region makes the high Yb3+-doping strategy applicable to facilitate UCL without sensitizer concentration quenching. A series of NaY(Gd)F4:x% Yb@NaYF4:2% Er (x = 20, 30, 50, 70) UCNPs were synthesized and their UCL spectra under 980 nm excitation were recorded. The Gd3+ ions, which reportedly do not affect UC energy transfer pathways,44 were incorporated into the host matrix to provide better control of the nanoparticle size (see the ESI, Fig. S3†). As shown in Fig. 2a, when increasing the Yb3+ concentration from 20% to 70%, the UCL intensity of the Yb@Er UCNPs increased steadily. In stark contrast, the effect of a high Yb3+ concentration can easily become adverse for Er@Yb UCNPs, with the highest UCL resulting for a Yb3+ concentration of 10%, while a further increase in Yb3+ concentration led to severe UCL quenching (Fig. 2b and ESI, Fig. S2†). It was reported that high Yb3+ doping also results in UCL quenching in conventional homogeneously doped UCNPs.45 This is ascribed to back energy transfer from the excited activator to sensitizer ions, energy hopping in the Yb3+ sublattice, and eventual excitation-energy transfer from the UCNP core to the particle surface, favoring nonradiative de-excitation.45 A high concentration of Yb3+ ions in the core boosts harvesting of excitation energy and subsequent fast Yb3+–Yb3+ energy migration,46 providing the basis for accumulating a high concentration of excited Yb3+ ions in a spatially confined region. This well-protected excitation-energy reservoir turned out to be very beneficial for pumping UCL. As shown in Fig. 2a, the UCL intensity was remarkably enhanced when a high concentration of Yb3+ ions was incorporated into the particle core. In order to provide a deeper insight into the enhancement mechanism, the excitation power dependencies of the green UCL of NaY(Gd)F4:20% Yb@NaYF4:2% Er and NaY(Gd)F4:70% Yb@NaYF4:2% Er UCNPs were investigated. In general, the number of photons (n) required to populate an emissive energy level can be obtained from the dependence of the UCL intensity (IUC) on the excitation power density (Iex) under unsaturated excitation conditions, i.e., IUC ∝ Iexn.47 With increasing Iex, the onset of saturation leads to a gradually decreasing apparent n value until n eventually reaches 1.48 As shown in Fig. 2c, the n values of the green emission in both samples are close to ∼2.0, indicating a two-photon UC mechanism. NaY(Gd)F4:70% Yb@NaYF4:2% Er UCNPs exhibit a noticeably smaller slope value (∼1.6) than NaY(Gd)F4:20% Yb@NaYF4:2% Er UCNPs (∼1.8) in the same excitation power density range, indicating a higher effective Iex for the former, caused by the higher Yb3+-doping level.49 In addition, the constant slopes which significantly exceed 1 up to an Iex of 300 W cm−2 point to highly unsaturated systems, the emission intensity of which can be further boosted by increasing Iex.48 The confined high content of Yb3+ was found to be especially in favor of multi-photon promoting UCL (Fig. 2a and d). As shown in Fig. 2d, three/four-photon-promoted blue/violet UCL bands at 410/380 nm (4G11/2/2H9/2 → 4I15/2) and a five-photon UCL band at 316 nm (2P3/2 → 4I15/2) of NaY(Gd)F4:70% Yb@NaYF4:2% Er UCNPs are 12, 25, and 36 times higher than those of NaY(Gd)F4:20% Yb@NaYF4:2% Er UCNPs, while the green emission bands are merely enhanced by a factor of 5. This result is in line with the findings reported in previous studies, where multi-photon UCL was drastically promoted by confining Yb3+ ions in the arrays of clusters at the sublattice level.50
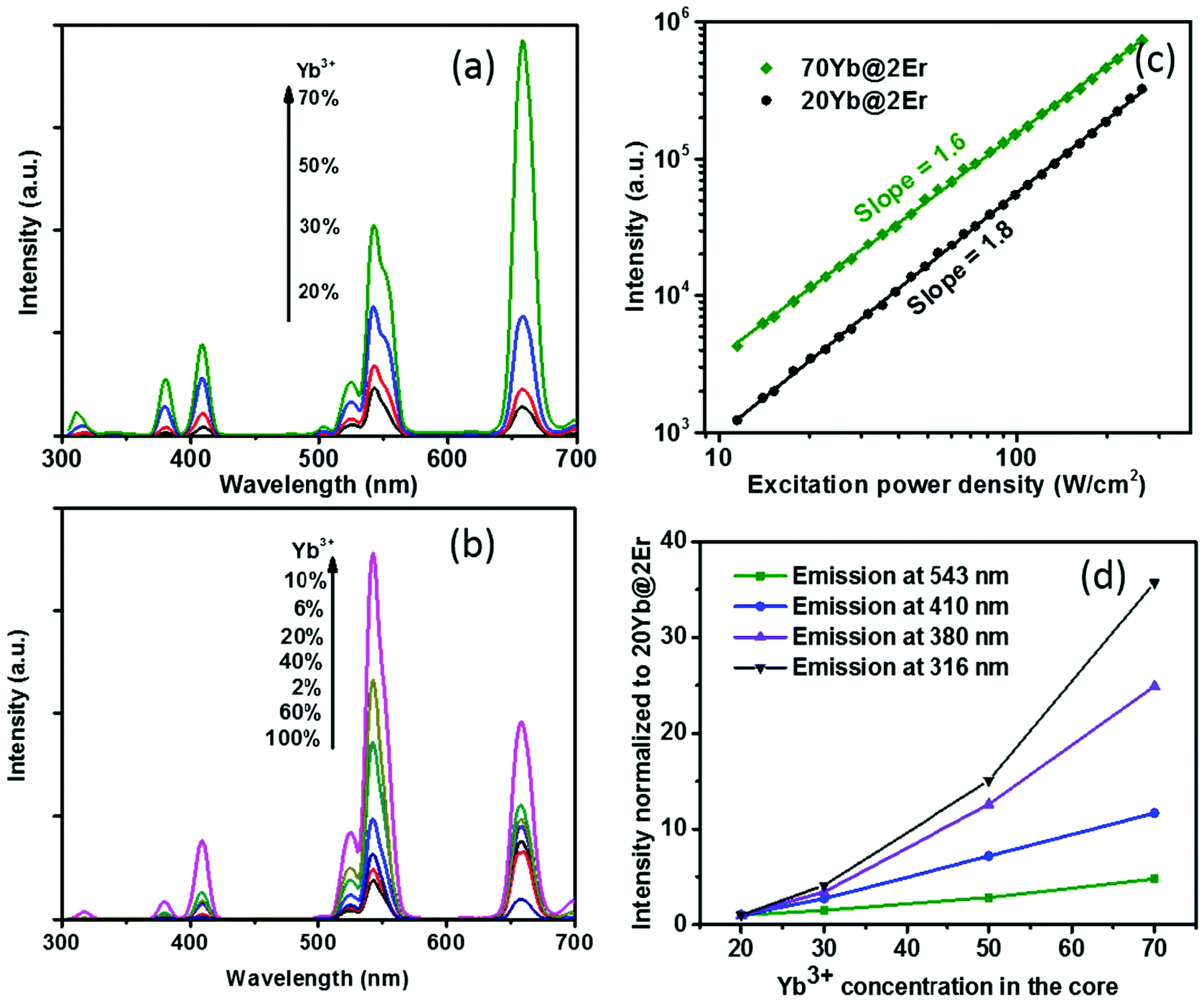 | ||
| Fig. 2 (a) Influence of the concentration of Yb3+ ions confined in the core on the UC spectra of Yb@Er UCNPs. (b) Influence of the concentration of Yb3+ ions confined in the shell on the UC spectra of Er@Yb UCNPs. The spectra were always normalized to the total amount of rare earth ions in the sample. (c) Logarithmic plot of the dependence of the intensity of the green UCL band of NaY(Gd)F4:20% Yb@NaYF4:2% Er and NaY(Gd)F4:70% Yb@NaYF4:2% Er UCNPs on the excitation power density Iex. (d) Influence of the concentration of Yb3+ ions confined in the core on the UC intensities of Yb@Er UCNPs (excitation intensity: 114 W cm−2, at 980 nm). The spectra were acquired in cyclohexane. | ||
The proposed Yb@Er UCNP architecture can also alleviate concentration quenching for higher activator concentrations. It was previously reported that the optimal dopant concentration of Er3+ ions is ∼2 mol% for Yb3+/Er3+ homogeneously co-doped NaYF4 UCNPs, with dopant concentrations exceeding this optimal concentration threshold resulting in a diminution of UCL intensity.31 With the Yb@Er particle architecture, we could realize significantly higher Er3+ doping levels with steadily enhanced UCL as revealed by spectroscopic studies of a series of NaY(Gd)F4:70% Yb@NaYF4:x% Er (x = 2, 6, 10, 14) UCNPs. As follows from a comparison of the UCL spectra of this series shown in Fig. 3a and in Fig. S4 in the ESI,† the green emission increased with increasing Er3+ concentration from 2% to 6% and reached a plateau for Er3+ concentrations exceeding 6%. The blue/violet UCL band was steadily enhanced even for an Er3+ concentration as high as 14%. Aside from suppressing Er3+ → Yb3+ back energy transfer and Yb3+ → environment energy dissipation, the high concentration of excited Yb3+ ions in the core region can also contribute to the reduction of activator concentration quenching, provided that a high excitation power density can alleviate activator concentration quenching in UCL.51
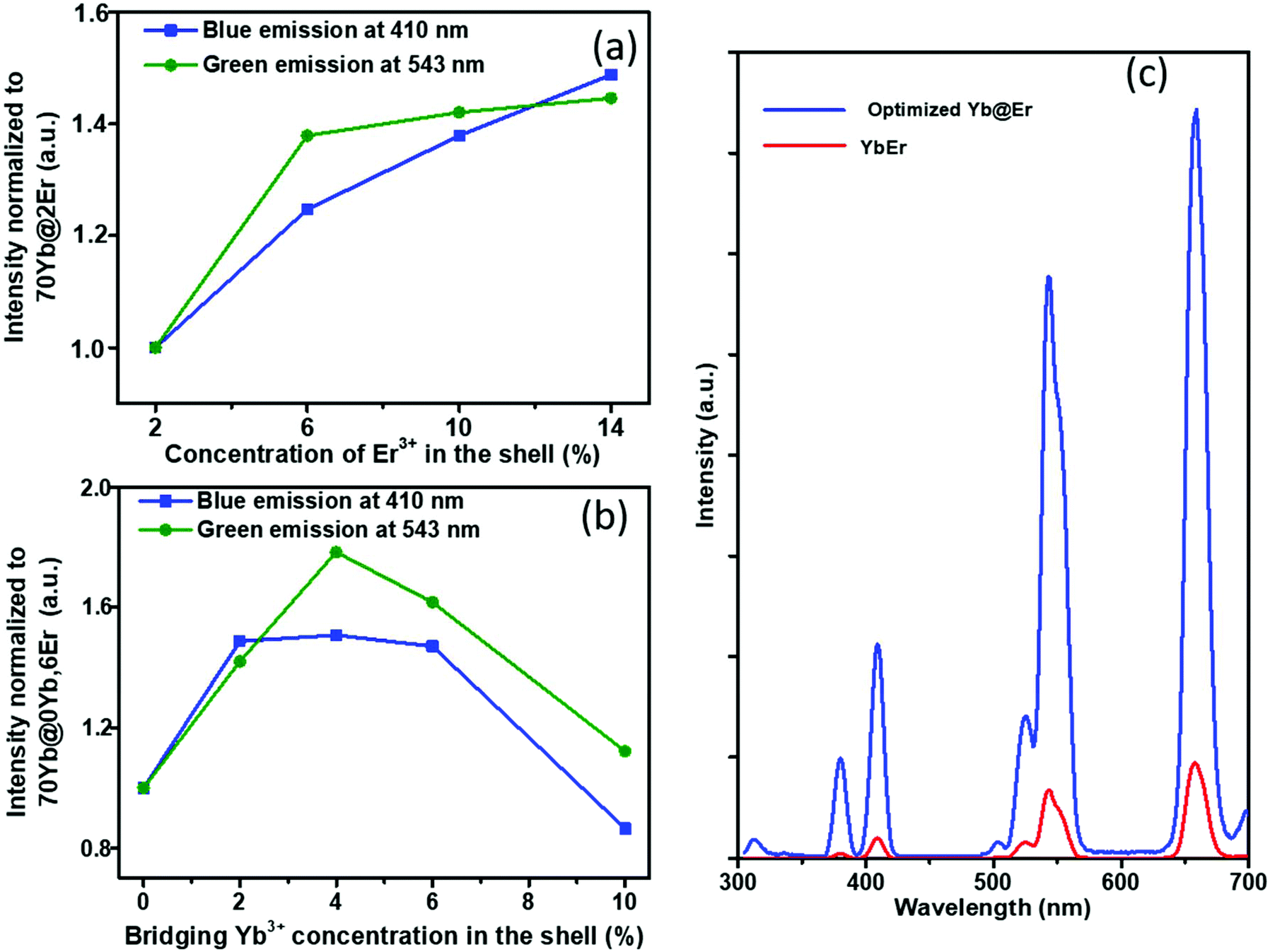 | ||
| Fig. 3 (a) Influence of Er3+ concentration in the shell on the UC intensities of the green emission band (543 nm) and blue emission band (410 nm) of Yb@Er UCNPs. (b) Influence of bridging Yb3+ ions in the shell on the intensities of the green (543 nm) and blue emission bands (410 nm) of Yb@(Yb)Er UCNPs. (c) Comparison of the UCL spectra of the optimized Yb@(Yb)Er (Yb3+ 70% in the core, and Yb3+ 4% and Er3+ 6% in the shell) and YbEr (homogeneously doped with Yb3+ 20% and Er3+ 2%) UCNPs (excitation intensity: 114 W cm−2, at 980 nm). The spectra were normalized to the total amount of rare earth ions and were acquired in cyclohexane. | ||
Along with the merit of inhibiting the excited state quenching pathway of the Er3+ ions, the spatial separation of Yb3+ and Er3+ ions nevertheless increases inevitably the Yb3+⋯Er3+ interionic distances, leading to less efficient UC pumping by Yb3+ → Er3+ energy transfer (ETU). In our proposed UCNP structure, it is very likely that only the Er3+ ions near the core–shell interface can be efficiently excited directly by ETU from the excited Yb3+ ions confined in the core. More distant Er3+ ions are then excited through Er3+–Er3+ interactions, which have a lower directional UC efficiency due to the complex energy level structure of Er3+ ions. In order to increase the overall pump efficiency of the Er3+ ions, small amounts of bridging Yb3+ (0–10%) ions were added, homogeneously co-doped with the Er3+ ions in the outer shell. Comparative studies of UCNPs with different concentrations of bridging Yb3+ ions show that the intensity of the green Er3+ emission increases first with increasing Yb3+ concentration and reaches a maximum at a Yb3+ ion concentration of 4%. A further increase in Yb3+ ion concentration leads to a decrease of the green Er3+ emission (Fig. 3b and ESI, Fig. S5†). This confirms the positive effect of a small number of bridging Yb3+ ions co-doped in the shell and reflects at the same time again the deleterious effect of a high concentration of Yb3+ ions in the immediate neighborhood of Er3+ ions due to creating an excitation-energy dissipation pathway to the environment via the Yb3+ network. Eventually, by adopting the novel core–shell architecture with a sensitizer-rich core derived by us and optimizing the activator- and bridging-ion concentrations, we could achieve an overall enhancement of the green Er3+ emission by a factor of ∼8.5 (Fig. 3c).
In UC bionanotechnology with UCNPs as energy relay materials, the efficiency of (nonradiative) resonant energy transfer from UCNPs to acceptors like singlet oxygen producing dyes or stimuli-responsive fluorophores plays a critical role.52 The rate of energy transfer depends strongly on the distance between the donors (UCNPs) and acceptors (usually organic molecules bound to or adsorbed onto the UCNP surface).14,15 In our proposed Yb@Er UCNP architecture where all emitting activator ions are located in the shell region, donor–acceptor distances can be minimized compared to other UCNP structures, which is expected to boost luminescence resonant energy transfer (LRET) processes. In order to validate the advantage of our nanoparticle architecture for LRET, a previously established LRET model employing the photodynamic therapy photosensitizer RB as an acceptor was used.14 The optimized Yb@Er (70% Yb@4% Yb,6% Er) nanoparticles with 70% Yb doped in the core and 6% Er and 4% Yb doped in the shell were employed in the LRET study. The conventional homogeneously doped YbEr (20% Yb, 2% Er) nanoparticles were used as the reference. The mechanism of LRET from UCNPs to RB photosensitizers is shown in Fig. 4a. The green emitting states (2H11/2/4S3/2) of Er3+ ions are first populated by sequential ETU processes, initiated by the absorption of 980 nm light of the sensitizer Yb3+, and RB molecules are subsequently excited nonradiatively by the green Er3+ emission,14 which overlaps with the RB absorption band (520–570 nm) (Fig. 4b). In order to realize such a LRET structure, RB molecules were incorporated into a silica shell (5 nm in thickness) deposited on the surface of our UCNPs (UCNP@SiO2 nanospheres) using a previously reported protocol with modifications.53 The dye molecules were first grafted onto (3-aminopropyl) triethoxysilane (APTES) to improve their stability. Then, the dye-modified APTES was co-hydrolyzed with tetraethyl orthosilicate (TEOS) to form a silica surface coating, using the reverse microemulsion method. The morphologies of RB-doped Yb@Er@SiO2 and YbEr@SiO2 nanospheres are shown in Fig. 4c and d. Their photoluminescence (PL) spectra upon 980 nm excitation are given in Fig. 4e, together with those of the corresponding control samples obtained by photobleaching RB molecules using mild UV light irradiation of 0.8 W cm−2 for 2 hours. All samples contained very similar amounts of RB molecules, as revealed by absorption spectroscopy (Fig. S6a†) as well as PL studies using a 25 mW 532 nm continuous-wave laser (Fig. S6b†) to directly excite the dye molecules. As follows from Fig. 4e, the characteristic UCL peaks of Er3+ ions at 524 nm and 542 nm were reduced for the RB-modified samples, compared to bleached RB-modified samples, and a new emission peak of RB at ∼583 nm appeared (an enlarged spectrum of the wavelength range of 560–630 nm is given in the ESI in Fig. S7†). This suggests LRET from UCNPs to RB molecules. In addition, the luminescence decay of the green 543 nm emission of the RB-modified UCNPs is shortened compared to the control samples (see the ESI, Fig. S8a†). This shortening of the donor emission decay time is a clear sign for LRET from UCNPs to RB molecules. In contrast, for example, simple mixing of RB solutions with undoped silica coated UCNPs did not alter the decay behavior of the green Er3+ emission (see the ESI, Fig. S8b†). It is worth mentioning that the red Er3+ emission band at 657 nm of both RB-coated test samples was decreased relative to the UV-bleached RB controls. Considering that the red emitting state (4F9/2) of Er3+ ions is partially populated from upper green emitting states (2H11/2/4S3/2) through nonradiative decay,54,55 this also provides evidence for the occurrence of nonradiative resonant energy transfer from UCNPs to RB molecules, otherwise (i.e., if RB is solely excited by absorbing the green UCL) the red UCL would not be altered.
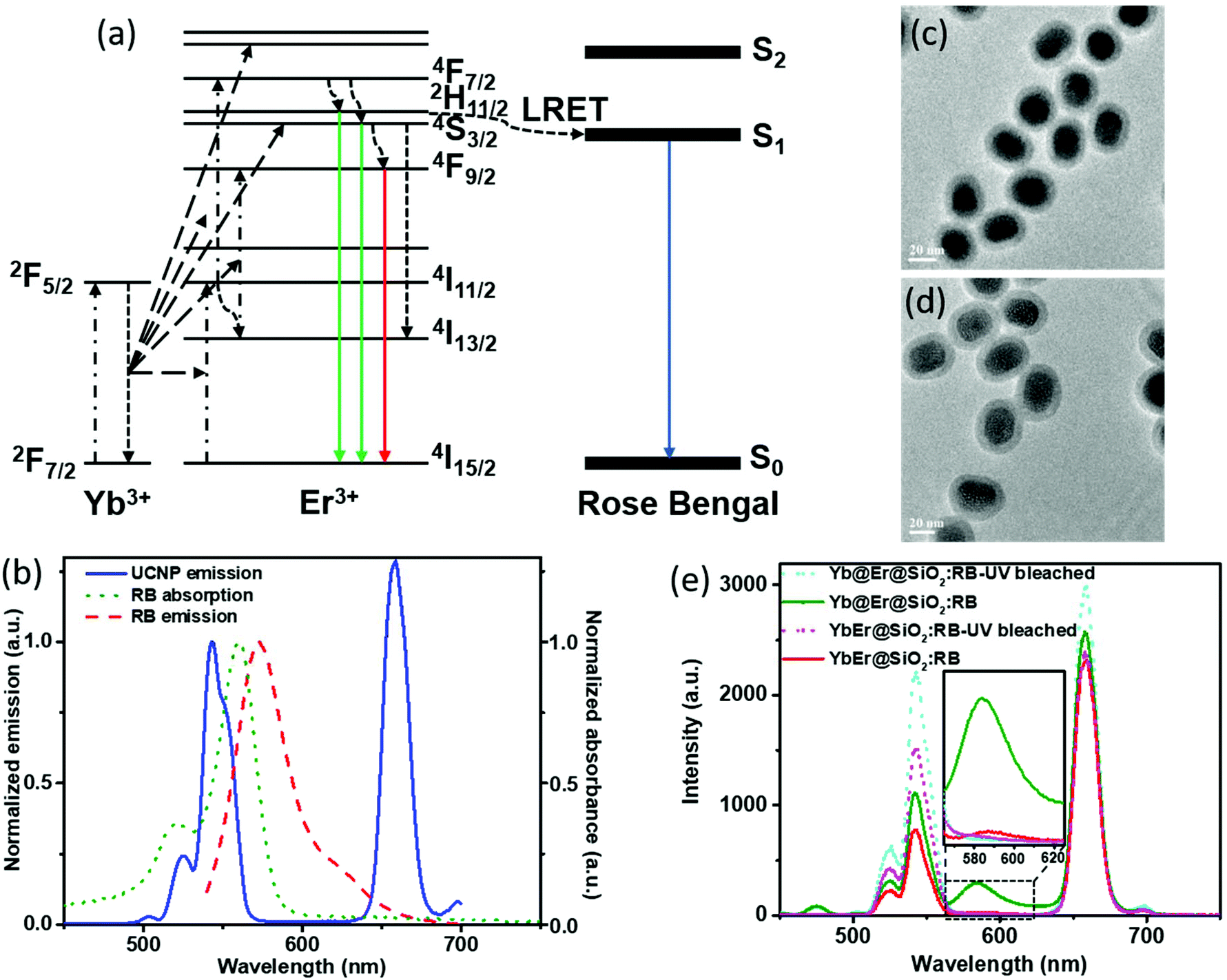 | ||
| Fig. 4 (a) Mechanism of UCL of Er3+ ions and energy transfer from UCNPs to RB in RB-doped UCNP@SiO2 nanospheres excited at 980 nm. (b) UCL spectrum of NaYF4:Yb,Er UCNPs and the absorption and emission spectrum of RB (λexc = 532 nm). TEM images of RB-doped (c) 70% Yb@4% Yb,6% Er@SiO2 and (d) 20% Yb,2% Er@SiO2 nanospheres. (e) Photoluminescence spectra of RB-coated UCNP@SiO2 nanospheres with unbleached RB and UV-bleached RB excited at 980 nm. | ||
In order to evaluate the overall UCNP → RB LRET efficiency, the figures-of-merit (FOMs) for both RB-modified UCNP samples, defined as the ratio of the integral of the RB emission (SRB), i.e., the emission of the acceptor and the decrease of the green and red Er3+ emission bands of the UCNPs (ΔSgreen + ΔSred), acting directly as an LRET donor or being indirectly linked to this energy transfer process, were calculated, see eqn (3):
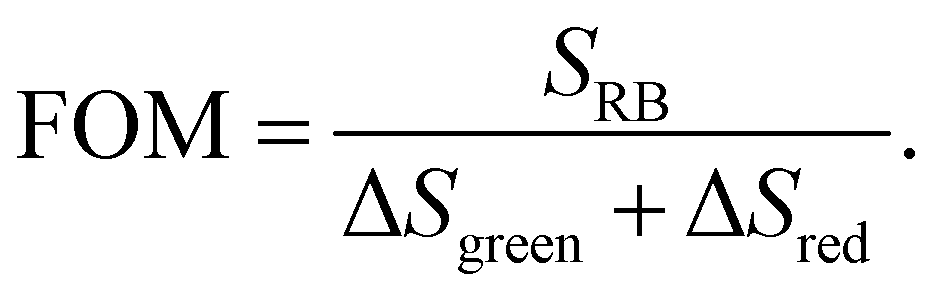 | (3) |
ΔSgreen and ΔSred were derived from the integral intensities of the green and red Er3+ emission bands without and with RB surface modification. The FOMs for RB-coated Yb@YbEr@SiO2 and YbEr@SiO2 nanospheres were determined to be 38.2% and 6.7%, respectively, yielding an about 6 times more efficient energy transfer from UCNPs to surface-bound acceptor dye molecules by our proposed Yb@YbEr@SiO2 particle architecture. Omitting the intensity change of the red Er3+ emission band in the calculation yielded FOMs of 47.6% and 7.0% for the RB-coated Yb@YbEr@SiO2 and YbEr@SiO2 nanospheres, respectively. These differences are related to the coupling of the energy levels. Here, contributions from the nonradiative resonant energy transfer from nanoparticles to RB molecules and the absorption of the donor emission by the acceptors (reabsorption or inner filter effect) cannot be distinguished.
Conclusion
We have proposed an efficient UCNP architecture using high concentrations of the sensitizer and activator ions Yb3+ and Er3+ confined in the core and shell region, respectively. Since the sensitizer-rich core is shielded from surface quenching effects, the Yb3+ can act as an excitation-energy reservoir to pump the activator ions and generate a significantly enhanced upconversion luminescence (UCL). Additionally, the emitting Er3+ centers located in the shell region enable very efficient luminescence resonance energy transfer (LRET) to organic molecules bound to the surface of the UCNPs due to the minimum distance between LRET donors and acceptors. Our work provides new insights into the rational design of UCNPs and will facilitate the application of UCNPs in various areas in life and materials sciences, especially for all applications where UCNPs are employed as energy relay materials for surface-bound molecules like singlet oxygen producing dyes or stimuli-responsive fluorophores.Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
The financial support from the Singapore National Medical Research Council (NMRC, grant number CBRG13nov052, R-397-000-199-511) and Ministry of Education (MOE) AcRF Tier 3 Programme Grant (grant number MOE2016-T3-1-004) is acknowledged. H. L. gratefully acknowledges an International Postdoc grant (2015-00160) and a Starting Grant (2016-03804) provided by the Swedish Research Council (Vetenskapsrådet). M. K. gratefully acknowledges the financial support from the Ph.D. program of BAM and U.R. from the German Research Council (DFG; grant RE 1203/20-1). M. K. and U. R. would like to acknowledge the COST Action CM1403 funded by the European Union.References
- F. Auzel, Chem. Rev., 2004, 104, 139–173 CrossRef CAS PubMed.
- C. Wang, L. Cheng and Z. Liu, Biomaterials, 2011, 32, 1110–1120 CrossRef CAS PubMed.
- Q. Zhan, H. Liu, B. Wang, Q. Wu, R. Pu, C. Zhou, B. Huang, X. Peng, H. Ågren and S. He, Nat. Commun., 2017, 8, 1058 CrossRef PubMed.
- H. Liu, M. K. G. Jayakumar, K. Huang, Z. Wang, X. Zheng, H. Agren and Y. Zhang, Nanoscale, 2017, 9, 1676–1686 RSC.
- T. Rantanen, M.-L. Jarvenpaa, J. Vuojola, R. Arppe, K. Kuningas and T. Soukka, Analyst, 2009, 134, 1713–1716 RSC.
- K.-C. Liu, Z.-Y. Zhang, C.-X. Shan, Z.-Q. Feng, J.-S. Li, C.-L. Song, Y.-N. Bao, X.-H. Qi and B. Dong, Light: Sci. Appl., 2016, 5, e16136–e16136 CrossRef CAS.
- N. M. Idris, M. K. Gnanasammandhan, J. Zhang, P. C. Ho, R. Mahendran and Y. Zhang, Nat. Med., 2012, 18, 1580–1585 CrossRef CAS PubMed.
- M. K. G. Jayakumar, A. Bansal, K. Huang, R. Yao, B. N. Li and Y. Zhang, ACS Nano, 2014, 8, 4848–4858 CrossRef CAS PubMed.
- M. K. G. Jayakumar, N. M. Idris and Y. Zhang, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 8483–8488 CrossRef CAS PubMed.
- D. Yang, P. A. Ma, Z. Hou, Z. Cheng, C. Li and J. Lin, Chem. Soc. Rev., 2014, 1416–1448 Search PubMed.
- C. T. Xu, J. Axelsson and S. Andersson-Engels, Appl. Phys. Lett., 2009, 94, 251107 CrossRef.
- C. T. Xu, P. Svenmarker, H. Liu, X. Wu, M. E. Messing, L. R. Wallenberg and S. Andersson-Engels, ACS Nano, 2012, 6, 4788–4795 CrossRef CAS PubMed.
- H. Liu, C. T. Xu, G. Dumlupinar, O. B. Jensen, P. E. Andersen and S. Andersson-Engels, Nanoscale, 2013, 5, 10034–10040 RSC.
- Y. Wang, K. Liu, X. Liu, K. Dohnalová, T. Gregorkiewicz, X. Kong, M. C. G. Aalders, W. J. Buma and H. Zhang, J. Phys. Chem. Lett., 2011, 2, 2083–2088 CrossRef CAS.
- V. Muhr, C. Würth, M. Kraft, M. Buchner, A. J. Baeumner, U. Resch-Genger and T. Hirsch, Anal. Chem., 2017, 89, 4868–4874 CrossRef CAS PubMed.
- H. Wen, H. Zhu, X. Chen, T. F. Hung, B. Wang, G. Zhu, S. F. Yu and F. Wang, Angew. Chem., 2013, 125, 13661–13665 CrossRef.
- G. Chen, H. Ågren, T. Y. Ohulchanskyy and P. N. Prasad, Chem. Soc. Rev., 2015, 44, 1680–1713 RSC.
- J. A. Damasco, G. Chen, W. Shao, H. Ågren, H. Huang, W. Song, J. F. Lovell and P. N. Prasad, ACS Appl. Mater. Interfaces, 2014, 6, 13384–13893 Search PubMed.
- C. Yuan, G. Chen, L. Li, J. A. Damasco, Z. Ning, H. Xing, T. Zhang, L. Sun, H. Zeng, A. N. Cartwright, P. N. Prasad and H. Ågren, ACS Appl. Mater. Interfaces, 2014, 6, 18018–18025 CAS.
- W. Shao, G. Chen, T. Y. Ohulchanskyy, A. Kuzmin, J. Damasco, H. Qiu, C. Yang, H. Ågren and P. N. Prasad, Adv. Opt. Mater., 2015, 3, 575–582 CrossRef CAS.
- G. Chen, J. Damasco, H. Qiu, W. Shao, T. Y. Ohulchanskyy, R. R. Valiev, X. Wu, G. Han, Y. Wang, C. Yang, H. Ågren and P. N. Prasad, Nano Lett., 2015, 15, 7400–7407 CrossRef CAS PubMed.
- J. Zhao, Z. Lu, Y. Yin, C. McRae, J. A. Piper, J. M. Dawes, D. Jin and E. M. Goldys, Nanoscale, 2013, 5, 944–952 RSC.
- F. Wang, J. Wang and X. Liu, Angew. Chem., Int. Ed., 2010, 49, 7456–7460 CrossRef CAS PubMed.
- C. Wurth, M. Kaiser, S. Wilhelm, B. Grauel, T. Hirsch and U. Resch-Genger, Nanoscale, 2017, 9, 4283–4294 RSC.
- S. Han, R. Deng, X. Xie and X. Liu, Angew. Chem., Int. Ed., 2014, 53, 11702–11715 CrossRef CAS PubMed.
- G. Chen, H. Liu, H. Liang, G. Somesfalean and Z. Zhang, J. Phys. Chem. C, 2008, 112, 12030–12036 CAS.
- J.-C. Boyer and F. C. J. M. van Veggel, Nanoscale, 2010, 2, 1417–1419 RSC.
- S. Heer, K. Kömpe, H. U. Güdel and M. Haase, Adv. Mater., 2004, 16, 2102–2105 CrossRef CAS.
- E. M. Chan, C. Xu, A. W. Mao, G. Han, J. S. Owen, B. E. Cohen and D. J. Milliron, Nano Lett., 2010, 10, 1874–1885 CrossRef CAS PubMed.
- J. He, W. Zheng, F. Ligmajer, C.-F. Chan, Z. Bao, K.-L. Wong, X. Chen, J. Hao, J. Dai, S.-F. Yu and D. Y. Lei, Light: Sci. Appl., 2017, 6, e16217–e16217 CrossRef CAS.
- X. Liu, X. Kong, Y. Zhang, L. Tu, Y. Wang, Q. Zeng, C. Li, Z. Shi and H. Zhang, Chem. Commun., 2011, 47, 11957–11959 RSC.
- M. Wang, Y. Tian, F. Zhao, R. Li, W. You, Z. Fang, X. Chen, W. Huang and Q. Ju, J. Mater. Chem. C, 2017, 5, 1537–1543 RSC.
- M. Kaiser, C. Wurth, M. Kraft, I. Hyppanen, T. Soukka and U. Resch-Genger, Nanoscale, 2017, 9, 10051–10058 RSC.
- H. S. Qian and Y. Zhang, Langmuir, 2008, 24, 12123–12125 CrossRef CAS PubMed.
- N. Bogdan, F. Vetrone, G. A. Ozin and J. A. Capobianco, Nano Lett., 2011, 11, 835 CrossRef CAS PubMed.
- Z. Q. Li, Y. Zhang and S. Jiang, Adv. Mater., 2008, 20, 4765 CrossRef CAS.
- R. Arppe, I. Hyppanen, N. Perala, R. Peltomaa, M. Kaiser, C. Wurth, S. Christ, U. Resch-Genger, M. Schaferling and T. Soukka, Nanoscale, 2015, 7, 11746–11757 RSC.
- Z. Li and Y. Zhang, Nanotechnology, 2008, 19, 345606 CrossRef PubMed.
- D. Gamelin and H. Gudel, in Transition Metal and Rare Earth Compounds, ed. H. Yersin, Springer, Berlin/Heidelberg, 2001, vol. 214, pp. 1–56 Search PubMed.
- N. Bogdan, F. Vetrone, G. A. Ozin and J. A. Capobianco, Nano Lett., 2011, 11, 835–840 CrossRef CAS PubMed.
- F. Vetrone, R. Naccache, V. Mahalingam, C. G. Morgan and J. A. Capobianco, Adv. Funct. Mater., 2009, 19, 2924–2929 CrossRef CAS.
- Q. Zhan, J. Qian, H. Liang, G. Somesfalean, D. Wang, S. He, Z. Zhang and S. Andersson-Engels, ACS Nano, 2011, 5, 3744–3757 CrossRef CAS PubMed.
- A. Beer, Ann. Phys., 1852, 162, 78–88 CrossRef.
- F. Wang, Y. Han, C. S. Lim, Y. Lu, J. Wang, J. Xu, H. Chen, C. Zhang, M. Hong and X. Liu, Nature, 2010, 463, 1061–1065 CrossRef CAS PubMed.
- T. Sun, R. Ma, X. Qiao, X. Fan and F. Wang, ChemPhysChem, 2016, 17, 766–770 CrossRef CAS PubMed.
- F. Wang, R. Deng, J. Wang, Q. Wang, Y. Han, H. Zhu, X. Chen and X. Liu, Nat. Mater., 2011, 10, 968–973 CrossRef CAS PubMed.
- M. Pollnau, D. R. Gamelin, S. R. Lüthi, H. U. Güdel and M. P. Hehlen, Phys. Rev. B: Condens. Matter Mater. Phys., 2000, 61, 3337–3346 CrossRef CAS.
- H. Liu, C. T. Xu, D. Lindgren, H. Xie, D. Thomas, C. Gundlach and S. Andersson-Engels, Nanoscale, 2013, 5, 4770–4775 RSC.
- J. F. Suyver, A. Aebischer, S. Garcia-Revilla, P. Gerner and H. U. Güdel, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 71, 125123 CrossRef.
- J. Wang, R. R. Deng, M. A. MacDonald, B. L. Chen, J. K. Yuan, F. Wang, D. Z. Chi, T. S. A. Hor, P. Zhang, G. K. Liu, Y. Han and X. Liu, Nat. Mater., 2014, 13, 157–162 CrossRef CAS PubMed.
- J. Zhao, D. Jin, E. P. Schartner, Y. Lu, Y. Liu, A. V. Zvyagin, L. Zhang, J. M. Dawes, P. Xi, J. A. Piper, E. M. Goldys and T. M. Monro, Nat. Nanotechnol., 2013, 8, 729–734 CrossRef CAS PubMed.
- Y. Ding, F. Wu, Y. Zhang, X. Liu, E. M. L. D. de Jong, T. Gregorkiewicz, X. Hong, Y. Liu, M. C. G. Aalders, W. J. Buma and H. Zhang, J. Phys. Chem. Lett., 2015, 6, 2518–2523 CrossRef CAS PubMed.
- Z. Li, Y. Zhang and S. Jiang, Adv. Mater., 2008, 20, 4765–4769 CrossRef CAS.
- G. Chen, G. Somesfalean, Y. Liu, Z. Zhang, Q. Sun and F. Wang, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 75, 195204 CrossRef.
- J. Zhang, Z. Hao, J. Li, X. Zhang, Y. Luo and G. Pan, Light: Sci. Appl., 2015, 4, e239–e239 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details, TEM images and XRD patterns of UCNPs, UCL spectra, absorption spectra and photoluminescence spectra of RB-loaded UCNPs, and temporal behavior of UC at 543 nm. See DOI: 10.1039/c7nr06900f |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2018 |