
Enhanced and synergistic downregulation of oncogenic miRNAs by self-assembled branched DNA†
S.
Nahar
ab,
A. K.
Nayak
c,
A.
Ghosh
ab,
U.
Subudhi
 *bc and
S.
Maiti
*abd
*bc and
S.
Maiti
*abd
aCSIR-Institute of Genomics & Integrative Biology, Mathura Road, Delhi 110025, India. E-mail: souvik@igib.res.in
bAcademy of Scientific & Innovative Research, Anusandhan Bhawan, New Delhi 110001, India
cDNA Nanotechnology & Application Laboratory, CSIR-Institute of Minerals & Materials Technology, , Bhubaneswar, 751013, India. E-mail: usubudhi@immt.res.in
dCSIR-National Chemical Laboratory, Dr. Homi Bhabha Road, Pune 411008, India
First published on 21st November 2017
Abstract
miRNAs, a group of small non-coding RNA molecules, regulate the expression of many genes involved in various cellular processes. Acute evidence suggests that one miRNA can regulate many genes as its targets, while one gene can be targeted by many miRNAs that co-operatively regulate the gene. Thus, targeting a single miRNA is not sufficient enough to rescue the disease phenotype but it is also essential to target multiple miRNAs simultaneously. This inspired us to design a novel DNA nanostructure that can concurrently downregulate multiple oncomiRNAs. Here we designed a programmable antimiR branched DNA (antimiR-bDNA) nanostructure having antimiRNAs for selective binding to oncomiRNAs miRNA-27a, 96 and 182 which collectively downregulate FOXO1a expression. The antimiR-bDNAs show enhanced stability compared to naked antimiRNAs in serum and are able to knockdown these miRNAs with up to ∼50% greater repression as compared to antimiRNAs. This synergistic miRNA repression leads to the restoration of FOXO1a protein levels which in turn inhibit G1-S traversion in cancer cells. To the best of our knowledge, this is the first study harnessing the ability of bDNA structures to silence multiple miRNAs simultaneously.
Introduction
DNA, the core molecule for storing genetic information can be modified to produce versatile self-assembled branched DNA (bDNA) nanostructures,1,2 DNA nanomachines,3,4 and assembled nanomaterials.5,6 Using the power of Watson–Crick complementarity rules, several DNA strands can be pre-assembled under controlled conditions to achieve predetermined 2- or 3-dimensional structures. Since the pioneering work of Seeman in the 1980s,7 numerous bDNA nanostructures with diverse geometry ranging from simple one dimensional to convoluted three dimensional configurations have been developed that offer great potential for materials science, nanotechnology and biology. In the recent past, such structures have been successfully used in applications including photonics, bioimaging, sensing, DNA-based materials, modular primers for polymerase chain reaction and biomedicine.8Gene expression can be regulated at many levels. Several thousand genes, estimated to be nearly one-third of the human genome, are potentially regulated by an abundant class of small (∼19–24 nucleotides) non-coding RNAs called miRNAs. Evolutionarily conserved, miRNAs represent a new layer of the gene regulatory network and primarily function as post-transcriptional repressors of complementary mRNA.9,10 miRNAs are expressed as long hairpin forming primary transcripts which get processed to mature miRNAs by the sequential action of Drosha and Dicer enzymes. miRNAs serve as guides for the RNA induced silencing complex (RISC) to get recruited to the 3′UTR of cognate mRNA, resulting in either degradation or translation inhibition,11,12 thereby affecting various cellular outcomes including differentiation, cell growth, proliferation and apoptosis.13 The aberrant expression pattern and function of miRNAs are associated with pathogenesis of many diseases, including cancer.14 Depending on the target genes, deregulated miRNAs act as tumor suppressors or oncomiRs15 and represent therapeutically important modalities for their manipulation and rescuing cancer phenotypes. This is highlighted by the increasing number of recent studies on developing either miRNA replacement or inhibition by antisense strategies or small molecule modifiers.16,17
One of the most rational approaches to target oncomiRs is via anti-miRNA oligonucleotide (AMO) technology. The administration of synthetic AMOs which are antisense to the endogenous miRNA, results in sequence specific inhibition of miRNA function. Over the past decade, AMOs have been subjected to various chemical modifications to enhance the efficacy of miRNA interference.18–21 The inhibition of single miRNA using AMO can result in efficient knockdown of the miRNA. However, robustness to any biological phenomena governed by miRNAs is also conferred by the synergistic and cooperative action of multiple miRNAs on a single transcript. For example, in the case of breast cancer, multiple oncomiRs like miR-27a, miR-96 and miR-182 act in concert to target and reduce FOXO1a tumor suppressor gene expression.22 FOXO1a is a transcription factor that regulates multiple genes involved in cell cycle progression, apoptosis and cellular metabolism. The reduced levels of FOXO1a contribute to malignant transformation and an oncogenic state.23 Thus, it must be stressed that targeting individual miRNAs may not be adequate in restoring FOXO1a expression. A previous report had employed a multiple targeting AMO unit which can repress two or three miRNAs together.24 However, the efficiency and stability of the linear AMO unit was not optimal. This provoked us to devise a novel, yet simple strategy of utilizing the bDNA nanostructure as a tool to silence multiple miRNAs simultaneously. In the present study we utilized four single stranded oligonucleotides which increased the stability and efficiency of self-assembly leading to a 100% yield.25 We remodelled the bDNA monomeric structure to integrate anti-miRNA sequences against oncomiRNAs – miRNA 27a, 96 and 182 (Fig. 1). These antimiR sequences were incorporated into the overhangs, enabling them to bind specifically to the cellular miRNAs.
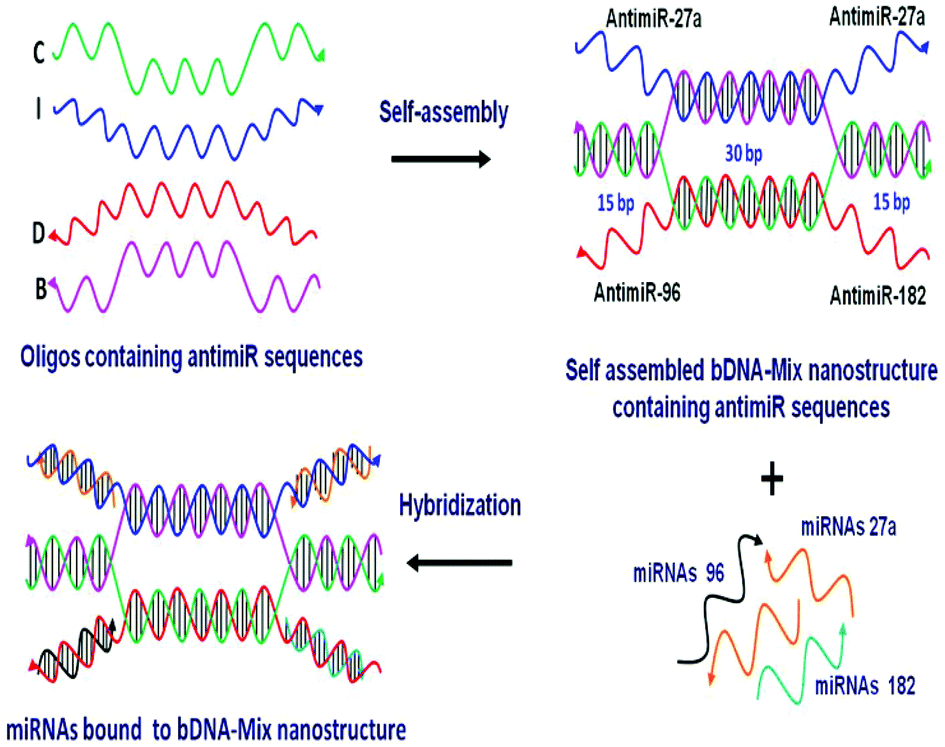 | ||
| Fig. 1 Illustration of the designed DNA nanostructure from oligonucleotides I, B, C and D having three different antimiR (antimiR 27a, 96 and 182) sequences at the four overhang regions and its hybridisation with target miRNAs. | ||
Methods
Designing of bDNA structures with antimiR sequences
The designing of oligonucleotides for the synthesis of the branched DNA scaffold has been elaborated in our previous report.25 However, bDNAs targeting specific miRNAs have been designed by replacing the four overhangs with the complementary sequence of target miRNA. The internal hybridizing sequences derived from catalase, SOD 1 and SOD 2 genes of Rattus norvegicus were kept intact without any modification. In this present study 11 distinctively designed oligonucleotides (oligo B, C, I-scramble, I-antimiR-27a, I-antimiR-96, I-antimiR-182, D-scramble, D-antimiR-27a, D-antimiR-96, D-antimiR-182, D-antimiR-96 and 182) have been used in an appropriate composition and stoichiometric ratio to form four different anti-miRNA containing bDNA structures (bDNA-27a, bDNA-96, bDNA-182 and bDNA-Mix). In the case of the bDNA scaffold, the overhangs have been replaced with scramble sequences (16 nts long), which have no sequence complementarity with any miRNA present in the cellular environment. The bDNA-27a structure has been designed by replacing all four overhangs with the antimiR-27a sequence to target miRNA-27a. Similarly, bDNA-96 and 182 structures were designed by replacing all four overhangs with antimiR-96 and antimiR-182 sequences, respectively. The bDNA-Mix contains all three antimiR sequences with a double dose of antimiR-27a because of its abundance in the cellular system and a single dose of antimiR-96 and 182. To compare the efficacy of the bDNA structures, single stranded antimiR-27a, 96 and 182 were used as control samples with suitable scramble sequences. Oligonucleotides were purchased from Integrated DNA Technology, USA and used without further modification and purification (Table S1†).Self-assembly study
Self-assembly among oligonucleotides was carried out in TAE/Mg2+ buffer which consists of Tris-base (40 mM, pH 8.0), acetic acid (20 mM), EDTA (2 mM) and magnesium acetate (12.5 mM). In a 25 μl assembly reaction mixture, contributing oligos for specific bDNA were added in an equimolar ratio (25 pmol each), denatured at 95 °C and then cooled to 4 °C with a ramp rate of 0.3 °C s−1 using a thermal cycler (Bio-Rad). The self-assembled bDNA structures are then directly used for characterization and application in the cell culture system without further purification.Native polyacrylamide gel electrophoresis
10% polyacrylamide gel (acrylamide![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :bis acrylamide, 29:1) is prepared by adding 9.422 ml of MilliQ water, 450 μl 50× TAE buffer, 7.5 ml acrylamide solution, 2.815 ml of magnesium acetate (0.1 M), 300 μl APS (10%) and 15 μl TEMED to make the gel volume of 22.5 ml which is sufficient for making two gels (10 cm × 10 cm). The gels were run at 4 °C for 2.5 h in a SE260 vertical electrophoresis unit (Hoefer) at a constant voltage of 150 V by taking 1× TAE as running buffer. After electrophoresis the gels were stained with ethidium bromide solution (0.5 μg ml−1) for 45 min and then documented in the FluroChem E system (Cell Biosciences).
:bis acrylamide, 29:1) is prepared by adding 9.422 ml of MilliQ water, 450 μl 50× TAE buffer, 7.5 ml acrylamide solution, 2.815 ml of magnesium acetate (0.1 M), 300 μl APS (10%) and 15 μl TEMED to make the gel volume of 22.5 ml which is sufficient for making two gels (10 cm × 10 cm). The gels were run at 4 °C for 2.5 h in a SE260 vertical electrophoresis unit (Hoefer) at a constant voltage of 150 V by taking 1× TAE as running buffer. After electrophoresis the gels were stained with ethidium bromide solution (0.5 μg ml−1) for 45 min and then documented in the FluroChem E system (Cell Biosciences).
Circular dichroism spectroscopy
To study conformational stability, the circular dichroism (CD) spectra of self-assembled bDNA structures containing antimiRs were analyzed using a Chirascan spectrophotometer (Applied Photophysics). The spectra were recorded with a scan speed of 60 nm s−1, a bandwidth of 1 nm and a time per point of 0.5 s. All measurements were done at 25 °C between the wavelength of 320 nm and 200 nm by using a quartz cuvette of 1 mm path length. Three spectra per sample were averaged for obtaining the final spectrum.Gel-retardation assay
Sequence specific binding between miRNA sequences and antimiR regions of bDNA structures was evaluated using gel retardation assay. Single stranded miRNA-27a, 96 and 182 were used to study hybridization with corresponding anti-miRNA containing bDNA in vitro. The scaffold structure and bDNA-Mix were incubated with all three miRNA sequences whereas other bDNA structures were incubated with their respective miRNA sequences at 37 °C for 1 h. Then the samples were run in 1.5% agarose gel at 100 volts for 30 min.Serum stability study
The stability study of different bDNA structures containing antimiR sequences in the presence of serum was conducted in vitro by taking 1 μM of bDNA in 10% Fetal Bovine Serum (FBS, Gibco) as purchased from GIBCO, USA. After mixing, the samples were incubated at 37 °C for 0, 2, 4, 12, 24, 36 and 48 h. The incubated products were then analyzed by 1.5% agarose gel electrophoresis. In addition to this, serum stability assay for free antimiR oligos was also performed by taking 4 μM of three anti-miRNAs in different aliquots and then supplementing them with FBS at a final concentration of 10%. The samples were then incubated at 37 °C and run in 2% agarose gel at 100 volts for 20 min. After electrophoresis the gel images were obtained using the FluorChem E documentation system. The intensity of DNA bands in agarose gel was analyzed using ImageJ software and the band intensity of each band corresponded to its stability in serum.Cell viability assay
MCF-7 cells were seeded on a flat transparent 96 well plate (Corning) at a density of 8000 cells per well. After 24 h they were transfected with the bDNA samples and the respective anti-miRs. 24 h after transfection of cells, MTT (dimethyl thiazolyl diphenyl tetrazolium bromide) (Sigma-Aldrich) at a concentration of 0.5 mg ml−1 was added to each well. The cells with MTT were incubated for 4 h at 37 °C. Post incubation, violet formazan crystals could be seen at the bottom of each transparent well. MTT-containing medium was removed and 150 μl DMSO (Sigma) was added to each well to dissolve the crystals. The absorbance of the plate was recorded using a TECAN plate reader at 570 nm wavelength. The percentage of cell viability was then calculated.Quantitative uptake of bDNA
A fluorescently labeled bDNA-Mix structure was formed by incorporating an Alexa-Fluor 488 modified strand I and stepwise cooling as described above.25 MCF-7 cells were seeded in a 24 well plate and Alexa-488 labelled bDNA and its linear oligo strand were transfected in a concentration range of 12.5, 25 and 100 nM in serum free media. The MCF-7 cells were purchased from European Collection of Authenticated Cell Cultures (ECACC) and were maintained as described in the Methods section. Cells were then incubated at 37 °C in a humidified incubator (5% CO2) for 8 h. The transfected cells were washed twice with ice-cold PBS containing 1 mg ml−1 heparin, followed by one wash with trypan blue to quench the extracellular fluorescence. Following two washes with 1× PBS, the cells were harvested by trypsinization. The cells were collected by centrifugation at 700g for 5 min at 4 °C, the supernatant was removed, and the pellet was resuspended in ice-cold PBS and FACS analysis was performed using a BD Accuri instrument. 10000 counts per sample was analyzed using BD Accuri C6 software.
Quantitative reverse transcription-PCR (qRT-PCR)
Total RNA was extracted from the cells using the TRIzol reagent (Invitrogen) following the manufacturer's instructions. The relative expression level of miR-27a, miR-96, and miR-182 was determined by quantitative real-time RT-PCR using the QuantimiR kit (SBI, catalog number RA660A-1) following the manufacturer's instructions. The specific primer pairs were as follows: miR-27a, 5′-TTCACAGTGGCTAAGTTCCGC-3′, miR-96 5′-TTTGGCACTAGCACATTTTTGCT-3′, miR-182 5′-TTTGGCAATGGTAGAACTCACACT-3′ and U6, 5′-CGCAAGGATGACACGCAAATTC-3′. The relative expression of these three miRNAs was quantified using the 2−ΔΔCt method and plotted using GraphPad Prism 5.0 software (GraphPad Software, San Diego, CA, USA).Western blot
Protein lysates were prepared from the transfected MCF-7 cells in 12 well plates after 48 h. The cells were trypsinized and pelleted down in microcentrifuge tubes. Cell pellets were washed with 1× PBS and centrifuged. To each cell pellet 50 μl of RIPA buffer (Pierce™ Thermo Scientific) was added along with 1× Protease inhibitor cocktail (Roche). Buffer treated cells were then incubated in ice for 30 min (with frequent vortexing after every 5–10 min). After incubation, each tube was centrifuged at 12000 rpm for 10 min and the supernatant of protein lysate was collected in fresh tubes. Protein estimation was done by using the Pierce™ BCA Protein Assay Kit (ThermoFisher). According to BCA estimation, 30 μg of protein from each sample was taken for sample preparation and heated to 95 °C for 5 min along with 1× SDS dye. Protein was run in 10% SDS-PAGE gel and was transferred to a 0.45 μm pore size PVDF Membrane (EMD Millipore). Wet transfer was performed at 80 V for 2.5 h. After performing the transfer, the membranes were kept for blocking in 5% BSA dissolved in 1× TBST at room temperature on a rocker for three hours. After blocking, antibodies FOXO1a (abcam) and Alpha-Tubulin (CST) were added to the respective blots at 1:1000 dilution dissolved in 5% BSA and the blots were incubated overnight at 4 °C on a rocker. Primary antibody binding was followed by three consecutive washing steps of 15 min each with 1× TBST on an orbital shaker at 170 rpm. After washing, blots were incubated with secondary antibodies (rabbit origin) conjugated with HRP at 1:10000 dilution dissolved in 1× TBST for 3 h at room temperature. Secondary antibody binding was followed by three consecutive washes with 1× TBST for 15 min each on an orbital shaker at 170 rpm. The blots were then further processed for developing on a Syngene Gel Doc instrument using an EMD Millipore™ Immobilon Western Chemiluminescent HRP Substrate (ECL).
Cell cycle analysis
MCF-7 cells were seeded in a 12-well plate (1 × 105 cells per well) and transfected with bDNA-Mix and bDNA scaffold at 25 nM, antimiR mixture of 27a:96:182 (2:1:1) and scramble control at 100 nM concentration. Upon 48 h incubation, the cells were trypsinized, centrifuged and washed twice with 1× PBS and fixed overnight in 70% ethanol at −20 °C. The pellet was resuspended in 200 μl of 4 mM sodium citrate buffer containing 0.1% Triton X-100 and RNase A at a final concentration of 100 μg ml−1 and incubated for 2 h at 37 °C. Following incubation, the cells were stained with propidium iodide (30 μg ml−1) and incubated at room temperature in the dark for 15–20 min. Cell cycle analysis was carried out using a BD Accuri C6 flow cytometer. The acquired data were processed using the FCS express software and BD CellQuest™ Pro software (Becton Dickinson, Mountain View, CA).
Results and discussion
Design and characterization of miRNA targeting self-assembled bDNA
A bDNA monomer was constructed from four single stranded oligonucleotides, strand-I (blue), strand-B (pink), strand-C (green) and strand-D (red). The sequences were designed in such a way that strands I and D comprised of overhangs (∼22 mer) complementary to miRNAs. On the other hand, the end regions of strands B and C are complementary to each other to enable hybridization. The central core region (30 nt) of I strand hybridized with strand-B and similarly strand-C hybridized with internal nucleotides of strand-D. To target miR-27a, bDNA-27a was constructed having 4 overhangs of antimiR-27a at the 5′ and 3′ ends of strands I and D (I-antimiR-27a and D-antimiR-27a). Each monomer contains overhangs with antimiR sequences which bind to the respective miRNAs (Table 1 and Table S1†).| bDNA structures | Composition of oligos | Targeting to |
|---|---|---|
| Scaffold | I-scramble, B, C, D-scramble | Control: not targeting |
| bDNA-27a | I-antimiR-27a, B, C, D-antimiR-27a | miRNA-27a |
| bDNA-96 | I-antimiR-96, B, C, D-antimiR-96 | miRNA-96 |
| bDNA-182 | I-antimiR-182, B, C, D-antimiR-182 | miRNA-182 |
| bDNA-Mix | I-antimiR-27a, B, C, D-antimiR-96, and 182 | miRNA-27a, 96, and 182 |
Hence, three different antimiR–bDNA structures were generated namely, bDNA-27a, bDNA-96 and bDNA-182 which can target three miRNAs: miR-27a, miR-96 and miR-182, respectively. The bDNA scaffold that contains scramble sequences in its overhangs was used as a negative control in all experiments for negating the effects of the bDNA structure itself. bDNA-27a contains 4 overhangs of antimiR-27a, similarly bDNA-96 and bDNA-182 possess 4 overhangs of antimiR-96 and antimiR-182, respectively. Apart from creating these homogeneous bDNAs targeting one miRNA, we envisaged the ability of a single bDNA nanostructure to simultaneously inhibit four miRNAs having sites in FOXO1 3′ UTR.
However, it was shown by Guttilla et al.22 that miR-183 did not interact with its predicted site and was devoid of suppressive activity for FOXO1a in MCF-7 cells. Based on the expression profile of miR-27a, miR-96 and miR-182 (data not shown), we found miR-27a to be expressed higher than miR-182 and miR-96. Thus, we designed bDNA-Mix having two overhangs of antimiR-27a and one overhang each of antimiR-96 and 182 that can target all three miRNAs simultaneously.
A quick self-assembly process was followed for the generation of bDNA structures.26 The self-assembled products and their stability have been demonstrated through native polyacrylamide gel electrophoresis. To observe the differential migration of di, tri, and tetra-oligo complexes, oligos with equimolar concentrations (B, C, I-scramble, I-antimiR-27a, I-antimiR-96, I-antimiR-182, D-scramble, D-antimiR-27a, D-antimiR-96, D-antimiR-182 and D-antimiR-96, 182) were allowed to hybridize among each other in different combinations. Since the electrophoretic mobility of a nucleic acid oligomer or its assembly under non-denaturing conditions is a function of its size, shape, and the extent of base pairing, both the molecular mass and conformational based retarded mobility of DNA complexes have been observed in 10% native PAGE suggesting the formation of the intended structure (Fig. 2 and Fig. S1†).
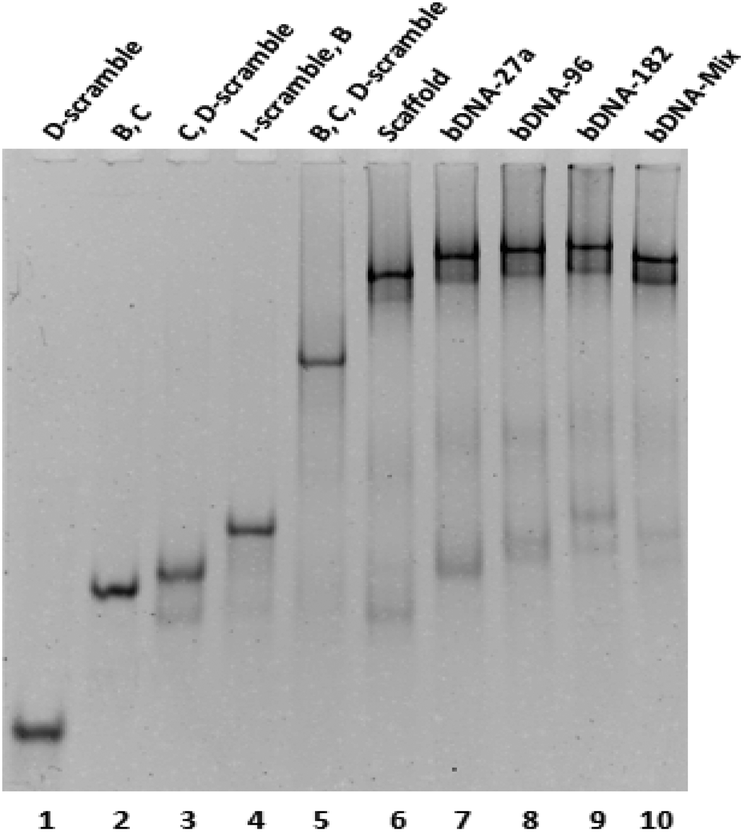 | ||
| Fig. 2 Characterization of self-assembled bDNA structures having antimiRNA sequences in overhangs through 10% native PAGE. The sample composition in each lane is mentioned on top of the lane. bDNA structures (lanes 6 to 10) showed decreased electrophoretic mobility with respect to their tri-oligo and di-oligo complexes. The presence of a single intense band in each lane showed precise base-pairing among designed oligos. | ||
Moreover, sharp and dominant band intensities in each lane confirmed the formation of a unimolecular structure. Once bDNA structures were synthesized, circular dichroism (CD) spectroscopy was employed to examine the structural properties of the self-assembled bDNA structures. All the bDNA structures showed canonical B-conformation characterized by the positive peaks at ∼280 nm and ∼220 nm and the negative peak at ∼250 nm (Fig. S2†). Next, we characterized the binding of bDNA structures with the respective complementary miRNA sequences through gel shift assay (Fig. S3†). bDNAs with specific binding to complementary miRNA sequences showed retarded electrophoretic mobility compared to bDNA alone. Importantly, no migration was observed with scaffold bDNA containing scramble sequences incubated with each miRNA.
Enhanced stability conferred by bDNA to nucleolytic degradation
bDNA nanostructures should be stable and intact long enough in the cytoplasm of the cells to carry out their predefined functions of targeting miRNAs. Although a few reports have shown successful delivery of DNA nanostructures carrying siRNA, miRNA or peptide into the cells,27–32 the details about their intracellular stability of DNA nanostructures are poorly understood. We wanted to further elucidate the stability and integrity of bDNA structures in the presence of a strong nuclease environment as present in the cellular milieu. In a previous report, DNA origami based nanostructures underwent denaturation and degradation in cell culture medium containing 10% or more FBS, which contains greater than 256 U L−1 equivalents of DNAse I activity.33 Hence, we incubated bDNA nanostructures in 10% FBS over a period of 0–48 h. The stability of these structures was compared with single stranded antimiRs under the same serum conditions (Fig. 3, Fig. S4 and S5†). It is evident that bDNA structures are 60% stable even after 36 h incubation in cell culture media as compared to only 10–15% stability of single stranded antimiRNAs up to 24 h. Their superior structural stability is bestowed upon by the steric hindrance conferred by the bDNA structures that could reduce the binding and hence the activity of endonucleases.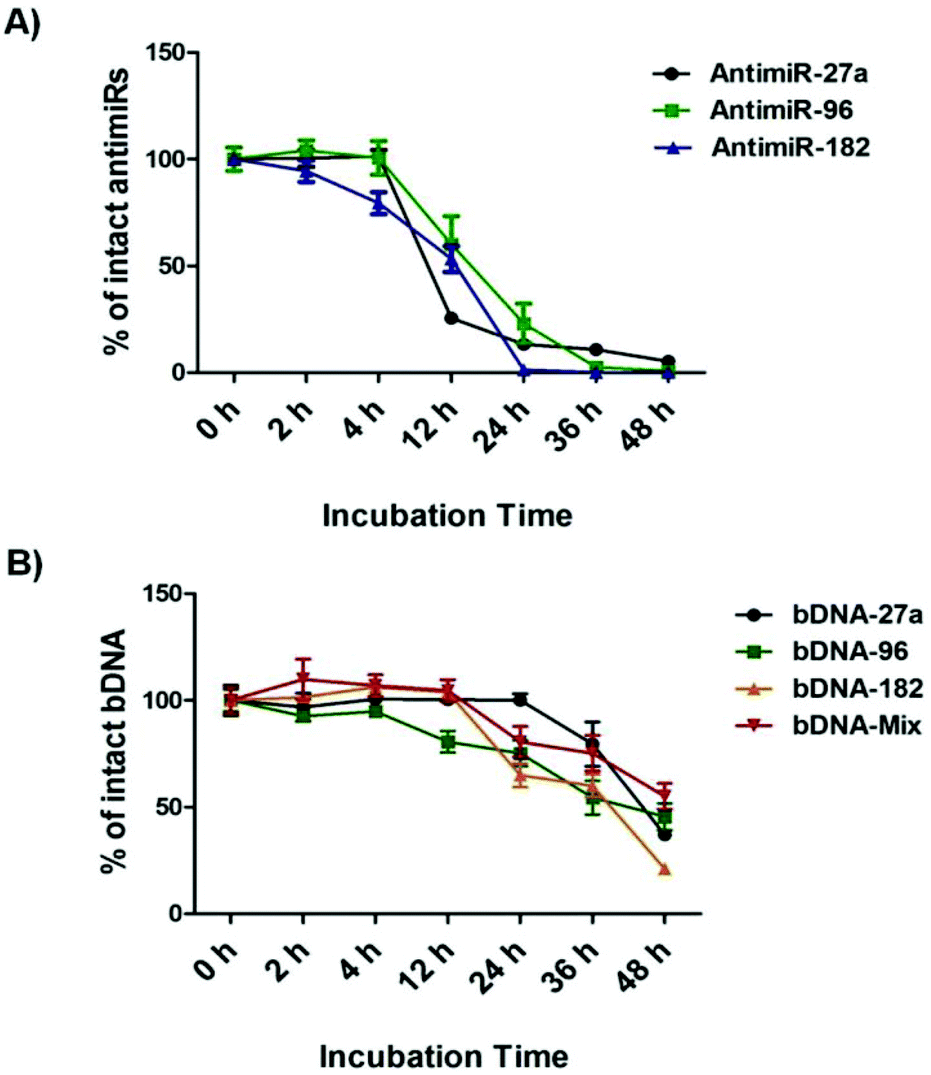 | ||
| Fig. 3 Stability study of antimiRNA and bDNA structures through agarose gel electrophoresis after incubation at 37 °C for 0 to 48 h in serum. (a) Serum stability of antimiR-27a, 96 and 182. (b) Serum stability of bDNA-27a, bDNA-96, bDNA-182 and bDNA-Mix structures. | ||
Investigating the effect of bDNA in targeting multiple miRNAs cooperatively
The remarkable stability of bDNA structures prompted us to investigate their utility in cultured cell lines. Firstly, we started to evaluate the cytotoxicity of these structures in the breast cancer cell line, MCF-7.We employed the MTT assay for assessing the metabolic activity of the cells upon transfection with bDNA. We observed that the bDNA structures did not induce measurable loss in cell viability up to 100 nM (Fig. S6†). Moreover, we compared this to the cytotoxic potential of the antimiRs and found minimal change in viability between both the treatments (Fig. S7†). Additionally, we measured the transfection efficiency of Alexa-fluor 488 tagged bDNA and linear single stranded oligonucleotide in a concentration dependent manner ranging from 12.5 nM to 100 nM (Fig. S8†). We observed a similar cellular uptake of bDNA as compared to its linear counterpart with the aid of a standard transfection agent lipofectamine 2000. In order to test the ability of bDNA structures to knockdown miRNA levels, we transfected scaffolds, bDNA-27a, bDNA-96 and bDNA-182 at 25 nM in the MCF-7 cell line for 48 h. Since each bDNA comprises of four overhangs of miRNA, we correspondingly treated cells with 100 nM of antimiR-27a, 96 and 182 individually and then the relative miRNA expression was studied (Fig. 4).
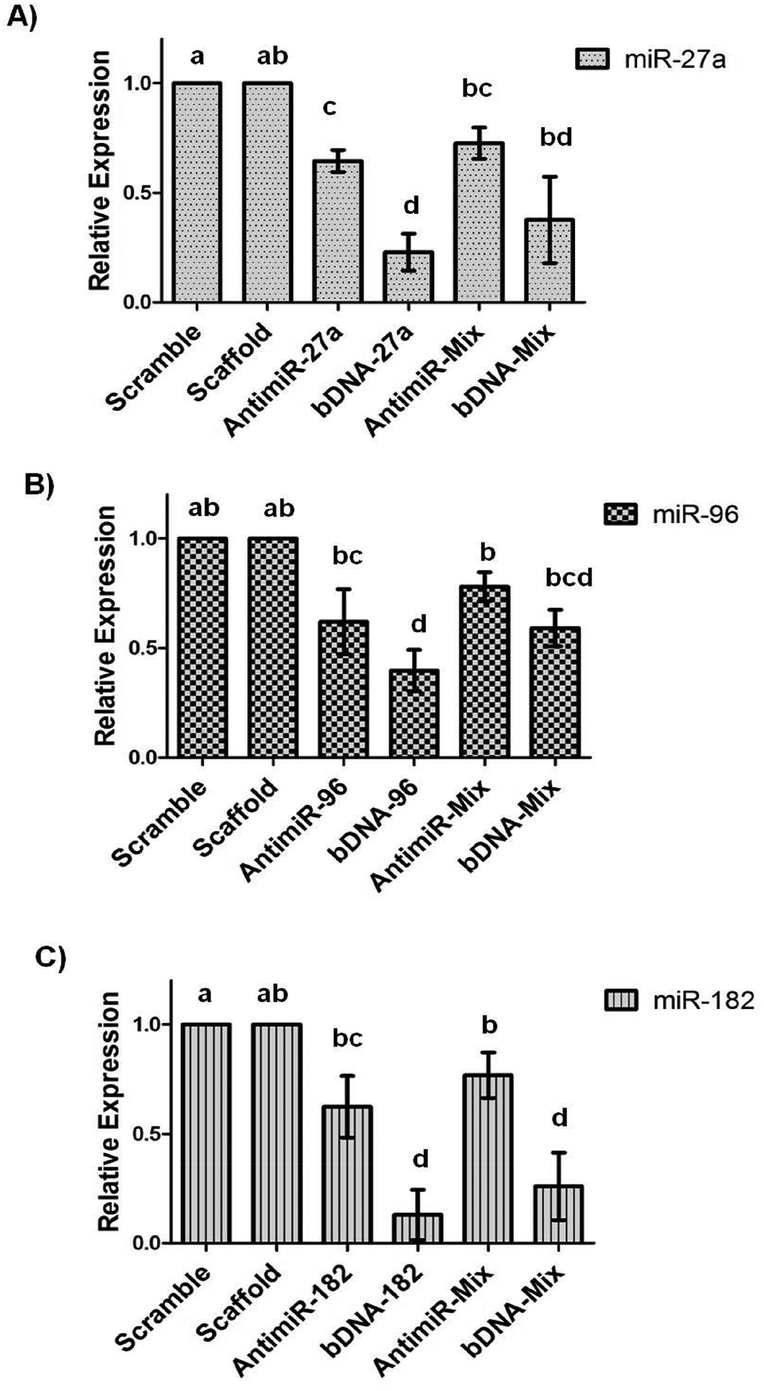 | ||
| Fig. 4 Quantitation of miRNA levels by RT-qPCR upon transfection with 100 nM of unmodified antimiRs or mixed antimiRs-27a, 96 and 182 (2:1:1) and 25 nM of bDNA-27a, 96, 182 or bDNA-Mix nanostructures. The effect of bDNAs was compared to scaffold and antimiRs were compared with scramble oligonucleotides. Further bDNAs were also compared with their respective antimiRs. miRNA expression was normalized by U6 RNA. Data shown here represent mean ± SEM, n = 3. The level of significance was determined by one-way ANOVA followed by Duncan's New multiple range test. Data having different superscripts (a–d) are significantly different at p < 0.05. | ||
We observed that compared to the bDNA-scaffold, bDNA-27a reduced miR-27a levels by ∼77% (p < 0.001) whereas the extent of reduction by stoichiometrically matched antimiR-27a was ∼35% (p < 0.005). Similarly, bDNA-96 aptly downregulated miR-96 to ∼60% (p < 0.001) and antimiR-96 could inhibit miR-96 only to ∼38% (p < 0.05). The transfection of bDNA-182 conferred a significant repression of 87% (p < 0.001) as compared to ∼38% by antimiR-182 (p < 0.01). The enhanced mitigation caused by antimiR-bDNA-27a (p < 0.005), bDNA-96 (p < 0.05) and bDNA-182 (p < 0.001) as compared to its respective linear single stranded antimiR counterparts highlights the significance of the higher stability of the bDNA structures, being available for inhibiting miRNAs with a long half-life. Since the bDNA structure encompasses four overhangs, we next sought to downregulate different oncogenic miRNAs by a single nanostructure unit. The relative levels of miR-27a are higher than miR-96 and miR-182 in the MCF-7 cell line, and thus we examined the silencing effect of bDNA-Mix as compared to co-transfection of a mixture of these three antimiRs (antimiR-mix) in 2:1:1 ratio (Fig. 4). We observed ∼35% and ∼20% improvement in the inhibition of miR-27a and miR-96 respectively by bDNA-Mix as compared to antimiR-Mix. A significantly greater repression was seen for miR-182 targeted by the bDNA-Mix (∼52%) as compared to antimiR-mix (p < 0.001).
However, a non-significant higher downregulation of individual miRNA by the respective bDNA was observed compared to bDNA-mix. The reason being 25 nM of individual bDNA selectively binds to the respective miRNA, whereas 25 nM of the bDNA-mix binds to all the three miRNAs. For example bDNA-27a contains four overhangs of antimiR-27a, whereas the bDNA-mix contains two overhangs of antimiR-27a and one overhang each of antimiR-96 and 182. When they are applied with equal concentration (25 nM) the bDNA-27a is two-fold more efficient in targeting miR-27a than the bDNA-mix, which results in better downregulation of miR-27a by bDNA-27a than bDNA-mix. Similarly bDNA-96 or 182 is four-fold more efficient in targeting miR-96 or 182 than bDNA-mix. Nevertheless, the bDNA-mix controls the three miRNAs (miR-27a, 96 and 182) simultaneously in one plate, hence synergistic regulation of all antimiRNAs over FOXO1a mRNA can be evident from the expression of FOXO1a. The strengthened ability of the bDNA nanostructure in reducing miRNAs can be attributed to the spatial orientation and enhanced intracellular stability as compared to naked single stranded antimiRs.
Synergistic downregulation of miRNAs by bDNA effectively upregulates endogenous FOXO1a protein levels and arrests cancerous cells at G1-S transition
We subsequently evaluated the effect of bDNA nanostructures on endogenous FOXO1a protein levels which harbor sites for all three miR-27a, miR-96 and miR-182. Henceforth, we checked its levels after the respective transfections with homogeneous bDNA nanostructures (bDNA-27a, bDNA-96 and bDNA-182) and heterogeneous bDNA-Mix (Fig. 5). A significant increase in the expression of FOXO1a was observed when treated with either antimiR-96 or bDNA-96 (p < 0.001). The FOXO1a protein levels increased up to 1.4-fold when treated with bDNA-96 as compared to antimiR-96. A similar effect was also seen in the protein level for the bDNA-182 as compared to antimiR-182. Most importantly the bDNA-Mix produced greater rescue effects than the co-transfected antimiR mix (∼2 fold) or individual bDNA nanostructures suggesting synergistic downregulation of these miRNAs. FOXO1a is a tumor suppressor gene, which plays a key role in inhibiting cell proliferation and disruption of its function leads to cellular transformation.23,34 The increment in FOXO1a levels prompted us to investigate its effect on different cell cycle stages upon transfection with bDNA nanostructures. Since the bDNA-Mix was designed to target all three miRNAs simultaneously, we transfected it at 25 nM and analyzed the cell cycle distribution by flow cytometry (Fig. 6).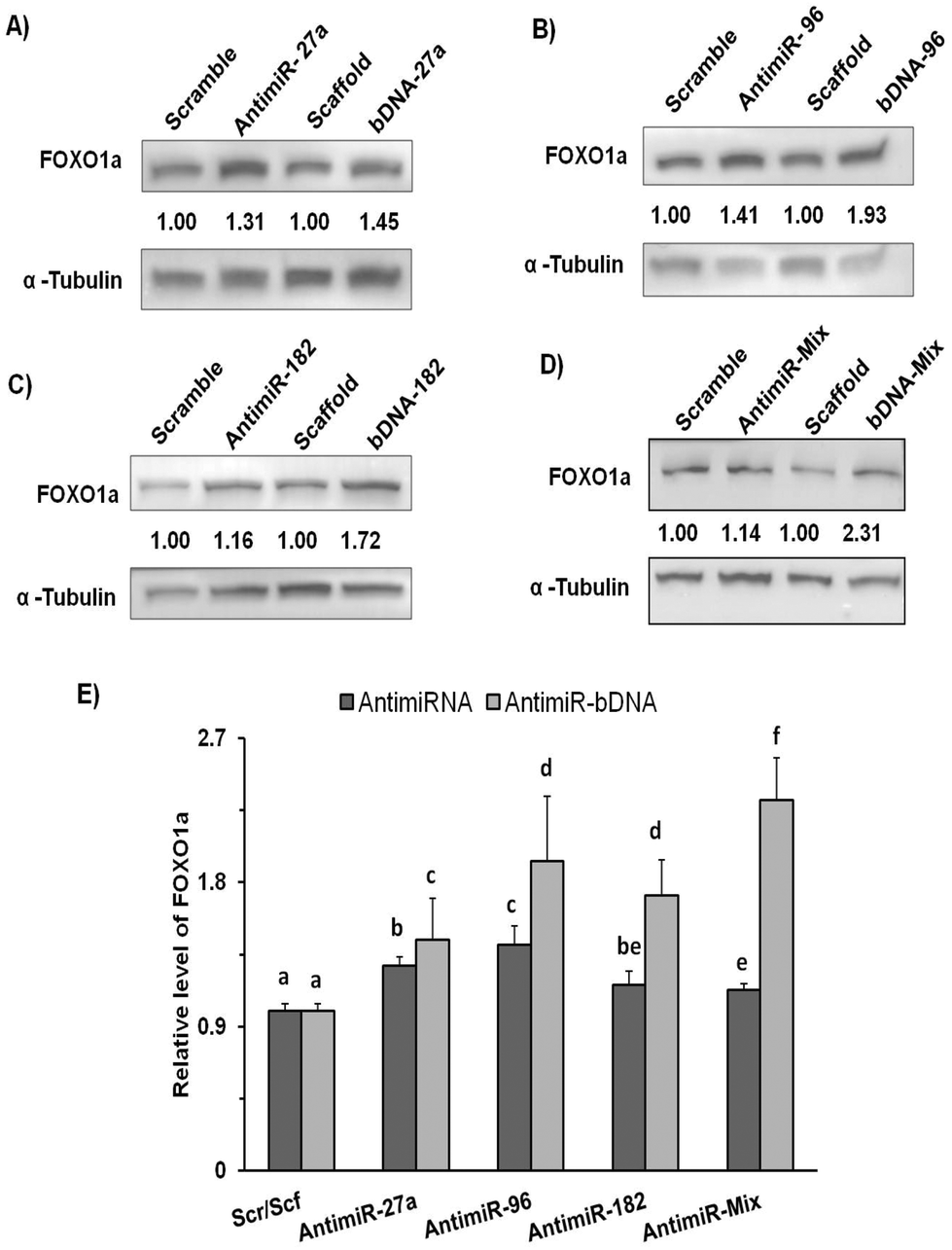 | ||
| Fig. 5 Measurement of endogenous FOXO1a levels by bDNA nanostructures and antimiRs targeting miR-27a, miR-96 and miR-182. MCF-7 cells were treated with 25 nM homogeneous bDNA-27a, bDNA-96 and bDNA-182 and compared to 100 nM of antimiR-27a (A), antimiR-96 (B) and antimiR-182 (C) respectively. Heterogeneous bDNA-Mix was compared to co-transfected antimiR-mix (D). Cells were harvested and lysed 48 h post transfection, and FOXO1a and α-tubulin levels (normalizing control) were measured by western blot. Numbers represent the mean fold change from 3 independent experiments. It was observed that the expression of FOXO1a was significantly elevated in MCF cells treated with antimiR-bDNA compared to their corresponding antimiRNAs (E). Data are expressed as mean ± SD of three independent experiments. Data having different superscripts (a–e) are significantly different at p < 0.05. | ||
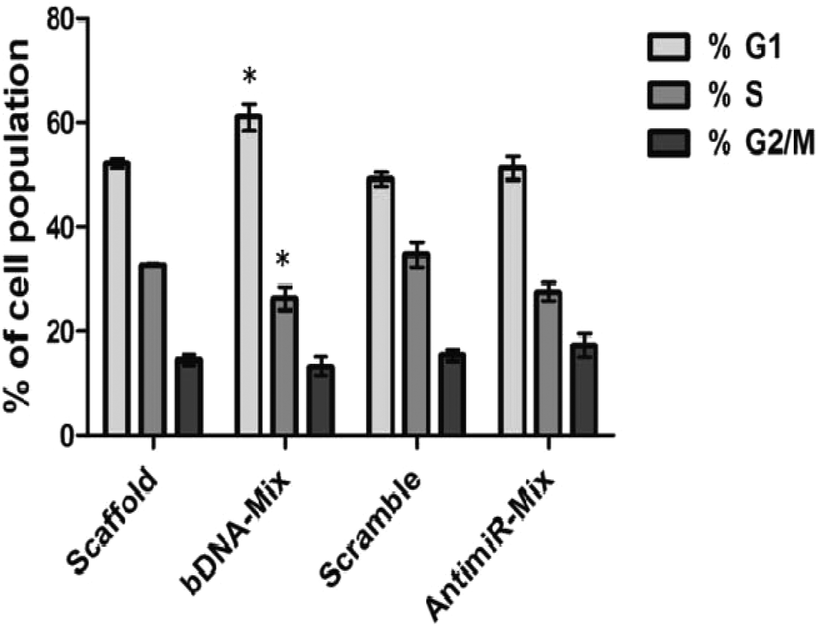 | ||
| Fig. 6 Cell cycle distribution detected by flow cytometry of MCF-7 cells upon transfection with mixed antimiRs-27a, 96 and 182 (2:1:1) (100 nM) and bDNA-Mix nanostructures (25 nM) after 48 h. Error bar represents the mean ± SEM, n = 3, *p < 0.05. | ||
With respect to the bDNA scaffold, bDNA-Mix caused significant cell cycle arrest at the G1 phase by ∼11% and decreased the number of cells in the S phase by ∼7%. However, co-transfection with different antimiRs did not lead to a significant difference in cell cycle population as compared to the scramble control.
Conclusions
In summary, we have developed a simple and low-cost strategy to prepare bDNA nanostructures that are non-toxic. DNA strands are easily programmable for molecular self-assembly and could be generically used for targeting miRNAs as well as mRNAs. This study demonstrated the use of four single stranded DNA fragments to self-assemble in a one-step reaction and self-organize into bDNA nanostructures having functional single stranded overhangs. The overhangs allowed specific binding to complementary oncomiRs miR-27a, miR-96 and miR-182 simultaneously. The four antimiR overhangs linked covalently in a single structural entity allow their co-delivery ensuring that the effected cells received all relevant miRNA antagonists at the same time in pre-decided stoichiometric ratios. The important factor of bDNA structure remains largely stable up to 48 h in biological fluids corresponding to higher availability as compared to naked antimiRs for performing the desired downstream processes. The enhanced nuclease resistance of the bDNA structure is reflected by the efficient down regulation of the target oncomiRs and thereby increment in their cognate target FOXO1a levels. The restoration of FOXO1a levels arrested the cell cycle progression, thus inhibiting cellular proliferation in cancerous cells.The precisely controlled expression of miRNAs ensures proper repression of cognate targets. Many miRNAs work in a combinatorial manner and act in concert to exert inhibition on a single transcript. Hence, inhibiting a single miRNA with conventional AMO may not result in the optimal interference of the target mRNA. The bDNA we designed acts as a single unit that has demonstrated its utility in targeting three different miRNAs synergistically. Moreover such nanostructured bDNAs have the potential to target practically many miRNAs. The ease of design and low cost of synthesis, combined with their high stability and low toxicity makes bDNA structures a superior tool compared to most of the conventional AMOs for silencing miRNAs. This multivalent approach has provided a new direction for miRNA targeting and is an opportunity to target cellular mRNAs.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Council of Scientific and Industrial Research (CSIR), Government of India (Project BSC-0123, EMPOWER Project OLP-19).Notes and references
- U. Feldkamp and C. M. Niemeyer, Angew. Chem., Int. Ed., 2006, 45, 1856–1876 CrossRef CAS PubMed.
- D. Han, S. Pal, Y. Yang, S. Jiang, J. Nangreave, Y. Liu and H. Yan, Science, 2013, 339, 1412–1415 CrossRef CAS PubMed.
- C. Mao, W. Sun, Z. Shen and N. C. Seeman, Nature, 1999, 397, 144–146 CrossRef CAS PubMed.
- B. Yurke, A. J. Turberfield Jr., A. P. Mills, F. C. Simmel and J. L. Neumann, Nature, 2000, 406, 605–608 CrossRef CAS PubMed.
- F. A. Aldaye, A. L. Palmer and H. F. Sleiman, Science, 2008, 321, 1795–1799 CrossRef CAS PubMed.
- R. J. Macfarlane, B. Lee, M. R. Jones, N. Harris, G. C. Schatz and C. A. Mirkin, Science, 2011, 334, 204–208 CrossRef CAS PubMed.
- N. C. Seeman, J. Theor. Biol., 1982, 99, 237–247 CrossRef CAS PubMed.
- L. A. Lanier and H. Bermudez, Curr. Opin. Chem. Eng., 2015, 7, 93–100 CrossRef PubMed.
- D. P. Bartel, Cell, 2004, 116, 281–297 CrossRef CAS PubMed.
- B. P. Lewis, C. B. Burge and D. P. Bartel, Cell, 2005, 120, 15–20 CrossRef CAS PubMed.
- J. Winter, S. Jung, S. Keller, R. I. Gregory and S. Diederichs, Nat. Cell Biol., 2009, 11, 228–234 CrossRef CAS PubMed.
- M. Ha and V. N. Kim, Nat. Rev. Mol. Cell Biol., 2014, 15, 509–524 CrossRef CAS PubMed.
- V. Ambros, Nature, 2004, 431, 350–355 CrossRef CAS PubMed.
- W. Wu, M. Sun, G. M. Zou and J. Chen, Int. J. Cancer, 2007, 120, 953–960 CrossRef CAS PubMed.
- A. Esquela-Kerscher and F. J. Slack, Nat. Rev. Cancer, 2006, 6, 259–269 CrossRef CAS PubMed.
- J. Stenvang, A. Petri, M. Lindow, S. Obad and S. Kauppinen, Silence, 2012, 3, 1 CrossRef CAS PubMed.
- G. G. Jayaraj, S. Nahar and S. Maiti, Chem. Commun., 2015, 51, 820–831 RSC.
- G. Meister, M. Landthaler, Y. Dorsett and T. Tuschl, RNA, 2004, 10, 544–550 CrossRef CAS PubMed.
- J. Krutzfeldt, N. Rajewsky, R. Braich, K. G. Rajeev, T. Tuschl, M. Manoharan and M. Stoffel, Nature, 2005, 438, 685–689 CrossRef PubMed.
- S. Davis, B. Lollo, S. Freier and C. Esau, Nucleic Acids Res., 2006, 34, 2294–2304 CrossRef CAS PubMed.
- S. Nahar, V. Kotikam, V. A. Kumar and S. Maiti, Nucleic Acid Ther., 2016, 26, 327–334 CrossRef CAS PubMed.
- I. K. Guttilla and B. A. White, J. Biol. Chem., 2009, 284, 23204–23216 CrossRef CAS PubMed.
- Z. Fu and D. J. Tindall, Oncogene, 2008, 27, 2312–2319 CrossRef CAS PubMed.
- Y. Lu, J. Xiao, H. Lin, Y. Bai, X. Luo, Z. Wang and B. Yang, Nucleic Acids Res., 2009, 37, e24 CrossRef PubMed.
- A. K. Nayak and U. Subudhi, RSC Adv., 2014, 97, 54506–54511 RSC.
- A. K. Nayak, A. Mishra, B. S. Jena, B. K. Mishra and U. Subudhi, Sci. Rep., 2016, 6, 26855 CrossRef CAS PubMed.
- H. Lee, A. K. Lytton-Jean, Y. Chen, K. T. Love, A. I. Park, E. D. Karagiannis, A. Sehgal, W. Querbes, C. S. Zurenko, M. Jayaraman and C. G. Peng, Nat. Nanotechnol., 2012, 7, 389–393 CrossRef CAS PubMed.
- X. Zhang and V. K. Yadavalli, Nanoscale., 2012, 4, 2439–2446 RSC.
- Y. Lv, R. Peng, Y. Zhou, X. Zhang and W. Tan, Chem. Commun., 2016, 52, 1413–1415 RSC.
- H. Qian, C. Y. Tay, M. I. Setyawati, S. L. Chia, D. S. Lee and D. T. Leong, Chem. Sci., 2017, 8, 1062–1067 RSC.
- R. Q. Li, Y. Ren, W. Liu, W. Pan, F. J. Xu and M. Yang, Nanoscale, 2017, 9, 2521–2530 RSC.
- A. Ora, E. Jarvihaasto, H. Zhang, H. Auvinen, H. A. Santos, M. A. Kostiainen and V. Linko, Chem. Commun., 2016, 98, 14161–14164 RSC.
- J. Hahn, S. F. Wickham, W. M. Shih and S. D. Perrault, ACS Nano, 2014, 8, 8765–8775 CrossRef CAS PubMed.
- K. K. Ho, S. S. Myatt and E. W. Lam, Oncogene, 2008, 27, 2300–2311 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7nr06601e |
| This journal is © The Royal Society of Chemistry 2018 |